
ABSTRACT
Circular RNAs (circRNAs) are broadly identified from precursor mRNA (pre-mRNA) back-splicing across various species. Recent studies have suggested a cell-/tissue- specific manner of circRNA expression. However, the distinct expression pattern of circRNAs among species and its underlying mechanism still remain to be explored. Here, we systematically compared circRNA expression from human and mouse, and found that only a small portion of human circRNAs could be determined in parallel mouse samples. The conserved circRNA expression between human and mouse is correlated with the existence of orientation-opposite complementary sequences in introns that flank back-spliced exons in both species, but not the circRNA sequences themselves. Quantification of RNA pairing capacity of orientation-opposite complementary sequences across circRNA-flanking introns by Complementary Sequence Index (CSI) identifies that among all types of complementary sequences, SINEs, especially Alu elements in human, contribute the most for circRNA formation and that their diverse distribution across species leads to the increased complexity of circRNA expression during species evolution. Together, our integrated and comparative reference catalog of circRNAs in different species reveals a species-specific pattern of circRNA expression and suggests a previously under-appreciated impact of fast-evolved SINEs on the regulation of (circRNA) gene expression.
Introduction
Circular RNAs (circRNAs) formed by back-spliced (circularized) exons were sparsely identified over 20 y ago,Citation1,2 and have been recently re-discovered genomewide in thousands of gene loci by taking advantage of deep sequencing of non-polyadenylated transcriptomes and specific bioinformatic pipelines that identify reads anchoring back-splicing junctions in a reversed order (for reviews, see refsCitation3-5). Although inefficiently catalyzed by the spliceosome and generally expressed at low levels,Citation6-8 circRNA formation can be facilitated by both RNA pairing of orientation-opposite complementary sequences across flanking introns and protein factors that are capable of binding flanking introns to bridge unfavorable back-splice sites to a close proximity presumably.Citation6,9-12
Expression profiling showed diverse expression patterns of circRNAs among cell types/tissues13-17 with a significant enrichment of circRNAs in neurons/brains.Citation8,18-20 Interestingly, a single gene locus can produce multiple circRNAs, referred to as alternative circularization.Citation10 Our recent study further suggests that both alternative back-splice site selection and alternative splicing site selection within circRNAs are involved in alternative circularization and contribute to circRNA complexity.Citation17 The competition of putative RNA pairs across introns that bracket different sets of alternative back-splice sites leads to diverse back-splice site selectionCitation17; whereas the regulation of alternative splicing within circRNAs is largely unknown.
CircRNAs were also found to be expressed across different species.Citation12-16,19 Comparison of circRNAs in several human and mouse data sets revealed that only a small portion of mouse circRNAs were orthologous to those in human,Citation13 suggesting a species-specific manner of circRNA expression. However, a detailed view of this species-specificity of circRNA expression and its underlying mechanism remained largely uncharacterized. Since circRNA expression is associated with orientation-opposite complementary sequences across their flanking introns and that most of these complementary sequences are abundant primate-specific Alu elements in human, we speculate that the evolvement of complementary sequences and their variable distribution in different species may largely contribute to the species-specific expression of circRNAs.
We hereby systematically compared circRNA expression patterns between human and mouse, and found that only a small portion of human circRNAs could be determined in parallel mouse samples and that the majority of circRNAs are species-specifically expressed. Further analysis revealed that the conserved circRNA expression between human and mouse is correlated with the existence of orientation-opposite complementary sequences in introns that flank back-spliced exons in both species, but not the circRNA sequences themselves. Among these complementary sequences, SINEs (short interspersed nuclear repetitive DNA elements), especially Alu elements in human, contribute the most for boosting circRNA formation. The diverse distribution of SINEs across species may lead to the increased complexity of circRNA expression during species evolution.
Results
Comparative analysis of circRNAs from parallel human and mouse data sets
Multiple human and mouse RNA-seq data sets (Table S1) of ribosomal-depleted (Ribo–), non-polyadenylated (poly(A)–) or RNase R-treated poly(A)– (poly(A)–/RNase R) RNAs from a spectrum of tissues were analyzed using CIRCexplorer2 pipelineCitation17 with the aligner of TopHat2/TopHat-Fusion (version 2.0.9, Materials and Methods). Approximately 100,000 and 36,000 circRNAs were identified in examined human or mouse samples, respectively (Tables S2 and S3). In addition, the majority (> 90%) of these identified circRNAs could be accordingly detected in human or mouse brain/neuron tissues ( and S1), consistent with the previous reports that circRNAs were highly enriched in brains/neuronsCitation8,18-20.
Figure 1. Comparison of human and mouse circRNAs. (A) More circRNAs are detected in human than in mouse. The highly-expressed circRNAs with RPM ≥ 0.2 in RNase R-treated samples or RPM ≥ 0.1 in all other samples are marked in dark gray. (B) More circRNAs are detected in human (blue) than in mouse (yellow) after normalized by sequencing depth. (C) The expression level of top 100 circRNAs in human (blue) is higher than those in mouse (yellow). ** p value < 0.01, *** p value < 0.001, Wilcoxon rank-sum test. (D) The expression level of top 100 circRNA cognate mRNAs is similar between human (blue) and mouse (yellow). ** p value < 0.01, *** p value < 0.001, Wilcoxon rank-sum test.
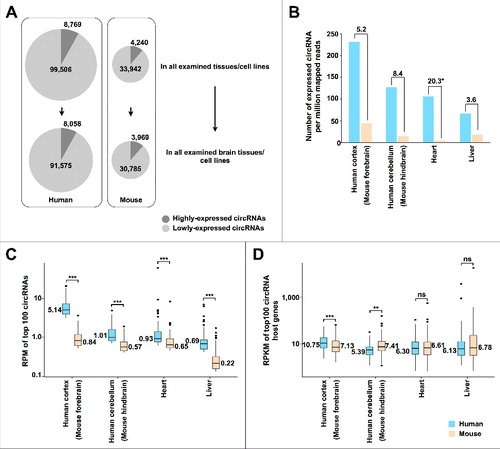
In addition to the tissue-specificity, comparison of circRNA expression in human and mouse samples suggested that the total number of expressed circRNAs is much higher in human than that in mouse samples ( and S1). Strikingly, much more circRNAs could be identified in human than in parallel mouse samples after normalized by sequencing depth (), further indicating the enrichment of expressed circRNAs in human. In addition, the expression levels of the top 100, 500 and 1000 circRNAs in human are also much higher than those in mouse samples ( and S2). Of note, the expression levels of their cognate mRNAs are comparable between human and mouse ( and S2). Together, these results indicate that circRNAs are more prevalently and highly expressed in human than in mouse (Table S2 and S3).
Genomic feature analysis suggested that most circRNAs contain multiple exons, commonly two to three exons, in both human and mouse (Fig. S3A), and their flanking introns are much longer than randomly selected ones (Fig. S3B), as previously reported.Citation10
The majority of circRNAs are species-specifically expressed
We next applied LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver) to identify conserved circRNAs between human and mouse (Materials and Methods). For each human circRNA, on the one hand, if there were an expressed circRNA identified in the mouse orthologous locus, this circRNA could be suggested as a conserved circRNA between human and mouse; on the other hand, if no mouse circRNA ortholog were found, this human circRNA could be suggested as a human-specific one. The same LiftOver analysis was also performed with all identified mouse circRNAs.
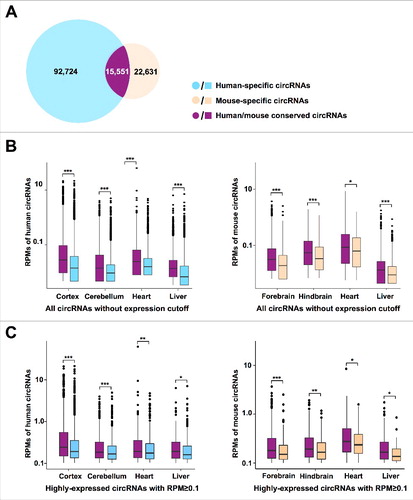
Using this method, about 15,000 of circRNAs could be identified in both human and mouse, representing about 15% or 40% of total circRNAs in human or mouse, respectively (), while the majority (about 85% in human and 60% in mouse) of circRNAs could be only found in one of the two species, indicative of their species-specific expression. Further analysis revealed that the expression level of conserved circRNAs is higher than that of species-specific circRNAs in all examined human and mouse circRNAs ( and S4A). Similar results were obtained by using highly-expressed circRNAs for analysis ( and S4B).
Figure 2. Species-specific expression of circRNAs. (A) A venn diagram shows conserved (purple), human-specific (blue), or mouse-specific (yellow) circRNAs. Of note, due to 5 nt difference to define circRNA orthologs between human and mouse, 15,551 conserved circRNAs were identified in human (Table S2) and 15,517 conserved circRNAs were identified in mouse (Table S3), respectively. (B), (C) The expression level of conserved (purple) circRNAs is higher than species-specific circRNAs in human (blue) and mouse (yellow) when detected in all (B) or highly-expressed (C) circRNAs. * p value < 0.05, *** p value < 0.001, Wilcoxon rank-sum test.
We then asked what factor(s) determines a circRNA to be conserved or species-specific. Genomic feature analysis suggested that the sequence conservation of conserved circRNAs is only slightly higher (PhastCons score: 0.95 vs 0.93) than that of species-specific circRNAs (Fig. S4C). Interestingly, however, flanking introns of conserved circRNAs are much longer than those of species-specific ones (Fig. S4D). It is well known that intronic base pairing across circRNA-flanking introns facilitates circRNA formation,Citation9,10 and therefore it was appealing to speculate that the species-specific expression of circRNAs might be correlated with species-specific base pairing across circRNA-flanking introns.
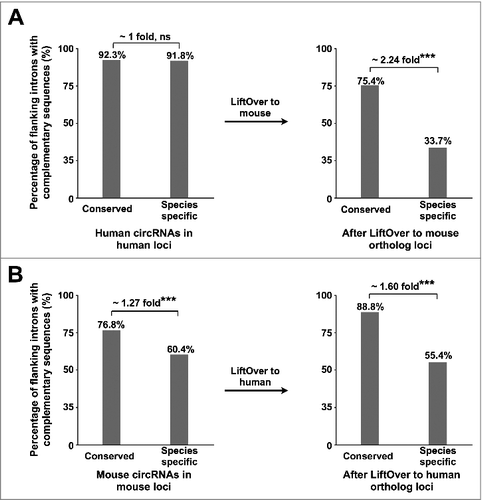
Our analyses showed this speculation is largely true. When compared in human genome context, intronic complementary sequences bracketing either conserve or human-specific circRNAs exhibited no difference (); however, a striking difference was observed after LiftOver human circRNAs to mouse genome: about 75% of conserved circRNA-flanking introns contain orientation-opposite complementary sequences in the mouse genome, while only 34% of human-specific circRNA-flanking introns contain orientation-opposite complementary sequences in the mouse orthologous loci (). Similar observation was also found with the analysis of mouse circRNAs (). This global analysis thus supports the view that conserved circRNAs preferentially contain orientation-opposite complementary sequences across their flanking introns in both human and mouse orthologous loci.
Figure 3. Species-specific distribution of orientation-opposite complementary sequences across circRNA-flanking introns is correlated with species-specific expression of circRNAs. (A) The percentage of conserved or human-specific highly-expressed circRNAs with complementary sequences is calculated in human (left panel) or in their mouse orthologous loci after LiftOver (right panel). *** p value < 0.001, Fisher's exact test. (B) The percentage of conserved or mouse-specific highly-expressed circRNAs with complementary sequences is calculated in mouse (left panel) or in their human orthologous loci after LiftOver (right panel). ***p value < 0.001, Fisher's exact test.
SINEs, especially Alus in the human genome, contribute the most for species-specific circRNA expression
To compare the effect of complementary sequences on circRNA formation, we developed Complementary Sequence Index (CSI) to quantitate RNA pairing capacity of orientation-opposite complementary sequences across circRNA-flanking introns (, and Materials and Methods). In this evaluation, we considered many factors that may affect the RNA pairing formation, including sequence pairing strengths (Blast Score), distances (Symmetry length) and competition with other complementary sequences. A maximum CSI was selected to represent the strongest RNA pairing potential for each given flanking intron set (Materials and Methods). By plotting CSIs of introns flanking randomly-selected 500 highly-expressed circRNAs (with RPM ≥ 0.1 in Ribo– and poly(A)– samples or RPM ≥ 0.2 in poly(A)–/RNase R samples) and those of 500 control flanking introns with (lengths ≥ 8,000 nt, which is the median length of flanking introns of circRNA-producing genes in human) or without intron length limitation, the method of CSI achieved an AUC (area under the curve) as 71.4% or 86.4% ( and S5A), respectively. Here, the AUC of the receiver operating characteristic (ROC) curve of CSI was used to evaluate the specificity and sensitivity of this index. Similar results were obtained by plotting CSIs of 1,000 or 5,000 highly-expressed circRNAs and those of 1,000 or 5,000 control flanking introns with lengths ≥ 8,000 nt (Fig. S5B)
Figure 4. SINEs contribute the most for RNA pairing across circRNA-flanking introns. (A) Complementary Sequence Index (CSI) was developed to quantitate RNA pairing potential of orientation-opposite complementary sequences across circRNA-flanking introns. The complementary sequences are indicated by red arrows. Complementary sequence pairing across circRNA-flanking introns is indicated by red arc. Complementary sequence pairs within circRNA-flanking intron are indicated by blue arc lines. See Materials and Methods for details. (B) ROC (receiver operating characteristic) curve was plotted to evaluate CSI performance and the AUC (area under the curve) of the ROC curve was used to evaluate the specificity and sensitivity of CSI. Flanking introns from 500 randomly-selected highly-expressed circRNAs and control intron pairs with lengths ≥ 8,000 nts from 500 randomly-selected non-circRNA producing genes were plotted. (C) Distribution of different types of complementary sequences that contribute RNA pairing across circRNA-flanking introns in human. Of note, ∼93.3% of the complementary sequences are Alu elements in human. (D) Boxplots of CSI values of different types of complementary sequences in human. *** p value < 0.001, Wilcoxon rank-sum test. (E) Distribution of different types of complementary sequences that contribute RNA pairing across circRNA-flanking introns in mouse. Note that, ∼77.4% of the complementary sequences are B1, B2 or B4 elements in mouse. (F) Boxplots of CSIs of different types of complementary sequences in mouse. *** p value < 0.001, Wilcoxon rank-sum test.
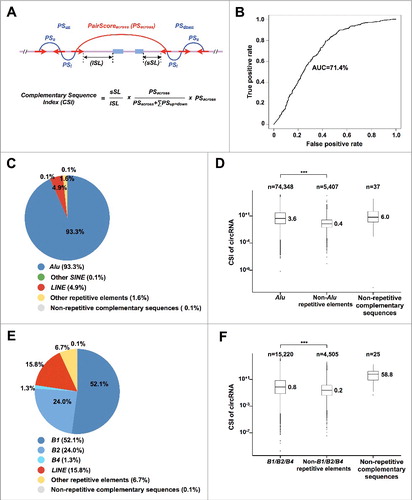
A large number of different types of repetitive elements could potentially form complementary sequences when reversely embedded across flanking introns to facilitate circRNA formation (Fig. S5C). Remarkably, calculation of CSI revealed that about 93.3% of circRNA-flanking introns in human exhibited the strongest RNA pairing capacity from inverted repeated Alu sequences (IRAlus) and only a very small portion from other non-Alu repetitive sequences, such as LINEs (long interspersed nuclear repetitive DNA elements), and other non-repetitive but complementary sequences (). Furthermore, these IRAlus showed much higher CSIs than those by other non-Alu repetitive sequences (). In the mouse context, about 77.4% of circRNA-flanking introns showed the strongest RNA pairing capacity from SINE elements, mostly B1, B2 and B4 (). Similarly, inverted repeated SINE sequences (IRSINEs) showed higher CSIs than those by other non-SINE repetitive sequences in mouse (). Importantly, we found that CSIs of IRAlus in human (the median CSI is 3.6, ) are dramatically higher than those of IRSINEs in mouse (the median CSI is 0.8, ). This observation suggests that the higher pairing capacity of IRAlus in human than that of IRSINEs in mouse may lead to the elevated circRNA expression in human. It was notable that the non-repetitive but complementary sequences showed even higher CSIs in both human and mouse, but they are only sparsely present (37 in human and 25 in mouse, ), indicating that they play at best limited role in circRNA formation.
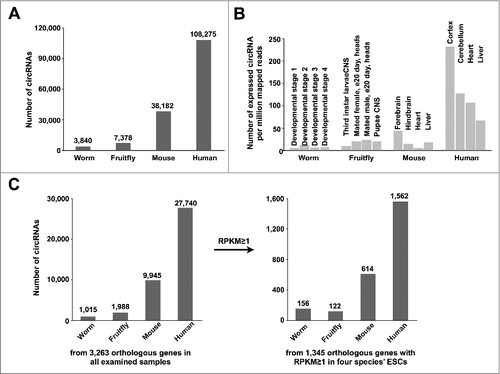
To further examine circRNA expression along species evolution, we extended the comparison of expressed circRNAs in model organisms from human and mouse to fruitfly and worm. As shown in , the total number of circRNAs was significantly increased from worm and fruitfly to mouse and human with currently available samples (Table S1). In addition, much more circRNAs could be identified in human than in mouse, fruitfly and worm samples after normalized by sequencing depths (). Similar result was observed from 3,263 orthologous genes () and 1,345 orthologous genes with their linear mRNA expression at RPKM ≥ 1 in ESCs from all four species ().
Figure 5. Increased circRNA expression during species evolution. (A) Increased numbers of circRNAs could be detected in mouse and human that in worm and fruitfly. (B) More circRNAs could be detected in human than in other species after normalized by sequencing depth. (C) CircRNA numbers are increased during species evolution. 3,263 orthologous genes from all examined samples (left panel) and 1,345 orthologous genes with their linear mRNAs expressed at RPKM ≥ 1 in ESC lines of worm, fruitfly, mouse and human (right panel) were analyzed.
Correlation of the increased complexity of circRNA expression and the accumulated number of SINE elements during species evolution
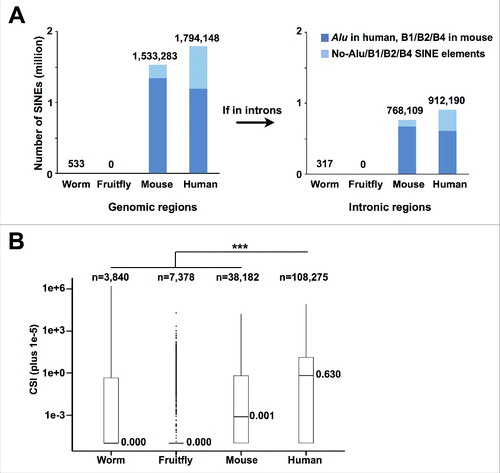
Since SINE elements, especially Alus in human, contribute the most to circRNA formation among all examined complementary sequences in human and mouse (), we suspect that the increasing SINEs (Alus in human) during species evolution () could result in the increased complexity of circRNAs in metazoan. In addition to the dramatic increase of absolute numbers of SINEs in primates, the RNA pairing capacity, which is mainly reflected by SINEs (Alus in human) and represented by CSI, is also significantly increased along with species evolution (). Taken together, these results suggest that the species-specific distribution of SINE elements and their distinct pairing capacity may play an important role in increasing circRNA complexity during species evolution.
Figure 6. Accumulated numbers and increased pairing potential of SINE elements during species evolution. (A) The numbers of SINE (B1, B2 and B4 in mouse or Alu in human) elements in genomic (left panel) and intronic regions (right panel) across worm, fruitfly, mouse and human. Note that the SINE elements are accumulated during species evolution. (B) Boxplots of CSIs of all identified circRNAs in worm, fruitfly, mouse or human, respectively. *** p value < 0.001, Wilcoxon rank-sum test.
Interestingly, we found that alternative circularizationCitation10 is generally more prevalent in examined human orthologs than in other examined species in our analysis (Fig. S6A). For example, 16 circRNAs, mainly produced by alternative back-splicing selection,Citation17 can be found in the human RERE (Arginine-Glutamic Acid Dipeptide Repeats) locus in the human embryonic stem cell H9 line (Fig. S6B); whereas only 9 in mouse, 2 in fruitfly, and none in worm stem cells could be determined in the relevant RERE locus with examined stem cell data sets (Fig. S6B).
Discussion
In addition to canonical splicing that sequentially joins exons to produce linear RNAs with the 5′ to 3′ polarity, split exons in eukaryotes can also be linked by back-splicing, by which downstream 5′ splice (donor) sites are connected with upstream 3′ splice (acceptor) sites in a reversed orientation for circRNA formation (for reviews, see refs.Citation3,5). Recently, a large number of circRNAs have been predicted in a variety of cell lines/tissues and across various species.Citation12-20 In addition, several studies have shown that circRNAs are tissue-specifically expressed, with an enrichment in neuron/brain tissues.Citation18-20 The neuron-enriched circRNA expression is possibly resulted from multiple layers of regulation, including transcription rate, RNA turnover and rate of cell division.Citation8 An early study has suggested a species-specific expression of circRNAs by showing that about 20% of mouse circRNAs are orthologous to those in human.Citation13 In this study, we further confirmed the species-specific expression of circRNAs (, and ) and identified that SINEs, especially Alu elements in human genome, significantly contribute circRNA formation in mammals (, and ). Importantly, we also suggest that the diverse distribution of SINEs across species may lead to the increased complexity of circRNA expression during species evolution ().
Back-splicing can be thought as one specific type of alternative splicing. In this scenario, it is not a surprise to identify the species-specific expression of circRNAs, since alternative splicing is frequently species-specific.Citation21,22 The species-specific expression of circRNAs is correlated with species-specific base pairing across circRNA-flanking introns. With a quantitative score by CSI, we showed that the primate-specific Alu elements generally contribute to circRNA formation ( and ). Correspondingly, IRSINEs play the most important role in mouse circRNA formation (), although much weaker than IRAlus. Extended analysis suggested that the complexity of circRNA expression is significantly increased along species evolution, which is highly correlated with the accumulated SINE elements (Alus in human) and their enhanced pairing capacity from worm and fruitfly to mouse and human ( and ). Thus, the lack of strong complementary sequences, such as IRAlus and IRSINEs, in lower species may lead to the observation that flanking intronic complementarity may not be a critical feature for circRNA formation in fruitfly.Citation19
Apparently, circRNA expression among species is also regulated by other factors. For example, binding of RBPs to circRNA-flanking introns could affect back-splicing;Citation6,11 so it is possible that differentially expression of RBPs among species may lead to species-specific circRNA expression. Furthermore, extended flanking intron lengths, but not complementary sequences, may function as a major mechanistic determinant for circRNA formation in fruitflyCitation19; thus different intron lengths across species may affect circRNA formation in distinct species. Moreover, circRNA formation is influenced by transcription speed of circRNA-producing genes and fast-transcribed genes tend to produce more circRNAs.Citation8 Finally, although inefficiently back-spliced,Citation6-8 circRNAs are resistant to exonucleolytic degradation due to their covalently closed circle structure,Citation8 which allows their accumulation to relatively high levels over time.Citation8,19,23 In this case, the elevated circRNA expression in human could be also in part explained by the fact that human tissues are significantly older than samples from other species. Taken together, RNA complementarity determined by CSI may play a limited role in the evaluation of circRNA expression among different species. Although failed to draw a positive correlation between CSIs and circRNA expression, we observed that CSIs of highly-expressed circRNAs are much higher than those of lowly-expressed ones in both human and mouse (Fig. S6C).
Collectively, the complexity of circRNA expression is remarkably increased along with accumulated SINE elements (Alus in human) during species evolution, suggesting a regulatory role of fast-evolved SINEs (especially Alus in human) in (circRNA) gene expression. However, it is also possible that some of bioinfomatically-identified circRNAs could be possibly generated as splicing artifacts in gene loci with complementary SINEs without any evolutionary advantage.
Materials and methods
Identification of back-splicing junction reads from multiple RNA-seq databases
A number of RNA-seq data sets, including samples from multiple tissues and cell lines across different species (Table S1), were applied to retrieve back-splicing junction reads for circRNA prediction using CIRCexplorer2 pipeline.Citation17 Briefly, RNA-seq reads from each sample were mapped by TopHat2 (2.0.9; parameters: -a 6 -g 1 –microexon-search -m 2) against GRCh37/hg19 human reference genome, GRCm38/mm10 mouse reference genome, BDGP R5/dm3 fruitfly reference genome and WS220/ce10 worm reference genome with known gene annotations (human: knownGene.txt updated at 2013/06/30, mouse: knownGene.txt updated at 2015/06/01, fruitfly: refFlat.txt updated at 2015/11/22, worm: refFlat.txt updated at 2013/03/18), respectively. The unmapped reads were then mapped to the relevant reference genome using TopHat-Fusion, and back-splicing junction reads were further retrieved to determine the back-splicing sites according to known gene annotations (human: knownGene.txt updated at 2013/06/30, refFlat.txt updated at 2013/10/13 and ensGene.txt updated at 2014/04/06; mouse: knownGene.txt updated at 2015/06/01, refFlat.txt updated at 2015/07/29 and ensGene.txt updated at 2014/04/06; fruitfly: refFlat.txt updated at 2015/11/22, ensGene.txt updated at 2014/04/06; worm: refFlat.txt updated at 2013/03/18, ensGene.txt updated at 2012/01/05). Finally, RPM (Reads Per Million mapped reads) was calculated to quantitate circRNA expression as previously reported.Citation10 Highly-expressed circRNAs were defined by RPM ≥ 0.1 in poly(A)– or Ribo– samples or RPM ≥ 0.2 in poly(A)–/RNase R samples from at least one sample for each species.
Genomic feature of circRNA-flanking introns
Sequences of circRNA-flanking introns were extracted from known gene annotations, described as above. The length distribution of all or highly-expressed circRNA-flanking introns was individually analyzed. Control introns were randomly selected from 5,000 intron pairs with known gene annotations in human or mouse.
Conservation analysis of circRNAs between human and mouse
Sequences of back-spliced exons were extracted from known gene annotations, described as above. To decipher conserved circRNAs between human and mouse, LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver) was used to identify orthologous coordinates between human and mouse (parameters: -bedPlus = 3 -tab -minMatch = 0.1 -minBlocks = 1). For each human circRNA, on the one hand, if there were an expressed circRNA identified in mouse orthologous locus within 5nt difference, this circRNA was suggested as a conserved circRNA between human and mouse; on the other hand, if no mouse circRNA ortholog were found, this human circRNA was suggested as a human-specific one. Similar strategy was also applied to mouse circRNAs. To examine the sequence conservation, PhastCons scores of circRNA-producing exons and their flanking introns were retrieved from UCSC and plotted for comparison.
Complementary sequence analysis
Complementary sequences of circRNA-flanking introns were detected by BLASTn (parameters: -word_size 11 -gapopen 5 -gapextend 2 -penalty -3 -reward 2 -outfmt 6 -strand minus). Orientation-opposite paired sequences with at least 50 nts on each side of circRNA flanking introns in human and mouse or 20 nts on each side of circRNA flanking introns in fruitfly and worm were defined as complementary sequences.
Complementary Sequence Index (CSI)
CSI was used to quantitate RNA pairing capacity of each orientation-opposite complementary sequence pair across circRNA-flanking introns. Three basic elements including symmetry length between the complementary sequences and their proximal back-splicing sites, pairing ability detected by Blast and competition of this paired complementary sequences with other complementary sequences, were all considered in the CSI estimation.
Symmetry length
Symmetry length (SL) is the distance from one side of complementary sequence pairs to its proximal back-splicing site. There are two SLs for each complementary sequence pair, a short one (sSL) and a longer one(lSL).
Pairing ability
The pairing ability of each given orientation-opposite complementary sequence pair across circRNA-flanking introns is determined by Pair Score (PSacross) that is quantitated by BlastScore/L2. Here, “L” is the distance between the pair of orientation-opposite complementary sequences, excluding the distances between back-splicing sites, which equals to “sSL+lSL.”
Competition
For a given orientation-opposite complementary sequence pair across circRNA-flanking introns, it could also be paired with suppressing complementary sequences within the same individual introns to inhibit (PSi) its pairing across circRNA-flanking introns. At the same time, the suppressing complementary sequences could also be competed by other sequences to enhance (PSe) the RNA pairing potential of this given orientation-opposite complementary sequence pair across circRNA-flanking introns. Competition of the upstream circRNA-flanking intron of this given orientation-opposite complementary sequence pair is calculated as PSup = ∑((PSi/(PSi+∑(PSe))xPSi), and competition in the downstream circRNA-flanking intron is accordingly determined as PSdown. Taken together, the competition of this given orientation-opposite complementary sequence pair from their hosting introns is summed up as PSup+down = PSup+PSdown. Thus, the potential complementarity of each given orientation-opposite complementary sequence pair across circRNA-flanking introns was competed by other pairing and could be calculated as PSacross/(PSacross+PSup+down).
Finally, the CSI of each given orientation-opposite complementary sequence pair is calculated by aggregation of all these factors and shown in . A maximum CSI was obtained from a strongest pair of orientation-opposite complementary sequences in each circRNA-flanking intron and was used to represent the strongest RNA pairing potential to enhance the relevant circRNA formation. The source codes of CSI will be accessed from https://github.com/YangLab/CSI.
CSI performance evaluation
To evaluate CSI performance, R software and the pROC R libraryCitation24 were used for ROC curve construction and AUC estimation. Flanking introns from 500 randomly-selected highly-expressed circRNAs and control intron pairs from 500 randomly-selected non-circRNA producing genes (: intron length ≥ 8,000 nts; Fig. S5: intron length ≥ 0 nt) were chosen for ROC curve construction. AUC of the ROC curve was calculated by pROC R library. The AUC of the ROC curve of CSI was used to evaluate the specificity and sensitivity of this index.
Repetitive element distribution in different species
Orientation-opposite complementary sequence pairs with maximum CSIs were overlapped with known repetitive element annotation (human: rmsk.txt updated at 2009/02/27; mouse: rmsk.txt updated at 2012/05/07; fruitfly: rmsk.txt updated at 2007/07/11; worm: rmsk.txt updated at 2012/01/05) to further categorize the types of complementary sequences that contribute to RNA pairing (). Basically, IRAlus in human and IRSINEs in mouse play the most important role in circRNA formation. The distribution of different types of repetitive elements was calculated in both genomic and intronic regions (Fig. S5B and 5D).
Orthologous genes among worm, fruitfly, mouse and human
In total, 3,263 orthologous genes from worm, fruitfly, mouse and human were retrieved from OrthoDB.Citation25 Among them, 1,345 of orthologous genes have their linear mRNAs expressed at RPKM ≥ 1 in ESCs from all four species (H9 in human, R1 in mouse, S2 in fruitfly and N2 in worm). Accordingly, the total numbers of circRNAs generated from 3,263 or 1,345 orthologous genes were calculated from all samples or the ESCs in these four organisms. The numbers of circRNAs from 3,263 or 1,345 orthologous genes were individually plotted by Cluster (version 3.0) and shown by Java Treeview (version 1.1.6r4).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
L.Y. and L.-L.C. conceived and designed the project. R.D. and X.-K.M. performed the bioinformatic analysis. L.Y. and L.-L.C. wrote the paper with contributions from coauthors. L.Y. supervised the project.
Supplemental_Data.zip
Download Zip (31.6 MB)Acknowledgment
We are grateful to laboratory members for discussion.
Funding
This work is supported by grants 2014CB910601 from MOST and 91540115, 91440202, 31271390 and 31471241 from NSFC.
Related Research Data
References
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell 1991; 64(3):607-13; PMID:1991322; https://doi.org/10.1016/0092-8674(91)90244-S
- Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993; 73(5):1019-30; PMID:7684656; https://doi.org/10.1016/0092-8674(93)90279-Y
- Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol 2015; 12(4):381-8; PMID:25746834; https://doi.org/10.1080/15476286.2015.1020271
- Yang L. Splicing noncoding RNAs from the inside out. WIREs RNA 2015; 6(6):651-60; PMID:26424453; https://doi.org/10.1002/wrna.1307
- Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Bio 2016; 17:205-11; PMID:26908011; https://doi.org/10.1038/nrm.2015.32
- Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56(1):55-66; PMID:25242144; https://doi.org/10.1016/j.molcel.2014.08.019
- Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A. Exon circularization requires canonical splice signals. Cell Rep 2015; 10(1):103-11; PMID:25543144; https://doi.org/10.1016/j.celrep.2014.12.002
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL. The Biogenesis of Nascent Circular RNAs. Cell Rep 2016; 15(3):611-24; PMID:27068474; https://doi.org/10.1016/j.celrep.2016.03.058
- Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Gene Dev 2014; 28(20):2233-47; PMID:25281217; https://doi.org/10.1101/gad.251926.114
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell 2014; 159(1):134-47; PMID:25242744; https://doi.org/10.1016/j.cell.2014.09.001
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015; 160(6):1125-34; PMID:25768908; https://doi.org/10.1016/j.cell.2015.02.014
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 2015; 10(2):170-7; PMID:25558066; https://doi.org/10.1016/j.celrep.2014.12.019
- Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15(7):409; PMID:25070500; https://doi.org/10.1186/s13059-014-0409-z
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna 2013; 19(2):141-57; PMID:23249747; https://doi.org/10.1261/rna.035667.112
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495(7441):333-8; PMID:23446348; https://doi.org/10.1038/nature11928
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet 2013; 9(9):e1003777; PMID:24039610; https://doi.org/10.1371/journal.pgen.1003777
- Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, Chen LL, Yang L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 2016; 26(9):1277-87; PMID:27365365; https://doi.org/10.1101/gr.202895.115
- Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell 2015; 58(5):870-85; PMID:25921068; https://doi.org/10.1016/j.molcel.2015.03.027
- Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014; 9(5):1966-80; PMID:25544350; https://doi.org/10.1016/j.celrep.2014.10.062
- You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015; 18:603-10; PMID:25714049; https://doi.org/10.1038/nn.3975
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012; 338(6114):1587-93; PMID:23258890; https://doi.org/10.1126/science.1230612
- Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012; 338(6114):1593-9; PMID:23258891; https://doi.org/10.1126/science.1228186
- Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 2015; 5:8057; PMID:25624062; https://doi.org/10.1038/srep08057
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics 2011; 12(1):1; PMID:21199577; https://doi.org/10.1186/1471-2105-12-77
- Kriventseva EV, Tegenfeldt F, Petty TJ, Waterhouse RM, Simao FA, Pozdnyakov IA, Ioannidis P, Zdobnov EM. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res 2015; 43(D1):D250-6; PMID:25428351; https://doi.org/10.1093/nar/gku1220