
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Comparison of mRNA and protein structures shows that highly structured mRNAs typically encode compact protein domains suggesting that mRNA structure controls protein folding. This function is apparently performed by distinct structural elements in the mRNA, which implies ‘fine tuning’ of mRNA structure under selection for optimal protein folding. We find that, during evolution, changes in the mRNA folding energy follow amino acid replacements, reinforcing the notion of an intimate connection between the structures of a mRNA and the protein it encodes, and the double encoding of protein sequence and folding in the mRNA.
Introduction
Translation is the universal process of decoding the codon sequence in a mRNA into the amino acid sequence of a protein. The rate of translation however, can greatly vary depending on environmental conditions,Citation1-3 development states,Citation4 between species,Citation5,6 and between genes within the same organism.Citation6 Even along a particular mRNA molecule, the speed of translation changes depending on codon usage,Citation7,8 RNA secondary structureCitation9-11 and the distribution of charged amino acid residuesCitation12-15 in the produced protein. Translation rate is critical for the fitness of a cell, as one of the principal factors controlling protein production.Citation4 In particular, the slow 5′-terminal ‘ramp’ that is present in most mRNAs prevents ribosomal jammingCitation16-18 and thus serves to minimize the cost of protein synthesis. Variation of the translation rate also seems to contribute to protein quality control: under stress, translation slows down, minimizing protein misfolding.Citation1,19
Secondary structure of mRNA affects translation in more than one way. Highly stable structures including pseudoknots and long hairpins stall the ribosomeCitation10,20-22 and can trigger frameshiftCitation23 or abortCitation10 of translation. Conversely, specific synonymous mutations have been shown to cause protein misfolding.Citation21,24-26 Thus, modulation of the translation rate appears to regulate protein folding such that acceleration or deceleration of translation at particular sites can direct the nascent protein along dufferent folding pathways. Recent experimental evidence and analysis of mRNA structure and evolutionary conservation in different codon positions suggest that there is a tradeoff between selective pressure acting at the RNA and protein levels.Citation6,27,28
To gain insight into the role of mRNA structure in the control of protein folding, we recently investigated in detail the connections between the predicted secondary structure of mRNA and the structural features of its protein product for many diverse organisms.Citation29 Here, we further discuss the results of this analysis and focus on the selection pressure on mRNA that appears to stem from the mRNA structure encoding information on protein folding. We then show that conservation of mRNA energy profiles between orthologs inversely correlates with sequence divergence. The dependency of the conservation of mRNA structure on the non-synonymous substitution rate is about equally strong in bacteria and eukaryotes, whereas the dependency on synonymous substitution rate (dS) is significantly stronger in bacteria. This finding seems to indicate that amino acid replacements affecting protein folding trigger adjustment of mRNA structure, and in bacteria, certain synonymous codon changes exert a comparable effect.
mRNA structure encodes protein structure
A comparison of mRNA structures, inferred from mRNA folding energy estimates, to the protein structures, represented by measures of compactness extracted from crystal structures, revealed a strong negative correlation in both prokaryotes and eukaryotes. Thus, mRNAs containing stable local structures typically encode compact proteins ().Citation29
Figure 1. Model of mRNA structure controlling protein compactness. A: ribosome spends more time to unwind structured mRNA, leaving time for the nascent peptide to co-translationally fold into a compact conformation. B: Protein or domain linkers are encoded by either unstable mRNA or stable mRNA. In the latter case, stable structural elements in mRNA can slow down translation, leaving time for the linker to be targeted for post-translational modifications.
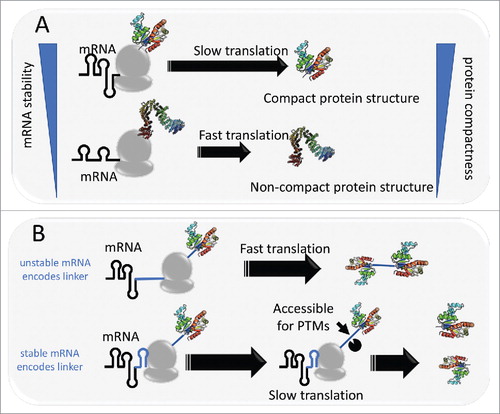
By dividing proteins into ordered and disordered parts,Citation30 we found that this correlation was effectively limited to the ordered regionsCitation29 (). This observation implies a direct link between mRNA structure and protein folding. Stable local structures in the mRNA can cause translational pausing.Citation10 Given that protein folding is currently thought to occur mostly co-translationally,Citation31 we proposed a model, in which structural elements in mRNA act as controlling devices for protein folding. Under this model, when translation slows down at stable structures in mRNA, the nascent protein has extra time to optimize folding by sampling the available configurations that involve contacts with the already synthesized portion of the polypeptide chain. Such a mechanism is expected to be particularly important for compact proteins with high contact density. Notably, the folding energy of the most stable structural element in an mRNA correlates with protein compactness considerably stronger than the mean folding energy, suggesting that mRNAs contain ‘special devices’ for regulation of protein folding. This model is compatible with numerous previous findings showing that slowdown of translation minimizes protein misfolding.Citation1,19
However, protein folding is more complicated than implied by this simple rule. It has been demonstrated that, for certain protein domains, acceleration of translation actually increases the probability of correct folding.Citation32 Conceivably, at high translation rate, the nascent peptide chain is less likely to follow the folding path of minimal local energy so that long-distance contacts can form. A comparison of protein contact order, i.e. the mean sequential distance between contacting amino acid residues,Citation33 and mRNA folding energy showed a positive correlation between contact order and mRNA folding energy (Spearman rank correlations from 0.29 to 0.33 in Prokaryotes (E. coli, B. subtillis, T. gammatolerans), and 0.12 to 0.20 in Eukaryotes (H. sapiens, S. ceverisiae)). Thus, proteins with many long-distance contacts are typically encoded by less stable mRNA. This observation reconciles our model, in which structural elements in mRNA control protein folding, with the results of O'Brien et al..Citation32
Furthermore, some proteins exhibit ‘paradoxical’ relationships between the RNA and protein structures. Watts et al.Citation34 have detected recurring stable elements of RNA structure in regions that encode linkers between domains in the HIV polyprotein. These findings seem to be best compatible with a model of post-translational domain folding and deviate from the general trend observed in our analysisCitation29 whereby disordered parts of proteins that are mostly linkers and terminal regionsCitation30 are not associated with any pattern of mRNA stability. It seems likely that, in the case of HIV, stable RNA structures in linker-encoding regions could be selected to facilitate co-translational polyprotein cleavage by slowing down translation around the cleavage sitesCitation22 (). Such a mechanism of pausing on disordered parts of protens for functional rather than structural reasons might not be limited to HIV and could facilitate polyprotein cleavage in other viruses as well as post-translational modification that typically occurs in disordered regions of proteins (). Validation of this hypothesis by computational and experimental methods could be a promising research direction.
To identify and examine such cases within our data set, we extracted mRNAs and their repective products that appeared to form a distinct, minor cloud of points in the plots of mRNA folding energy vs protein solvent accessibility for S. cereivisiae and H. sapiens, the 2 organisms for which the weakest correlations have been observedCitation29 (see supplementary Figure 1 and supplementary File 1). The distribution of the points on the plot is distinctly asymmetric: most of them feature a stable mRNA (ΔG < −22 kcal/mol) encoding a protein with high solvent accessibility ACC (>50) whereas there are few if any deviations in the other direction, i.e., low-stability mRNAs encoding compact proteins. These poorly structured proteins encoded by stable mRNAs include ribosomal proteins, histones, and subunits of various large complexes. Typically, these proteins co-crystallize with their interaction partners and (supplementary File 1) and conceivably also co-fold with them co-translationally. Such co-translational assembly of protein complexesCitation35,36 might require slow translation similarly to co-translational folding. Of particular interest is the presence, in the list of outliers, of several ribosomal proteins which might co-translationally interact with the rRNA. Experimental validation of this hypothesis could be a promising research direction. Deceleration of translation by secondary structure elements in mRNA potentially might be associated also with non-structured inter-domain linkers and PTM. We noted also that most of the proteins in the group have been reported to encompass PTMs (supplementary File 1) which seems to be copmpatibel with our hypothesis.
Combining these experimental dataCitation34 with molecular evolution analyses, Sanjuan and BorderiaCitation37 demonstrated that amino-acid sites encoded by structured regions of mRNA consistently evolve slower than those for which RNA-level selection appears to be absent. These observations suggest a trade-off between selection pressures at the RNA and protein levels,Citation27,37,38 whereby the ability of the mRNA to form stable structures is restricted by selection at the protein level.
Selection pressure on mRNA stability
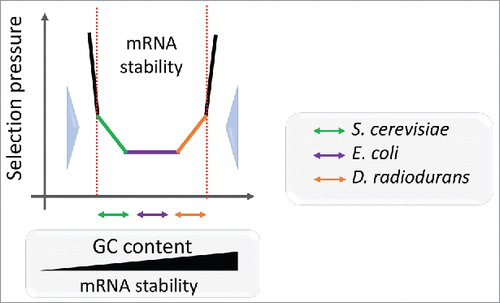
Obviously, the stability of mRNAs varies greatly with the GC content, i.e., GC-rich mRNAs are substantially more stable than AU-rich ones.Citation6,16 The GC content is largely uniform among the genes within the same genome, constraining mRNAs to evolve within a relatively narrow range of stability.Citation27,39 To explore the selection pressure on the mRNA energy landscape, we compared the estimated mRNA stability (ΔG) to the rates of synonymous (dS) and non-synonymous (dN) subsitutions in the respective genes.Citation29 For GC-rich genomes, such as Deinococcus radiodurans, a positive correlation between ΔG and evolutionary rates implies that mRNAs evolve under selection pressure against excessive stability that would impede translation (orange arrow in ). In contrast, in AU-rich species, e.g. Saccharomyces cerevisiae, mRNA seems to evolve under pressure to increase stability, which provides protection from nucleases as well as means for regulation of translation as discussed above (green arrow in ). These findings are in agreement with previous results on folding of mammalian transcriptomes,Citation16,40 which indicate that selection favors optimally stable and ordered mRNA secondary structures rather than the most stable or most relaxed structures.Citation16,27
Figure 2. Schematic representation of the mRNA stability landscape depending on GC content and selective pressure. The GC content defines the mRNA stability range for each species. Double arrows depict correlations between mRNA folding energy and dS for S. cerevisiae (orange), E. coli (violet) and D. radiodurans (green) transcripts. Large blue arrows show selection pressure on mRNA stability.
Information encoding protein folding within mRNA is conserved between orthologs
Because mRNA structure appears to control protein folding and protein folds are conserved between orthologs, information encoding protein folding within mRNA structure also can be expected to be conserved. To test this hypothesis, we aligned the mRNA energy profiles (i.e., ΔG for all 30 nucleotides windows; (see Ref. Citation29 for details) between orthologs from 2 bacteria, E. coli and S. typhi, and 2 eukaryotes, H. sapiens and M. musculus, using codon alignments as guides. For energy profile alignments for each pair of orthologous mRNAs, the profile energy distance (ΔΔG) was estimated as the mean difference between local energies estimated for each position of the alignment:
where ΔGsp1,i is local folding energy of the mRNA from species 1 at position i in the alignment, and ΔGsp2,i is the local folding energy for species 2 at the same codon position. The absolute difference between ΔGsp1,i and ΔGsp2,i was estimated only for ungapped positions of the alignment, where local energies are available for both codons. The sum of all differences is normalized by N, the number of codons aligned without gap. We then compared the ΔΔG for each pair of orthologs to the synonymous (dS) and non-synomyous evolutionary rates (dN) estimated from the same codon alignments. We observed a moderate but statistically significant positive correlation () between ΔΔG and both evolutionary rates (dN and dS) in bacteria and eukaryotes, indicating that mRNA energy profiles vary almost precisely linearly with dS and dN. This correlation is independent of differences in GC content () between orthologs and the mRNA size, 2 compounding variables for mRNA folding energy.Citation29 Notably, in bacteria, the slopes of the linear regression on ΔΔG were nearly identical for dN and dS, whereas in eukaryotes, the slope for dN was about the same as in bacteria, but that for dS was significantly smaller. Thus, in eukaryotes (at least in mammals), mRNA energy profiles between orthologs decay significantly faster with protein divergence compared with the mRNA divergence. Chursov et al.Citation41 have previously reported that mRNA structure distances between paralogous transcripts in yeast co-vary with the RNA sequence until a plateau of 85% sequence identity is reached, below which mRNA structure distance and sequence similarity are not correlated anymore; this observation suggests that below 85% sequence identity, dN and dS co-vary differently with the mRNA structure distance. Amino-acid substitutions can impact protein folding directly,Citation42,43 so we hypothesize that some of these substitutions require different translation pausing patterns to adjust the folding path of the protein, thus driving the evolution of the mRNA folding profiles. The stronger dependency of the mRNA structure on dS in bacteria implies that, due to the generally stronger selection in bacterial, compared with eukaryotic populations,Citation44 even some synonymous codon replacements could affect protein folding and drive adjustment of the mRNA structure.
Figure 3. Correlation between energy profile distance (ΔΔG) between orthologous mRNAs and evolutionary rates (estimated using PAML47) as synonymous and non-synonymous sites (dS and dN, respectively, shown on the logarithmic scale). The plots include 2,638 pairs of orthologs from E. coli and S. typhi, and 6,783 pairs of orthologs from H. sapiens and M. musculus. The sets of orthologous genes are from .29 Spearman rank correlation coefficients (SPR) between profile energy distance and evolulationary rates (dS in blue, dN in red) are significant (p < 0.005) as indicated in the corner. Slopes of the linear regression (S) are indicated next to the trend line.
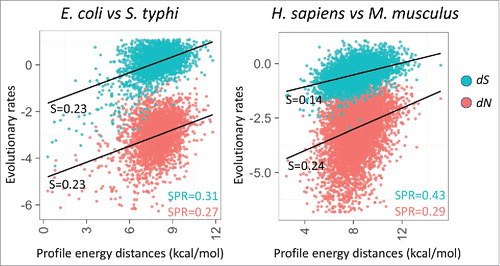
Table 1. Statistical analysis of the connections between mRNA energy profile distance (ΔΔG) and evolutionary rates (dS and dN).
Conclusions and future directions
Our comparison of mRNA and protein structures, along with a substantial body of experimental data, indicates that one of the important roles of mRNA structure is regulation of protein folding. The correlation of the RNA folding energy with protein compactness implies that stable structures in mRNA increase the probability of correct protein fold formation by allowing extra time for folding. Moreover, this function appears to be performed by distinct structural elements in the mRNA, suggesting that selection ‘fine tunes’ mRNA structure to modulate protein folding at critical junctures. Additional findings reported here reinforce this notion of mRNA structure fine tuning by showing that changes in the folding energy during evolution tend to follow amino-acid replacements, and especially in bacteria, also synonymous codon changes.Regulatory effects of mRNA structure are unlikely to be limited to modulation of protein folding. For instance, co-translational assembly, although intimately linked to protein folding, is likely to be a distinct phenomenon as suggested by the current analysis of outliers in the present analysis, i.e., non-compact proteins encoded by stable mRNAs. Furthermore, in some proteins, highly structured regions in mRNA could encode disordered segments, such as interdomain linkers, possibly, under selection pressure to slow down translation and facilitate protein cleavage or post-translational modification.
The partial encoding of protein folding in the mRNA sequence and structure is part of the general theme of multiple, overlapping molecular codes in nucleotide sequences.Citation45,46 Under the concept developed here, certain regions in mRNA can encode information on both the protein sequence and the folding of a different part of the protein (encoded upstream of a structural element). Purifying selection pressure on structural elements in mRNAs would bring about constraints not only on codon choice but also on the amino acid sequence. Thus, evolution of some segments of protein sequences is likely to be governed by factors unrelated to the actual structure or function of the given protein. Furthermore, the finding reported here and previously,Citation29 that the minimum free energy of a 30 bp window correlates better with protein compactness than the mean free energy, could be construed as an evolutionary strategy to restrict the constraints imposed by the folding code to relatively short sequence segments. Comprehensive investigation of the functions and evolution of structural elements in mRNAs can be expected to enrich the existing understanding of the factors that shape protein structure and function during translation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Materials.zip
Download Zip (285 KB)Acknowledgments
We thank Koonin group members for useful discussions.
Funding
Intramural funds of the US Department of Health and Human Services (to the National Library of Medicine).
Related Research Data
References
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. Rate-limiting steps in yeast protein translation. Cell 2013; 153:1589-601; PMID:23791185; https://doi.org/10.1016/j.cell.2013.05.049
- Kortmann J, Narberhaus F. Bacterial RNA thermometers: Molecular zippers and switches. Nat Rev Microbiol 2012; 10:255-65; PMID:22421878; https://doi.org/10.1038/nrmicro2730
- Subramaniam AR, Zid BM, O'Shea EK. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 2014; 159:1200-11; PMID:25416955; https://doi.org/10.1016/j.cell.2014.10.043
- Klumpp S, Scott M, Pedersen S, Hwa T. Molecular crowding limits translation and cell growth. Proc Natl Acad Sci U S A 2013; 110:16754-59; PMID:24082144; https://doi.org/10.1073/pnas.1310377110
- Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res 2014; 42:9171-81; PMID:25056313; https://doi.org/10.1093/nar/gku646
- Gorochowski TE, Ignatova Z, Bovenberg RA, Roubos JA. Trade-offs between tRNA abundance and mRNA secondary structure support smoothing of translation elongation rate. Nucleic Acids Res 2015; 43:3022-32; PMID:25765653; https://doi.org/10.1093/nar/gkv199
- Fredrick K, Ibba M. How the sequence of a gene can tune its translation. Cell 2010; 141:227-9; PMID:20403320; https://doi.org/10.1016/j.cell.2010.03.033
- Novoa EM, Ribas de Pouplana L. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet 2012; 28:574-81; PMID:22921354; https://doi.org/10.1016/j.tig.2012.07.006
- Meyer IM, Miklos I. Statistical evidence for conserved, local secondary structure in the coding regions of eukaryotic mRNAs and pre-mRNAs. Nucleic Acids Res 2005; 33:6338-48; PMID:16275783; https://doi.org/10.1093/nar/gki923
- Somogyi P, Jenner AJ, Brierley I, Inglis SC. Ribosomal pausing during translation of an RNA pseudoknot. Mol Cell Biol 1993; 13:6931-40; PMID:8413285; https://doi.org/10.1128/MCB.13.11.6931
- Dana A, Tuller T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLoS Comput Biol 2012; 8:e1002755; PMID:23133360; https://doi.org/10.1371/journal.pcbi.1002755
- Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol 2013; 11:e1001508; PMID:23554576; https://doi.org/10.1371/journal.pbio.1001508
- Artieri CG, Fraser HB. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res 2014; 24:2011-21; PMID:25294246; https://doi.org/10.1101/gr.175893.114
- Wilson DN, Arenz S, Beckmann R. Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr Opin Struct Biol 2016; 37:123-33; PMID:26859868; https://doi.org/10.1016/j.sbi.2016.01.008
- Ito K, Chiba S. Arrest peptides: Cis-acting modulators of translation. Annu Rev Biochem 2013; 82:171-202; PMID:23746254; https://doi.org/10.1146/annurev-biochem-080211-105026
- Shabalina SA, Ogurtsov AY, Spiridonov NA. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res 2006; 34:2428-37; PMID:16682450; https://doi.org/10.1093/nar/gkl287
- Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol 2010; 6:e1000664; PMID:20140241; https://doi.org/10.1371/journal.pcbi.1000664
- Tuller T, Zur H. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res 2015; 43:13-28; PMID:25505165; https://doi.org/10.1093/nar/gku1313
- Wolin SL, Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J 1988; 7:3559-69; PMID:2850168
- Ignatova Z, Narberhaus F. Systematic probing of the bacterial RNA structurome to reveal new functions. Curr Opin Microbiol 2017; 36:14-19; PMID:28160611; https://doi.org/10.1016/j.mib.2017.01.003
- Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 2006; 314:1930-3; PMID:17185601; https://doi.org/10.1126/science.1131262
- Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 2010; 329:1534-37; PMID:20847274; https://doi.org/10.1126/science.1191701
- Lopinski JD, Dinman JD, Bruenn JA. Kinetics of ribosomal pausing during programmed -1 translational frameshifting. Mol Cell Biol 2000; 20:1095-103; PMID:10648594; https://doi.org/10.1128/MCB.20.4.1095-1103.2000
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007; 315:525-8; PMID:17185560; https://doi.org/10.1126/science.1135308
- Czech A, Fedyunin I, Zhang G, Ignatova Z. Silent mutations in sight: Co-variations in tRNA abundance as a key to unravel consequences of silent mutations. Mol Biosyst 2010; 6:1767-72; PMID:20617253; https://doi.org/10.1039/c004796c
- Agashe D, Martinez-Gomez NC, Drummond DA, Marx CJ. Good codons, bad transcript: Large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. Mol Biol Evol 2013; 30:549-60; PMID:23223712; https://doi.org/10.1093/molbev/mss273
- Shabalina SA, Spiridonov NA, Kashina A. Sounds of silence: Synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res 2013; 41:2073-94; PMID:23293005; https://doi.org/10.1093/nar/gks1205
- Shabalina SA, Ogurtsov AY, Spiridonov NA, Koonin EV. Evolution at protein ends: Major contribution of alternative transcription initiation and termination to the transcriptome and proteome diversity in mammals. Nucleic Acids Res 2014; 42:7132-44; PMID:24792168; https://doi.org/10.1093/nar/gku342
- Faure G, Ogurtsov AY, Shabalina SA, Koonin EV. Role of mRNA structure in the control of protein folding. Nucleic Acids Res 2016; 44:10898-911; PMID:27466388; https://doi.org/10.1093/nar/gkw671
- Faure G, Callebaut I. Comprehensive repertoire of foldable regions within whole genomes. PLoS Comput Biol 2013; 9:e1003280; PMID:24204229; https://doi.org/10.1371/journal.pcbi.1003280
- Thommen M, Holtkamp W, Rodnina MV. Co-translational protein folding: Progress and methods. Curr Opin Struct Biol 2017; 42:83-89; PMID:27940242; https://doi.org/10.1016/j.sbi.2016.11.020
- O'Brien EP, Vendruscolo M, Dobson CM. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat Commun 2014; 5:2988; PMID:24394622; https://doi.org/10.1038/ncomms3988
- Bonneau R, Ruczinski I, Tsai J, Baker D. Contact order and ab initio protein structure prediction. Protein Sci 2002; 11:1937-44; PMID:12142448; https://doi.org/10.1110/ps.3790102
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr., Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009; 460:711-6; PMID:19661910; https://doi.org/10.1038/nature08237
- Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 2002; 12:54-60; PMID:11839490; https://doi.org/10.1016/S0959-440X(02)00289-0
- Natan E, Wells JN, Teichmann SA, Marsh JA. Regulation, evolution and consequences of cotranslational protein complex assembly. Curr Opin Struct Biol 2017; 42:90-7; PMID:27969102; https://doi.org/10.1016/j.sbi.2016.11.023
- Sanjuan R, Borderia AV. Interplay between RNA structure and protein evolution in HIV-1. Mol Biol Evol 2011; 28:1333-38; PMID:21135148; https://doi.org/10.1093/molbev/msq329
- Goz E, Tuller T. Evidence of a direct evolutionary selection for strong folding and mutational robustness within HIV coding regions. J Comput Biol 2016; 23:641-50; PMID:27347769; https://doi.org/10.1089/cmb.2016.0052
- Sharp PM, Averof M, Lloyd AT, Matassi G, Peden JF. DNA sequence evolution: The sounds of silence. Philos Trans R Soc Lond B Biol Sci 1995; 349:241-7; PMID:8577834; https://doi.org/10.1098/rstb.1995.0108
- Resch AM, Carmel L, Marino-Ramirez L, Ogurtsov AY, Shabalina SA, Rogozin IB, Koonin EV. Widespread positive selection in synonymous sites of mammalian genes. Mol Biol Evol 2007; 24:1821-31; PMID:17522087; https://doi.org/10.1093/molbev/msm100
- Chursov A, Walter MC, Schmidt T, Mironov A, Shneider A, Frishman D. Sequence-structure relationships in yeast mRNAs. Nucleic Acids Res 2012; 40:956-62; PMID:21954438; https://doi.org/10.1093/nar/gkr790
- Roderer DJ, Scharer MA, Rubini M, Glockshuber R. Acceleration of protein folding by four orders of magnitude through a single amino acid substitution. Sci Rep 2015; 5:11840; PMID:26121966; https://doi.org/10.1038/srep11840
- Taketomi H, Kano F, Go N. The effect of amino acid substitution on protein-folding and -unfolding transition studied by computer simulation. Biopolymers 1988; 27:527-59; PMID:3370293; https://doi.org/10.1002/bip.360270402
- Lynch M, Conery JS. The origins of genome complexity. Science 2003; 302:1401-4; PMID:14631042; https://doi.org/10.1126/science.1089370
- Ottolenghi C. Some traces of hidden codes. Riv Biol 1998; 91:515-42; PMID:10212571
- Trifonov E, Volkovich Z, Frenkel ZM. Multiple levels of meaning in DNA sequences, and one more. Ann N Y Acad Sci 2012; 1267:35-8; PMID:22954214; https://doi.org/10.1111/j.1749-6632.2012.06589.x
- Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007; 24:1586-91; PMID:17483113; https://doi.org/10.1093/molbev/msm088