
ABSTRACT
The pathogenesis of neuropathic pain (NP) is characterized by an increased responsiveness of nociceptive neurons in the nervous system. However, the molecular mechanisms underpinning the NP still remain elusive. Recent data suggest that long non-coding RNAs (lncRNAs) regulate expression of NP-associated genes. Herein, we analyzed lncRNAs and mRNA profiles in the spinal cord of rats by RNA sequencing during the progression of NP in a spared nerve injury (SNI) model. Sprague-Dawley (SD) rats were employed for the establishment of the SNI models, and nociceptive responses to mechanical and thermal stimuli were measured 3 hours prior to surgery and on postoperative days 1, 3, 7 and 14, with L4-5 spinal cords extracted from three SD rats under deep anesthesia at each time point after behavioral test. SNI rats exhibited higher sensitivity to mechanical stimuli from days 1 to 14. Mechanical hyperalgesia reached a steady peak at day14 after surgery, whereas thermal allodynia did not develop. The results of second-generation sequencing suggested that the expression profiles of lncRNAs and mRNAs were significantly altered in spinal cords of SNI rats versus the control rats at different stages during NP. Differentially expressed (DE) lncRNAs and mRNAs were demonstrated at each stage during the NP course using Volcano Plot, Venn and Hcluster heatmap analyses. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathway analyses were performed to predict the functionalities of differentially expressed lncRNAs and target genes. Protein interaction networks were constructed based on the correlation analyses of DE lncRNA target proteins at 7 and 14 days after SNI, respectively. Taken together, our results revealed the profiles of lncRNAs and mRNAs in the rat spinal cord under an NP condition. These lncRNAs and mRNAs may represent new therapeutic targets for the treatment of NP.
Introduction
Neuropathic pain (NP) is a complicated, chronic pain state that is generally caused by tissue damage that affects the somatosensory nervous system.Citation1 The damaged, dysfunctional, or injured nerve fibers send incorrect signals to other pain centers, which induce abnormal sensations called dysesthesia or allodynia.Citation2-3 Notwithstanding the obscure mechanism underlying NP, multiple alterations across the nervous system, including ectopic generation of action potentials, facilitation and disinhibition of synaptic transmission, loss of synaptic connectivity and formation of new synaptic circuits and neuro-immune interactions, are involved in NP.Citation4-6 An improved knowledge of the pathogenesis of NP is crucial for the development of effective therapeutic strategies to prevent NP and improve curative effects.Long non-coding RNAs (lncRNAs) are a novel class of non-protein coding transcripts ranging from 200 nt to 100 kb in length and exhibit the functionality of gene regulation.Citation7-8 The expression of LncRNAs is tissue-specific and involved in diverse facets of cell biology and diseases.Citation9-11 However, the differential expression sites, binding sites, acting modes and complex network role of lncRNAs, which involve mRNAs, miRNAs and proteins,Citation12-13 cloud the functions of most lncRNAs.
Due to the difficulties of human trials, experimental animal models are crucial to elucidate mechanisms of NP.Citation14-15 LncRNAs are highly diversified in the spinal cord of mice after spinal nerve ligation (SNL), and the expression pattern of lncRNAs may regulate mRNAs and signaling pathways involved in neuropathic pain with gene microarray.Citation16 We analyzed lncRNAs at different time points during the NP process with sequencing method in spared nerve injury (SNI) rat models, which facilitate less tissue damage, simple and intuitive experimental steps, good behavior repeatability and more similar clinical manifestations of NP.Citation17-18 We performed a systematic analysis of the functionality of lncRNAs and mRNAs during the course of NP, which will shed new light on the effect of lncRNAs in NP. Nevertheless, the molecular and cellular functions of the predicted lncRNAs and mRNAs as well as the signaling pathways involved based on the present experiment await for further investigation.
Results
Model identification of neuropathic pain
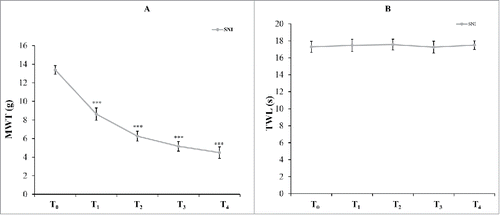
SNI-induced pain behaviors are more similar to the clinical symptoms of patients with NP.Citation18 18 rats were employed to test the mechanical allodynia and thermal sensitivity by von Frey and Hargreaves plantar tests, respectively. Three rats were randomly euthanized, and L4-5 spinal cords were extracted after the detection of pain thresholds. Another three rats were randomly euthanized, and specimens of the L4-5 spinal cord were collected after the detection of pain thresholds at 1, 3, 7 and 14 days after surgery (T1, T2, T3 and T4). Both mechanical allodynia and thermal sensitivity were determined in all SNI model rats at different stages of NP, with the numbers of rats being 18, 15, 12, 9 and 6 at T0, T1, T2, T3 and T4, respectively. The mechanical withdrawal thresholds were significantly decreased in the ipsilateral sides of rats at 1, 3, 7 and 14 day after SNI versus those 3 hours prior to surgery (), with the thermal sensitivity intact in both groups ().
Figure 1. Nociceptive behavior developed in SNI model rats. MWT (mechanical withdrawal threshold) in each time points (A), TWL (thermal withdrawal latency) in each time points (B). n = 18, *p < 0.001 compared T0.
Differentially expressed (DE) lncRNAs and mRNAs
We investigated significantly different expression of the transcripts in the L4-5 spinal cord samples in SNI-induced NP rats by variance analysis. Cuffdiff software was applied to analyze DE lncRNAs and mRNAs,Citation19 with the criteria q-value < 0.05 considered statistically significant. The readings of lncRNAs for each sample were 94210880–113312518. The clean bases of lncRNAs for each sample were 13.74G-16.43G. The error rates of lncRNAs for each sample were less than < 0.02%. The screened conditions of lncRNAs splicing transcription were as follows: the number of exon ≥ 2, length > 200 bp, FPKM ≥ 0.5 (Cuffquant) and to eliminate overlapping and coding potential transcription with annotation of database at exon region (Cuffcompare Software). The raw data in this study can be available in NCBI SRA database.
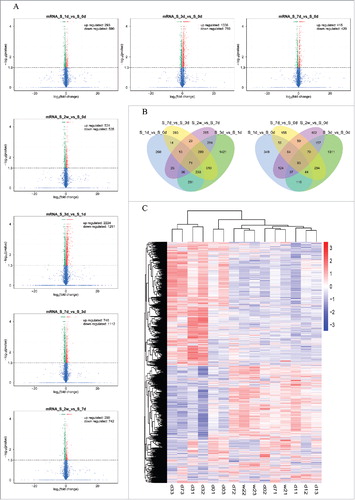
The results of DE lncRNAs revealed that 35, 44, 25 and 15 lncRNAs were up-regulated in rats at 1, 3, 7 and 14 days after SNI, and 59, 135, 101 and 129 lncRNAs were down-regulated as compared to the rats at 3 hours before SNI (). In addition, the results of DE mRNAs revealed 293, 1336, 415, and 531 mRNAs were significantly up-regulated in rats at 1, 3, 7 and 14 days after SNI, and 590, 786, 420 and 535 mRNAs were significantly down-regulated versus those at 3 hours before SNI (). and list the top 20 up-regulated and down-regulated lncRNAs and mRNAs at each time point, respectively. Others DE lncRNAs and mRNAs can be obtained from the raw data of SRA.
Table 1. The detailed information of the top 20 up-regulated and 20 down-regulated lncRNA.
Table 2. The detailed information of the top 20 up-regulated and 20 down-regulated mRNAs.
Figure 2. The expression profiling changes of lncRNAs during the different stages of SNI. Vocalno Plot indicated up-regulated and down-regulated lncRNAs at different time points of SNI model (); Venn diagram showed the number of overlap lncRNAs during the different stages of SNI in ; Heat map of lncRNAs showed hierarchical clustering of changed lncRNAs at the different time points. In clustering analysis, up-regulated and down-regulated genes are colored in red and blue, respectively. The sequencing samples at 0d, 1d, 3d, 7d and 2w were labeled d0, d1, d3, d7 and w2, respectively; the serial numbers of samples in each time point were labeled ending ().
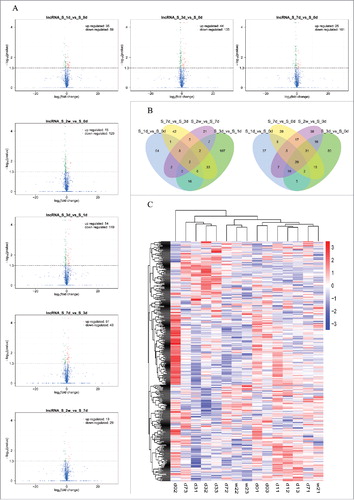
Statistics were performed for the different transcripts at each time point when the numbers of compared combinations ranged from 2 to 5, and the Venn Diagram of DE transcripts were drawn. These analyses intuitively revealed the number of common and special DE transcripts between the different compared combinations. showed the Venn Diagrams of DE lncRNAs, and show the Venn Diagrams of DE mRNAs. The same types of transcripts may exhibit similar functions or participate jointly in the same metabolic processes or cellular pathways. Clustering analysis of DE transcripts may help identify the function of unknown transcripts or the unknown function of known transcripts by gathering the same expression pattern or similar genes to a class. presented the clustering analysis of DE lncRNAs, and presented the clustering analysis of DE mRNAs.
Figure 3. The expression profiling changes of mRNAs during the different stages of SNI. Vocalno Plot indicated up-regulated and down-regulated mRNAs at different time points of SNI model (); Venn diagram showed the number of overlap mRNAs during the different stages of SNI in ; Heat map of mRNAs showed hierarchical clustering of changed mRNAs at the different time points. In clustering analysis, up-regulated and down-regulated genes are colored in red and blue, respectively. The sequencing sample at 0d, 1d, 3d, 7d and 2w were labeled d0, d1, d3, d7 and w2, respectively; the serial numbers of samples in each time point were labeled ending ().
Prediction of DE lncRNA target mRNA
LncRNAs may play a role in regulating adjacent protein-coding genes. We set the threshold of co-location to 100 kb and predicted the lncRNA co-expressed genes using the correlation or co-expression analysis of expression quantity between lncRNA and mRNA in the samples. 6 up-regulated and 6 down-regulated DE lncRNAs were significantly altered at different time points (), with seven DE lncRNAs randomly chosen for further analysis. and illustrated the annotation of the seven lncRNAs co-located and co-expressed mRNAs, respectively. The top changed lncRNAs and mRNAs listed in are more likely to play important roles in NP, and the data presented in and help us to understand how these lncRNAs act on the molecular mechanisms of mRNAs, which will be explored in our further research. However, those significant changed lncRNAs and mRNAs which are not listed in and may also play important roles in NP. The complete sequencing data can be obtained from the NCBI database (Accession: SRP105463) and will help other scholars in-depth study of the roles of lncRNAs in pathogenesis of NP in the future.
Table 3. The detailed information of seven significant changed lncRNAs co-located mRNAs.
Table 4. The detailed information of seven significant changed lncRNAs co-expressed mRNAs.
Real-time quantitative PCR (qPCR) validation of lncRNA expression
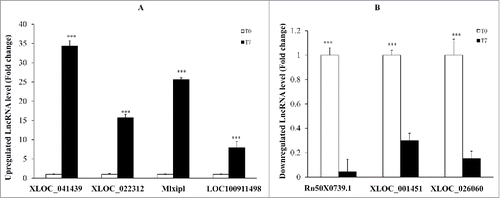
To validate the reliability of the sequencing results, the expression levels of lncRNAs were analyzed by qPCR. The expression of XLOC_041439, Mlxipl, XLOC_022312 and LOC100911498, all of which represent up-regulated lncRNAs, were significantly increased in rats at day 7 after SNI as compared to the control rats. The expression of Rn50_X_0739.1, XLOC_001451 and XLOC_026060, all of which exemplify the down-regulated lncRNAs, were significantly decreased at 7 days after SNI versus the control rats (). All the findings determined by qPCR were consistent with the results of the second-generation sequencing, potently validating their reliability.
Figure 4. QPCR validations of seven regulated lncRNAs in the spinal cord from SNI rats. The expressions of lncRNAs(A) were significantly up-regulated at 7 days after SNI. The expressions of lncRNAs(B) were significantly down-regulated at 7 days after SNI. One-way ANOVA followed by Tukey's multiple comparison test. ***P < 0.001.
Functional prediction of DE lncRNAs in SNI
The Gene Ontology (GO) is a major bioinformatics tool that unifies the representation of genes and gene product attributes across all species and serves as a good predictor of gene function and trend. KEGG pathway databases store the higher order functional information for systematic analysis of gene functions and are more widely used in current enrichment analysis platforms. Thus, we predicted the major function of DE lncRNAs in the spinal cords of rats at different stages of NP with GO and KEGG pathway analysis in the present study. The background set was rattus genome library under control conditions from adult/male rat.
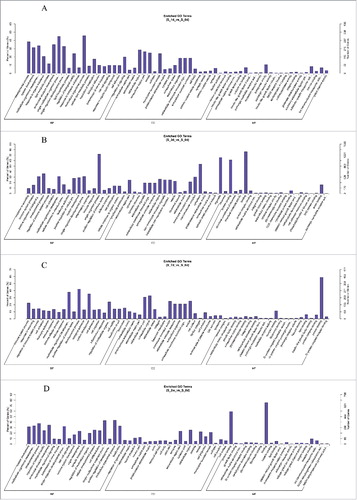
The most significantly enriched biological processes of regulated genes at 1 day after SNI were the multicellular organismal process, the single-multicellular organismal process, the developmental process and anatomical structure development. The most noteworthy enriched cellular components of the regulated genes were the extracellular region, the extracellular region part, the vesicle and the membrane-bounded vesicle. The most significantly enriched molecular functions of regulated genes were transporter activity, substrate-specific transporter activity, ion transmembrane transporter activity and calcium ion binding (). The most significantly enriched biological processes of regulated genes at 3 days after SNI were the single-organism process, the developmental process, multicellular organismal development and the positive regulation of the biological process. The most noteworthy enriched cellular components of regulated genes were organelles, cytoplasms, macromolecular complexes and extracellular regions. The most significantly enriched molecular functions of regulated genes were binding, protein binding, anion binding and structural molecular activity (). The most significantly enriched biological processes of regulated genes at 7 days after SNI were the developmental process, the multicellular organismal process, system development and the regulation of multicellular organisms. The most noteworthy enriched cellular components of regulated genes were the extracellular region, the extracellular region part, the vesicle and the membrane-bounded vesicle. The most significantly enriched molecular functions of regulated genes were protein binding, lipid binding receptor binding, and protein complex binding (). The most significantly enriched biological processes of regulated genes at 14 days after SNI were the multicellular organismal process, the single-multicellular organismal process, the developmental process and anatomical structure development. The most noteworthy enriched cellular components of regulated genes were the extracellular region, the extracellular region part, the vesicle and the membrane-bounded vesicle. The most significantly enriched molecular functions of regulated genes were binding, protein binding, receptor binding and lipid binding (). The results indicated that the most strikingly changed category of biological processes, cellular components and molecular functions at 3 to 14 days after SNI were similar to and consistent with the behavioral trends of the SNI model. These most strikingly changed categories of function will be the focus of a future study.
Figure 5. Enriched GO Terms with SNI pathogenesis. The significant molecular function, biological process and cellular component changed mRNAs in different stages of SNI. SNI 1d vs. 0d (), SNI 3d vs. 0d (), SNI 7d vs. 0d () and SNI 2w vs. 0d ().
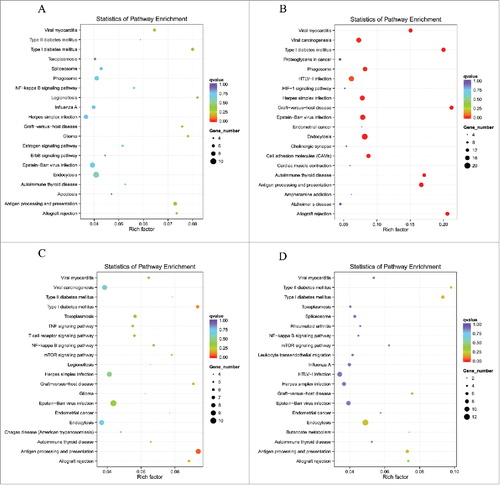
Scatter diagrams of KEGG measured the degree of enrichment by rich factor, q value and gene number of the enriched pathway. We selected 20 pathway items of the most significant enrichments and displayed these items in the KEGG scatter diagram. The results demonstrated that lncRNAs co-localized mRNAs in SNI pathogenesis were most significantly involved in viral myocarditis, type l diabetes mellitus, graft-versus-host disease, autoimmune thyroid disease, antigen processing and presentation, allograft rejection and antigen processing and presentation (–). The lncRNAs co-expressed mRNAs in SNI pathogenesis were most significantly involved in systemic lupus erythematosus, alcoholism, steroid hormone biosynthesis, spliceosome, metabolism of xenobiotics by cytochrome P450 and chemical carcinogenesis (–). The regulated mRNAs in SNI pathogenesis were most significantly involved in systemic lupus erythematosus, ribosomes, staphylococcus aureus infection, malaria, osteoclast differentiation and fc gamma R-mediated phagocytosis (–). Most of the involved pathways that were enriched 7 and 14 days after SNI reached peak expression, which remained in a steady stage of SNI-induced NP. It should be explained that the top biological functions and signaling pathways with GO and KEGG analysis presented in – will help us clarify the mechanism of downstream of lncRNAs. However, as noted above, the signaling pathways provided by complete data in NCBI SRA database are far greater than those listed in the paper. When other scholars determine the relevant targeted lncRNAs, the SRA database will help to explore its downstream signaling pathways in NP mechanism.
Figure 6. DE lncRNAs co-localized genes enriched KEGG pathway scatterplot with SNI pathogenesis. LncRNAs enriched KEGG pathway scatterplot showing statistics of pathway enrichment at different time points. Pathway of lncRNAs co-localized genes at different stage of SNI including SNI 1d vs. 0d (), SNI 3d vs. 0d (), SNI 7d vs. 0d () and SNI 2w vs. 0d ().
Protein interaction network analysis
To explore the molecular mechanisms of the lncRNAs involved in pathogenesis of NP, analyses of protein interaction network were further performed in different stages of NP in SNI rats versus the sham group. The results demonstrated that the DE lncRNAs-protein interactions in rats at 1 day after the SNI had no correlation with the those at 3 days after the SNI, whereas the DE lncRNAs-protein interaction was driving the complex interaction network at 7 () and 14 days () after SNI. These results are consistent with the trends in SNI-induced NP behaviors and explain the important role of the protein interaction networks that were targeted by lncRNAs in the pathogenesis of NP. Many novel lncRNAs that we screened may play an important role through regulation of protein expression in NP, but future in-depth studies are required. Although the mechanism of lncRNA is complex, the protein level regulated RNA play the biological effect. Therefore, the protein interaction nets we constructed not only verified the reliability of our sequencing data, but also provided a wealth of information of downstream target with lncRNAs for further study.
Figure 7. DE lncRNAs co-expressed genes enriched KEGG pathway scatterplot with SNI pathogenesis. LncRNAs enriched KEGG pathway scatterplot showing statistics of pathway enrichment at different time points. Pathway of lncRNAs co-expressed genes at different stage of SNI including SNI 1d vs. 0d (), SNI 3d vs. 0d (), SNI 7d vs. 0d () and SNI 2w vs. 0d ().
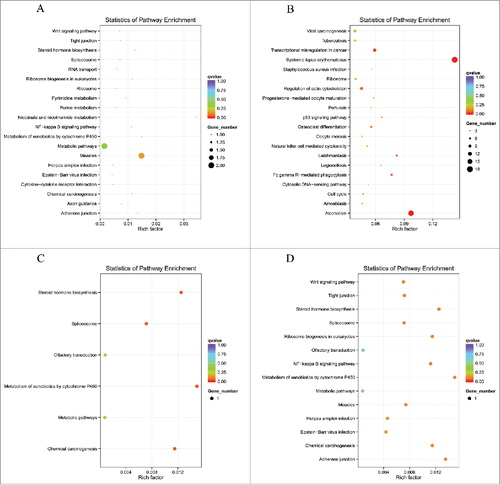
Figure 8. DE mRNAs enriched KEGG pathway scatterplot with SNI pathogenesis. LncRNAs enriched KEGG pathway scatterplot showing statistics of pathway enrichment at different time points. Pathway of lncRNAs co-expressed genes at different stages of SNI including SNI 1d vs. 0d (), SNI 3d vs. 0d (), SNI 7d vs. 0d () and SNI 2w vs. 0d ().
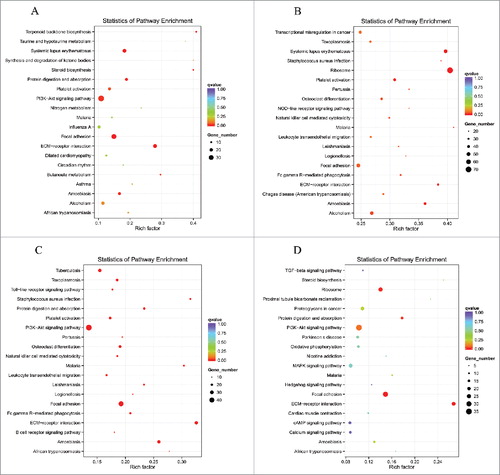
Figure 9. Protein interaction network analysis of DE mRNA in rats at 7d after SNI.
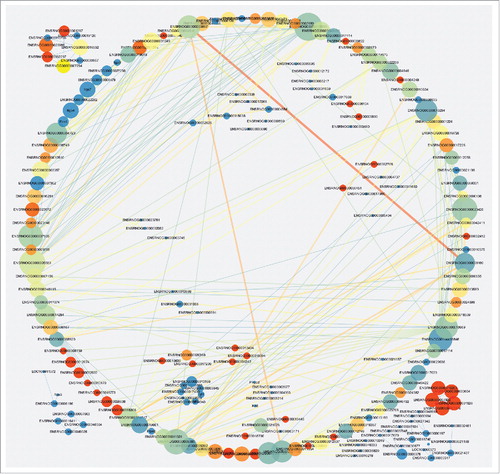
Figure 10. Protein interaction network analysis of DE mRNA in rats at 14d after SNI.
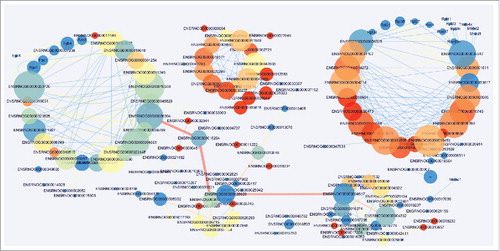
Discussion
NP is caused by disorders of or damage to the nervous system. The damaged nerve fibers in NP send incorrect signals to other pain centers and induce pain sensations in the central nervous system.Citation20 NP is often associated with dysesthesias, hyperesthesia, hyperalgesia, allodynia, and hyperpathia.Citation21 However, no effective therapies for NP are available,Citation22 thus a better understanding of the mechanism of NP is needed. Some lncRNAs participate in physiological processes that maintain cellular and tissue homeostasis. Additionally, the deregulated expression of lncRNAs is implicated in the onset and progression of multiple pathological conditions. Previous study indicated that some lncRNAs regulate gene expression and traffic cellular protein complexes, genes and chromosomes to appropriate locations.Citation23-24 LncRNA were differentially expressed in the spinal cord of mice following spinal nerve ligation-induced neuropathic pain by gene microarrays.Citation16 Notably, in present experiment, most DE lncRNAs in the SNI models were novel using second-generation sequencing technology. Furthermore, the mechanisms of gene expression regulation by lncRNAs during the pathogenesis of NP await to be unveiled. Our study presented more information of the lncRNAs in NP with a different species and model. We validated that some lncRNAs and mRNAs were significantly altered in the spinal cord in response to SNI-induced NP. The potential functions of differentially expressed lncRNAs were annotated using GO and KEGG pathway analyses. Protein interaction networks were also constructed. These results indicate that lncRNAs play key roles in the pathogenesis of NP via the regulation of some signaling pathways and gene expression.
Bao-Chun Jiang et alCitation16 detected the expression of lncRNAs in a mouse SNL model. Compared with the previous study, the novelty and advancement of our study can be elaborated as follows. Firstly, we analyzed DE lncRNAs and mRNAs in SNI rats using second-generation sequencing technology, which is more comprehensive than gene microarray.Citation25 Therefore, we confirmed a large number of novel lncRNAs that have significantly changed in the spinal cord of NP rats with this technology. Secondly, Bao-Chun Jiang's research was based on gene microarray, and all experimental data were derived from mice as well as rats. We used rats only in our sequencing analysis, avoiding errors caused by mixed analysis of different species. Finally, SNL model has been widely used in the investigation of the mechanisms underlying NP. However, the SNI model used in our study has more advantages, including less tissue damage, more intuitive and simpler experiment steps, better behavior repeatability and more similar to the clinical manifestations of NP. Furthermore, a recent study indicated that the pain sensitivity between male and female mice were regulated by different immune cells, suggesting that male rats could not be used as proxies for females in pain research.Citation26 Therefore, we have selected male rats in order to avoid the analysis error caused by gender in the present study. Sequencing analysis of gender differences in NP will also be an interesting topic, which is absolutely worth further studying in the future. The results revealed that a total of 24,833 lncRNAs were detected in the spinal cords in SNI rats, of which XLOC_041439, Mlxipl, XLOC_022312, LOC100911498, Rn50_X_0739.1, XLOC_001451 and XLOC_026060 were significantly altered at different stages of NP. It suggests a greater possibility that these lncRNAs were implicated in the pathogenesis of NP. In addition, the number of DE lncRNAs reached a peak at 7 days after SNI, suggesting the regulation of gene function and signaling pathways reaching a maximum at that time point. All data afore indicate a potent role of lncRNAs in NP and provide a research basis for the role of lncRNAs in the pathogenesis of NP.
LncRNA are a class of transcripts that are more than 200nt in length and do not encode proteins themselves, but rather regulate the expression of genes in RNA form on a variety of levels, such as epigenetic, transcriptional and post-transcriptional regulation. The content of body is quite rich, accounting for about 4–9% of mammalian RNA.Citation7 mRNA is direct template of synthetic protein which transcript the genetic information of DNA. The length of mRNA is different, it accounting about 1–2% of RNA in mammals. According to the present study, lncRNA mainly achieve to regulate the expression of mRNA or protein from three levels, including epigenetics, transcriptional and post-transcriptional regulation.Citation9,Citation27 The possible mechanisms are mainly included in the following ways: Coding promoter region transcription of the upstream genes and interfering with the expression of downstream genes; Inhibiting RNA polymerase II or mediating chromatin remodeling and histone modification to affect the expression of downstream genes; Forming complementary double-stranded with gene transcripts, on the one hand, to interfere with mRNA cleavage and produce different shear forms, also to produce endogenous siRNA under the action of Dicer enzymes on the other hand; Binding to specific proteins to regulate the activity of the corresponding protein; Forming a nucleic acid protein complex with protein as a structural component; Changing cell localization of protein to bind it to a specific protein; Playing roles as precursor molecules of small molecule RNA, such as miRNA, piRNA.
Burgeoning evidence reveals that lncRNAs play a critical role in cell biology and disease etiology.Citation28,Citation29 LncRNAs are differentially expressed in dorsal root ganglia following sciatic nerve resectionCitation30 and a functional lncRNA Kcna2 was recently found to contribute to NP,Citation31 hinting the involvement of lncRNAs in NP. However, the underlying mechanisms of lncNRA in NP are poorly understood, and the study of lncRNAs in NP is scarce. The higher orders of structure, differential splicing and alternative transcription initiation sites render lncRNAs more flexible in targeting proteins or gene sites.Citation32 The mechanism of lncRNA interaction with mRNA is not clear. The positional relationship and correlation of expression between lncRNA and protein coding gene are closely related to the biological function of lncRNA. Therefore, we hypothesized that the function of lncRNA-related genes and the corresponding pathways in the spinal cord of SNI-induced NP rats would be identified using GO and KEGG term enrichment analyses. We noted that the most strikingly changed category of biological processes, cellular components and molecular functions were similar from days 3 to 14 after SNI. These phenomena also demonstrated the reliability of the RNA sequencing results. The functional prediction of lncRNAs using GO and KEGG provide a research basis of lncRNAs in the mechanisms of NP for future work.
Protein-protein interaction and identification play important roles in molecular biology.Citation33 We constructed a protein interaction network of rat species during the progression of SNI-induced NP based on the predicted list of different genes in present study using the database of STRING protein interactions. Our analyses revealed complex network relationships among DE mRNA-encoded proteins at days 7 and 14 after SNI. These results provide a new research perspective of NP molecular mechanisms and increase our understanding of the important role and internal relationship of lncRNAs, mRNAs and related proteins in the pathogenesis of NP, which are potentially important novel targets for the treatment of NP. The protein interaction network modules also provide potentially new targets for treatment of NP.
NP is caused by noxious stimulation and produces a series of complex changes in the nervous system. Spinal cord is the primary center of information transfer and integration in nervous system, which integrates pain information from peripheral afferent nerve and downward projected nerve of brainstem and cerebral cortex. Multiple target genes in the spinal cord mediate complex biological signaling pathways and play an important role in the development and maintenance of NP.Citation34 Although multiple target genes and signaling pathways in spinal cord level were proved to be involved in pathogenesis of NPCitation35 and the vision field of study was expanded with the application of gene chip technology,Citation36 it is undeniable that there are still largely unknown about the specific regulatory mechanism of NP in spinal cord level. Therefore, we comprehensively analyzed the change of lncRNAs and mRNAs in spinal cord of SNI model rats with second generation sequencing technology. On the one hand, a large amount of genes and signaling pathways which had been reported to participate in NP were affirmed in our results, such as PI3K, NF-kB, MAPK, etc. On the other hand, we offered amount of information about lncRNAs of spinal cord in the pathogenesis of NP. For example, cell apoptosis and autophagy in spinal cord have been shown to participate in the mechanism of NP, and mTOR is an important signal pathway of autophagy and apoptosis, our data also accurately predicted the DE lncRNA (XLOC_035479), which is co-located with mTOR gene.
At present, there is considerable evidence to focus on the important role of microRNAs to play in the NP,Citation37 but little attention is given to the role of lncRNAs which interact with miRNAs by ceRNA mechanism.Citation38 This knowledge gap has motivated our work. We uploaded open data of DE lncRNAs in spinal cord of rattus to NCBI databases, which can help many researchers to in-depth study in physiology and pathogenesis of NP in overcoming the paucity of evidence due to limited experiments, high costs and technical difficulties. However, few lncRNAs have been studied in mechanism of NP up to now. For instance, inflammatory reaction of spinal cord plays an key role in the mechanism of NP,Citation39 but there is currently little evidence for direct interaction between lncRNAs and the co-located and co-expressed mRNAs which be connected with inflammatory factors. In our study, a mountain of lncRNAs were detected to be related with genes and signaling pathways associated with pain. These screened lncRNAs will be further validated and studied by our team and other scholars in the future.
It is worth mentioning that the dorsal and ventral horn of spinal cord have completely distinct role in the mechanism of neuropathic pain, and the spinal cord contains a variety of cell populations, including many kinds of neuron subtypes, astrocytes and microglia. It seems imprecise to state that GO and KEGG analyses of lncRNAs could just make the spinal cord as a whole. Therefore, lots of experiments both in vitro and in vivo need to be further verified and analyzed. Based on the data we provided in present study, the following research will be implemented as follows: (1) Further screening of the lncRNAs obtained from the present data which are closely related to the mechanism of NP, and verifying via multiple experimental methods; (2) Locating a certain number of lncRNAs for further screening; (3) Culturing neurons and glial cells of spinal cord respectively and combining the sequencing data in present study to confirm the expression and roles of lncRNAs in the spinal cord level in the pathogenetic process of NP; (4) Combining with signaling pathway prediction that we constructed in this study to explore the roles of the lncRNAs in NP and the specific mechanism with knockout rats models and cell models of NP.
In conclusion, the expression profiles of lncRNA and mRNA were detected using microarrays at different stages of NP. GO and KEGG pathway analyses were performed to annotate the potential functions of differentially expressed lncRNAs. Protein interaction network in the pathogenesis of NP were constructed and analyzed. This abundant datasets suggest that lncRNAs locally regulate their related protein-gene expression and play key roles in the pathogenesis of NP. The molecular and cellular functions of the screened lncRNAs in the present study will be further determined, and we hope to invite more researchers to study the role of lncRNAs in this field.
Methods
Animals
Adult male Sprague–Dawley rats, body weight 250–280g (The Laboratory Animal Center, The First People's Hospital of Foshan, Guangdong, China) were housed at a constant ambient temperature (22 ± 0.581°C) and humidity (60-70%) under a 12-hour light/dark cycle (lights on at 7 AM). All animals were fed a standard laboratory diet and tap water ad libitum. Prior to the behavioral testing, rats were acclimatized in the experimental room for 24 hours. All experiments were in accordance with the recommendations of national and international animal care and ethical guidelines, with the approval of the First People's Hospital of Foshan animal welfare committee.
SNI model
Rats with successful intrathecal cannula placement underwent SNI surgery after anesthesia.Citation40 The rats were fixed to an operating table with the femur at the right mid-thigh level as the incision landmark. The 3 peripheral branches (the sural, common peroneal, and tibial nerves) of the sciatic nerve were exposed at mid-thigh level distal to the trifurcation, without stretching of the nerve structures and freed of connective tissue. Subsequently, the tibial and common peroneal nerves were tightly ligated by two knots, 4 mm apart, using 6.0 silk suture (Ethicon, Johnson & Johnson Inc, Brussels, Belgium) and completely severed between the knots. The sural nerve maintains intact. Muscle and skin were closed in two distinct layers using 5.0-silk suture.Citation18
Nociceptive behavior
All behavioral tests were performed in a blinded manner. Nociceptive responses to mechanical and thermal stimuli were measured 30 minutes before and 1, 3, 7 and 14 days after surgery.
Mechanical withdrawal threshold (MWT) was measured as the right hind paw responded to von Frey filament stimulation.Citation41 The test area was the mid-plantar paw in the distribution area of the sciatic nerve. Von Frey filaments with logarithmically incremental stiffness (0.4-15.1 g) were serially applied to the paw using the up-down method. The filaments were presented in an ascending order of strength, perpendicular to the plantar surface, with sufficient force to cause a slight bending against the paw and held for 6–8s. A positive response was defined as an onset of sharp withdrawal of the hind paw. Flinching immediately upon removal of the filaments was also considered a positive response. The 15.1-g filament was selected as the upper cutoff for testing. Animals that exhibited no response to stimulation of the 15.1-g filament were assigned cutoff values. The bending force that evoked 50% of paw withdrawal occurrence was set as the MWT.
The Hargreaves test was used to measure thermal withdrawal latency (TWL).Citation42 The rats were placed on the surface of a 2 mm-thick glass plate covered with a plastic chamber (20 × 20 × 25 cm). TWLs were measured using a radiant thermal stimulator (BME410A, Institute of Biological Medicine, Academy of Medical Science, Tianjin, China). Heat was concentrated on the hind paw, which was flush against the glass, and radiant heat stimulation was delivered to the site. The stimulus was terminated with hind paw movement, and a 25-s cutoff was imposed to prevent tissue damage. The intensity of thermal stimulation remained constant throughout the experiment. Five TWL tests were performed to the same sites with the values from three thermal tests averaged as TWL.
Tissue collection and RNA isolation
Animals were deeply anesthetized with isoflurane at each time point and perfused through the ascending aorta with normal saline. The L4–5 spinal cord segments were collected. Total RNA was extracted from the spinal cord dorsal horn tissue using Trizol reagent (Invitrogen, Carlsbad) according to the manufacturer's protocol. RNA quantity and quality were measured using a NanoDrop ND-1000. RNA integrity was assessed using standard denaturing agarose gel electrophoresis.
Quantitative Real-Time RT-PCR
Quantitative analysis of the targeted mRNA expression was performed using real-time RT-PCR and the relative standard curve method. 12 rats underwent SNI and sham surgeries. The nerves were exposed but not ligated in rats with sham surgeries. Animals were deeply anesthetized with isoflurane at 7 days post-surgery and perfused through the ascending aorta with saline. Total RNA was extracted from snap-frozen L4-5 spinal cord tissues using TRIzol Reagent (Invitrogen), followed by RNase-free DNase I (Roche). Aliquots (1 μg) of total RNA were reverse transcribed and amplified in triplicate using IQ SYBR green supermix reagent (Bio-Rad, Herculus, CA, USA) in an Opticon real-time PCR machine (MJ Research, Waltham, MA, USA) according to the manufacturer's instructions. The specificity of real-time PCR was confirmed via routine agarose gel electrophoresis and melting-curve analysis. shows all the primer sequences.
Table 5. Primers designed for qRT-PCR validation of candidate lncRNAs.
Library preparation for lncRNA sequencing
A total amount of 3 μg RNA per sample was used as input material for RNA sample preparations. Ribosomal RNA was removed using an Epicentre Ribo-zero™ rRNA Removal Kit (Epicentre, USA), and rRNA-free residue was cleared using an ethanol precipitation. Sequencing libraries were generated from the rRNA-depleted RNA using a NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's recommendations. First-strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase. Second-strand cDNA synthesis was performed using DNA Polymerase I and RNase H. dNTPs with dTTP were replaced by dUTP and the reaction buffer. The 3′ ends of DNA fragments were adenylated, and a NEBNext Adaptor with a hairpin loop structure was ligated for hybridization. Library fragments were purified using an AMPure XP system (Beckman Coulter, Beverly, USA) to select cDNA fragments of the preferred length (150∼200 bp). Products were purified (AMPure XP system), and library quality was assessed on an Agilent Bioanalyzer 2100 system. The libraries were sequenced at the Novogene Bioinformatics Institute (Beijing, China) on an Illumina Hiseq 2500 platform, and 100 bp paired-end readings were generated.
Clustering and sequencing of lncRNA
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer's instructions. After cluster generation, the libraries were sequenced on an Illumina HiSeq 2500 platform, 125 bp paired-end and 50-bp single-end reads were generated. The error rates of lncRNAs for each sample were respectively less than < 0.02%. The transcription with splicing of each sample were combined and screened as lncRNAs with Cuffmerge Software, and the conditions were as follows: the number of exon ≥ 2, length > 200 bp, FPKM ≥ 0.5 (Cuffquant) and to eliminate overlapping and coding potential transcription with annotation of database at exon region (Cuffcompare Software).Citation43,Citation44 All sequencing program were performed by Novogene Company (China, Beijing).
GO & KEGG Pathways analysis
GO and KEGG pathway analysis were used to investigate the roles of all DE mRNAs. Briefly, GO analysis was used to elucidate genetic regulatory networks of interest by forming hierarchical categories according to the biological process (BP), cellular component (CC) and molecular function (MF) aspects of the differentially expressed genes (http://www.geneontology.org). Pathway analysis was performed to examine the significant pathways of the differentially expressed genes according to KEGG (http://www.genome.jp/kegg/).
Analysis of protein interaction network
We constructed protein interaction networks based on a correlation analysis of DE mRNAs to identify interactions among the DE gene proteins in NP pathogenesis. The network was constructed according to the protein interactions in the database (http://string-db.org/).
Statistical analysis
Data are presented as the means ± SEM. The results from the behavioral study were statistically analyzed using repeated measures analysis of variance. RT-PCR results were analyzed using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test. Significance was set at p < 0.05.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Author contributions
Jun Zhou, Youling Fan and Zhenxing Huang designed the study and wrote the paper. Sen Lin and Hanbing Wang contributed to acquisition,analysis and interpretation of data. Houtao Chen provided technical assistance and contributed to the preparation of all the figures. All authors analyzed the results and approved the final version of the manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (81300974), the Natural Science Foundation of Guangdong Province (2015A030313899) and the Medical Scientific Research Foundation of Guangdong Province (A2015013).
References
- Backonja MiM. Defining Neuropathic Pain. Anesth Analg. 2003;97(3):785–90. doi:10.1213/01.ANE.0000062826.70846.8D.
- Backonja MM, Galer BS. Pain assessment and evaluation of patients who have neuropathic pain. Neurol Clin. 1998;16(4):775–90. doi:10.1016/S0733-8619(05)70097-9.
- Ossipov MH, Lai J, Malan TP Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. doi:10.1111/j.1749-6632.2000.tb06673.x.
- Taylor BK. Pathophysiologic mechanisms of neuropathic pain. Curr Pain Headache Rep. 2001;5(2):151–61. doi:10.1007/s11916-001-0083-1.
- Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131(3):243–57. doi:10.1016/j.pain.2007.07.026.
- Zhuo M, Wu G, Wu LJ. Neuronal and microglial mechanisms of neuropathic pain. Mol Brain. 2011;4:31. doi:10.1186/1756-6606-4-31.
- Da Sacco L, Baldassarre A, Masotti A. Bioinformatics Tools and Novel Challenges in Long Non-Coding RNAs (lncRNAs) Functional Analysis. Int J Mol Sci. 2012;13(1):97–114.
- Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–61. doi:10.1038/nm.3981.
- Sun M, Kraus WL. From Discovery to Function: The Expanding Roles of Long Non-Coding RNAs in Physiology and Disease. Endocr Rev. 2015;36(1):25–64. doi:10.1210/er.2014-1034.
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, et al. RNA Exosome-Regulated Long Non-Coding RNA Transcription Controls Super-Enhancer Activity. Cell. 2015;161(4):774–89. doi:10.1016/j.cell.2015.04.034.
- Gong C, Maquat LE. LncRNA stransactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011;470 (7333):284–8. doi:10.1038/nature09701.
- Guo L, Zhao Y, Yang S, Zhang H, Chen F. An integrated analysis of miRNA, lncRNA, and mRNA expression profiles. Biomed Res Int. 2014:345605.
- Paraskevopoulou MD, Hatzigeorgiou AG. Analyzing MiRNA-LncRNA Interactions. Methods Mol Biol. 2016;1402:271–86. doi:10.1007/978-1-4939-3378-5_21.
- Yalcin I, Megat S, Barthas F, Waltisperger E, Kremer M, Salvat E, Barrot M. The sciatic nerve cuffing model of neuropathic pain in mice. J Vis Exp. 2014;(89):e51608. doi:10.3791/51608.
- Lau WK, Lau YM, Zhang HQ, Wong SC, Bian ZX. Electroacupuncture versus celecoxib for neuropathic pain in rat SNL model. Neuroscience. 2010;170(2):655–61. doi:10.1016/j.neuroscience.2010.07.031.
- Jiang BC, Sun WX, He LN4, Cao DL, Zhang ZJ, Gao YJ. Identification of lncRNA expression profile in the spinal cord of mice following spinal nerve ligation-induced neuropathic pain. Mol Pain. 2015;11:43. doi:10.1186/s12990-015-0047-9.
- E Park, D Youn. Rat Models of Neuropathic Pain: CCI, PSNL, SNL and SNI. Laboratory Animal Research. 2009;25(2):87–94.
- Bourquin AF, Süveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain. 2006;122(1–2):14.e1–14. doi:10.1016/j.pain.2005.10.036.
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi:10.1038/nbt.1621.
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353(9168):1959–64. doi:10.1016/S0140-6736(99)01307-0.
- Bouhassira D, Attal N. Translational neuropathic pain research: A clinical perspective. Neuroscience. 2016;338:27–35. doi:10.1016/j.neuroscience.2016.03.029.
- Gilron I, Baron R, Jensen T. Neuropathic pain: Principles of diagnosis and treatment. Mayo Clin Proc. 2015;90(4):532–45. doi:10.1016/j.mayocp.2015.01.018.
- Li J, Meng H, Bai Y, Wang K. Regulation of lncRNA and Its Role in Cancer Metastasis. Oncol Res. 2016;23(5):205–17. doi:10.3727/096504016X14549667334007.
- Ballantyne MD, McDonald RA, Baker AH. LncRNA/microRNA interactions in the vasculature. Clin Pharmacol Ther. 2016;99(5):494–501. doi:10.1002/cpt.355.
- LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152(8):1888–98. doi:10.1016/j.pain.2011.04.014.
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–3. doi:10.1038/nn.4053.
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. doi:10.1038/nrg.2015.10.
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA. 2008;105(2):716–21. doi:10.1073/pnas.0706729105.
- Zhang B1, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, Spector DL. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2(1):111–23. doi:10.1016/j.celrep.2012.06.003.
- Yu B, Zhou S, Hu W, Qian T, Gao R, Ding G, Ding F, Gu X. Altered long noncoding RNA expressions in dorsal root ganglion after rat sciatic nerve injury. Neurosci Lett. 2013;534:117–22. doi:10.1016/j.neulet.2012.12.014.
- Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. 2013;16(8):1024–31. doi:10.1038/nn.3438.
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi:10.1016/j.cell.2013.06.020.
- Chakrabarti KS, Agafonov RV, Pontiggia F, Otten R, Higgins MK, Schertler GF, Oprian DD, Kern D. Conformational Selection in a Protein-Protein Interaction Revealed by Dynamic Pathway Analysis. Cell Reports. 2015;14(1):32–42. doi:10.1016/j.celrep.2015.12.010.
- D'Mello R, Dickenson AH. Spinal cord mechanisms of pain. Br J Anaesth. 2008;101(1): 8–16. doi:10.1093/bja/aen088.
- Bee LA, Dickenson AH. Rostral ventromedial medulla control of spinal sensory processing in normal and pathophysiological states. Neuroscience. 2007;147:786–93. doi:10.1016/j.neuroscience.2007.05.004.
- Raju HB, Englander Z, Capobianco E, Tsinoremas NF, Lerch JK. Identification of potential therapeutic targets in a model of neuropathic pain. Front Genet. 2014;5:131. doi:10.3389/fgene.2014.00131.
- Hori N, Narita M, Yamashita A, Horiuchi H, Hamada Y, Kondo T, Watanabe M, Igarashi K, Kawata M, Shibasaki M, et al. Changes in the expression of IL-6-Mediated MicroRNAs in the dorsal root ganglion under neuropathic pain in mice. Synapse. 2016;70(8):317–24. doi:10.1002/syn.21902.
- Wang W, Zhuang Q, Ji K, Wen B, Lin P, Zhao Y, Li W, Yan C. Identification of miRNA, lncRNA and mRNA-associated ceRNA networks and potential biomarker for MELAS with mitochondrial DNA A3243G mutation. Sci Rep. 2017;7:41639.
- Schomberg D, Ahmed M, Miranpuri G, Olson J, Resnick DK. Neuropathic pain: Role of inflammation, immune response, and ion channel activity in central injury mechanisms. Ann Neurosci. 2012;19(3):125–132.
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87(2):149–58. doi:10.1016/S0304-3959(00)00276-1.
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi:10.1016/0165-0270(94)90144-9.
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi:10.1016/0304-3959(88)90026-7.
- Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. doi:10.1093/nar/gkr688.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.