
ABSTRACT
CRISPR-Cas systems provide an adaptive defence against foreign nucleic acids guided by small RNAs (crRNAs) in archaea and bacteria. The Type III CRISPR systems are reported to carry RNase, RNA-activated DNase and cyclic oligoadenylate (cOA) synthetase activity, and are significantly different from other CRISPR systems. However, detailed features of target recognition, which are essential for enhancing target specificity remain unknown in Type III CRISPR systems. Here, we show that the Type III-B Cmr-α system in S. islandicus generates two constant lengths of crRNA independent of the length of the spacer. Either mutation at the 3ʹ-end of crRNA or target truncation greatly influences the target capture and cleavage by the Cmr-α effector complex. Furthermore, we found that cleavage at the tag-proximal site on the target RNA by the Cmr-α RNP complex is delayed relative to the other sites, which probably provides Cas10 more time to function as a guard against invaders. Using a mutagenesis assay in vivo, we discovered that a seed motif located at the tag-distal region of the crRNA is required by Cmr1α for target RNA capture by the Cmr-α system thereby enhancing target specificity and efficiency. These findings further refine the model for immune defence of Type III-B CRISPR-Cas system, commencing on capture, cleavage and regulation.
Introduction
CRISPR-Cas systems are widespread in archaea and bacteria, which provide an adaptive immune defence against invading genetic elements, including viruses and plasmids [Citation1–Citation3]. As new CRISPR-Cas systems are uncovered, the rapid evolution of highly diverse Cas proteins and the genomic architecture of CRISPR-Cas loci has led to the development of the latest classification scheme, among which the ribonucleoprotein complexes with multiple protein subunits or single protein were classified into Class 1 (Type I, III and IV) and Class 2 (Type II, V and VI), respectively [Citation4–Citation6]. The adaptive antiviral immunity functions in three stages. In the adaptation stage, short fragments of invading nucleic acids are acquired as new spacers into a CRISPR locus by adaptation proteins [Citation7–Citation10]. The second stage is defined as CRISPR expression or crRNA processing, a CRISPR locus is transcribed and precisely processed into crRNA [Citation11,Citation12]. Finally, mature crRNA and Cas proteins form a ribonucleoprotein complex (RNP) that recognizes and then cleaves incoming foreign DNA (or RNA) via sequence complementarity [Citation13,Citation14].
As research continues, a more comprehensive understanding of interference mechanisms has been gained. DNA interference in Type I, II, and V systems distinguishes non-self target DNA from self chromosomal DNA by relying on short conserved sequences defined as protospacer adjacent motifs (PAMs) [Citation15–Citation18]. It is noteworthy that with the exception of the Cmr7-including Type III-B system in Sulfolobus solfataricus and the Type III-B Cmr-β in Sulfolobus islandicus that are known to solely mediate RNA interference [Citation19–Citation22], Type III systems seem to target both DNA and RNA since a Type III-B system in S. islandicus was reported to target invader plasmids dependent on directional transcription of the protospacer in vivo [Citation23]. Subsequently, a Type III-A system which cleaves both target DNA and its transcript in Staphylococcus epidermidis was described [Citation24,Citation25]. A unified mechanism of RNA-activated DNA targeting by Csm(III-A) and Cmr(III-B) effector complexes was uncovered in Streptococcus thermophiles and Thermus thermophilus, and in Pyrococcus furiosus and Thermotoga maritime respectively [Citation26–Citation29]. We previously described ssDNA degradation activity which is activated by RNA complementary to crRNA by a Type III-B Cmr-α complex in S. islandicus [Citation30]. Moreover, Type III systems can avoid self-targeting by detecting sequence complementarity between the 5ʹ-tag of the crRNA and transcript of the protospacer, that manifests as PAM-independent [Citation23,Citation31,Citation32]. Even more interestingly, Type III RNP complexes were found not only to have crRNA-guided RNase and DNase activity but also cyclic oligoadenylate (cOA) synthetase activity [Citation33–Citation35]. The DNase and cOA synthetase activities catalyzed respectively by the HD domain and Palm domain of Cas10 are activated uniformly by the interaction between the Type III RNP complex and target RNA [Citation35–Citation38]. Meanwhile, the cyclic oligonucleotide signaling pathway coordinates Type III RNP complexes and CARF domain ribonucleases of Csm6/Csx1 families to prevent virus infection and propagation [Citation33–Citation35]. Recently, Athukoralage and colleagues uncovered a novel nuclease that degrades cyclic oligoadenylate to regulate Csm6/Csx1 activity [Citation39]. At this point, the overall framework of Type III CRISPR-Cas systems from interference to regulation is largely clear.
Besides that, the general attention of the research community has been on target recognition, which is essential for the function of all CRISPR-Cas systems. Seed regions are common features identified in several RNA-based systems, which contribute to ensure target specificity despite the fact that non-target nucleic acids share a degree of complementarity [Citation40–Citation42]. In Type I, II, and V systems, DNA interference requires perfect protospacer-crRNA complementarity in the region located adjacent to the PAM named the seed sequence [Citation43–Citation49]. Analogously, target RNA binding requires a protospacer flanking site (PFS) and induces conformational changes for the Type VI system [Citation50–Citation52]. In contrast, little is known about how Type III CRISPR-Cas systems capture target RNA. Although early studies revealed that an S. epidermidis Csm system tolerates mutations in the protospacer [Citation24,Citation53], a multi-nucleotide mutation at the 3ʹ-end of the spacer completely abolished Cmr-mediated RNA interference in S. islandicus [Citation21]. Indeed, it was predicted to be facilitated by rear-end subunit Cmr1 and Cmr6 of the P. furiosus Cmr complex [Citation54,Citation55]. Hence, an adequate conception of the molecular mechanism for target RNA capture in type III systems remains to be uncovered.
Sulfolobus islandicus REY15A encodes one Type I-A and two Type III-B (Cmr-α and Cmr-β) CRISPR-Cas modules [Citation56]. The Cmr-α system includes six component subunits: Cmr1α, Cmr2α (also known as Cas10), Cmr3α, Cmr4α, Cmr5α and Cmr6α. In this study, we showed two constant lengths of crRNA were generated by the Type III-B Cmr-α system independent of length of the spacer and the downstream repeat element in S. islandicus. We found that the tag-proximal cleavage site of the Cmr-α RNP complex for target RNA is delayed in action, probably to help maintain the active state of Cas10 against invaders. Especially, we identified a capture motif located at the tag-distal region of the crRNA to play a key role for the Cmr1 in targeting RNA capture, which directly affected the invader defence efficiency of the Cmr-α system.
Results
30 nt is the minimum spacer length for efficient Cmr-α RNA interference in S. islandicus
In any given CRISPR cassette, the length of spacers is similar (30–48 nucleotides), while their sequences vary. This raised the question whether the longer or shorter spacers can also mediate the interference of CRISPR system. This prompted us to test in vivo RNA interference activity of the Cmr-α system guided by crRNA derived from truncated SS1 spacers (matching the protospacer SS1 in the lacS gene, a sequence following the pentanucleotide 5ʹ-GAAAG-3ʹ which can protect the fatal immune defence activities of Type I-A and Type III-B systems) ranging from 20 to 76 nt in S. islandicus. In short, mini-CRISPR arrays containing a single copy of spacers were generated by oligo alignment and cloned to the pSe-Rp vector as previously described [Citation21], giving a series of artificial mini-CRISPR plasmids (pSS1). Since we aimed to characterize the Cmr-α system, a Cmr-β deletion mutant was constructed using S. islandicus E233, yielding S. islandicus ΔβE233 carrying ΔpyrEF ΔCmr-β (Table S1). This mutant was used as the host for transformation with each of the constructed pSS1 plasmids.
The specific activity of β-glycosidase in the transformants was determined with the results shown in . Plasmid-borne mini-CRISPR arrays carrying a spacer of ≥32 nt (32–76 nt) guided Cmr-α to confer a similar level of RNA interference in S. islandicus, suggesting that there are no significant differences in the RNA interference activity resulting from 32–76 nt spacers. When the spacer was truncated, the mini-CRISPR containing ≤29 nt SS1 spacer led about an eight-fold increase in reporter gene activity. However, there was still quite strong RNA targeting activity when the pSS1 plasmid carried a 30 nt spacer. We also employed qRT-PCR to directly analyse the quantity of mRNAs of lacS, the uncut mRNA levels were basically in line with specific activity of β-glycosidase (Figure S1). These results demonstrate that there is a minimum limit but not a maximum limit in spacer length for RNA interference of the Cmr-α CRISPR system in S. islandicus for the range of spacer lengths tested, and the minimal effective length of a spacer is 30 nt.
Figure 1. Effect of spacer length on RNA interference efficiency by Cmr-α in S. islandicus in vivo.
Schematic of the sequence region used for constructing the artificial mini-CRISPRs with varying length of spacers. They were located on the antisense strand from 690 relative to the start codon (ATG) of the lacS gene. Followed by a pentanucleotides (5ʹ-GAAAG-3ʹ), and Ter, terminator of the lacS gene. Relative β-glycosidase activity assay of ΔβE233 harbouring pSeSD or pSS1(N) plasmids is shown. pSS1(N) plasmids expressed an artificial mini-CRISPR with varying individual lengths of the spacers from 20 to 76 nucleotides. The right panel represents β-glycosidase activity of pSeSD (100%) and pSS1(N) plasmids relative to the former. Three independent transformants were analyzed for each construct with bars indicating standard deviations.
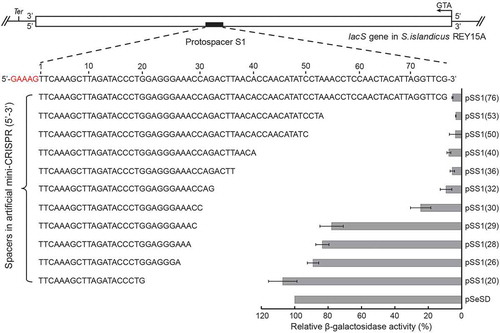
Cmr-α crRNA maturation strictly followed the ruler mechanism independent of spacer length
Previously, we established a Sulfolobus host-vector system from which native Cmr-α effector complexes can be purified from S. islandicus for biochemical characterization. The system uses an S. islandicus MF1 strain lacking both chromosomal CRISPR loci and only crRNAs expressed from plasmid-borne mini-CRISPRs will be assembled into the Cmr-α effector complex [Citation30]. Here we employed the system to investigate if the sizes of spacers could affect crRNA maturation in the S. islandicus Cmr-α effector complexes. A series of pSS1-cmr6α-10His expression plasmids (Table S2) each of which contained a single SS1 CRISPR array of a defined size (either 14, 20, 28, 40 or 76 nt) were generated. These plasmids were introduced into S. islandicus MF1 by electroporation, yielding transformants that were employed for purification of the native Cmr-α effector complex via nickel affinity purification of His-tagged Cmr-6α and size exclusion chromatography as previously described [Citation30].
Cmr-α complex was obtained from each strain. Analysing these purified Cmr-α complexes indicated that they had the same composition in their protein and crRNA components (Figure S2). All the Cmr-α complexes contained crRNAs of two constant lengths, 40 and 46 nt, regardless whether the length of the spacer in the plasmid-borne CRISPR array was 14 or 76 nt (Figure S2B). Otherwise, a construct containing only upstream repeat and a 40 nt spacer, also produced mature crRNAs which were exclusively 40 and 46 nt (Figure S3A). According to the correlation between the length of crRNA and the RNA cleavage patterns of our previous data [Citation30], we determined that the Cmr-α complexes carrying 46 nt crRNAs contain four Cmr4 and three Cmr5 subunits while three Cmr4 and two Cmr5 for the small ones with 40 nt crRNAs, besides one for each of other four subunits (Cmr1, 2, 3 and 6), consistent with the current model of Cmr systems deduced by the EM structure and native MS analysis [Citation57,Citation58]. Together, these results indicated that the Cmr-α system exhibited very stringent control on the size of the effector complexes, carrying two species of crRNAs with constant lengths followed the ruler mechanism measuring from the 5ʹ-handle of the crRNA.
Tolerance of Cmr-α to spacer truncation in RNA cleavage and binding in vitro
According to the RNA sequencing results previously obtained, crRNAs of Cmr-α complex started with the 8 nt repeat tag at the 5ʹ-end [Citation30], and we predicted the sequences of crRNAs from five RNP complexes. As shown in , both of the constructs harbouring 40 and 76 nt spacers had enough nucleic acid bases to be processed into the guide sequence of crRNAs, while the subsequent repeat sequence should be incorporated as crRNA in constructs with 28, 20 and 14 nt length spacers.
Figure 2. Influence of spacer length on in vitro RNA cleavage and RNA binding activity by the Cmr-α system.
(a) Predicted hairpin conformation of S. islandicus REY15A repeat elements (in red) and sequence of mature 40/46nt crRNAs with varying length of spacers. The primary processing site relying on Cas6 is indicated with a green triangle. Underlined bases indicate the 8 nt tag of the repeat sequence at the 5ʹ-end after primary processing. Two target RNAs used in the assay are shown in yellow. Predicted cleavage sites are indicated with red (for RNP complexes with 40/46 nt crRNAs) and black (only for RNP complexes with 46 nt crRNAs) triangles. (b) In vitro RNA cleavage assay using the Cmr-α complexes purified from each strain harbouring different lengths of spacers (76 nt, 40 nt, 28 nt, 20 nt and 14 nt). Fifty nanomolars of each Cmr-α complex was incubated with 25 nM labelled SS1-46 RNA for indicated time points. Then, the samples were analysed by denaturing PAGE. (c) Comparison of RNA cleavage on SS1-46 RNA and a trinucleotide mutation substrate (28–30 nt) by Cmr-α and Cmr-αΔ1 complexes. Cmr-αΔ1 – Cmr-α complex without Cmr1α, which was purified from S. islandicus ΔCmr1α. (d) Target RNA binding assay of Cmr-α complex with varying length of spacers (40 nt, 28 nt, 20 nt and 14 nt) for indicated amounts of SS1-46 RNA. Left: binding buffer without EDTA, Right: binding buffer with EDTA.
Each Cmr-α effector complex was then tested for its capability to cleave a radio-labelled SS1-46 target RNA using the cleavage assay. High RNA cleavage activity was observed for Cmr-α-76 and Cmr-α-40 effector complexes among which four cleavage products were produced, while Cmr-α-28 produced only three products, although the complementary region between the crRNA and the target RNA covered all four cleavage sites. Much weaker RNA cleavage activity was observed for Cmr-α-20 and Cmr-α-14, among which, Cmr-α-20 cleaved all three sites present in the crRNA/target RNA complementary region except for the fourth site that was located in the mismatched region, and only two sites were cut weakly by Cmr-α-14 (). Combined with existing research on RNA cleavage modules of Type III systems, we marked three cleavage sites belonging to both Cmr-α RNP complexes (red triangle) and another cleavage site which can only be cut by a long RNP complex (46 nt crRNA, black triangle; ). This prompted us the inaction at the fourth cleavage site of target RNA by Cmr-α-28 effector complex should be because of the long complex does not work resulting from mismatch between the 3ʹ-end of the 46 nt crRNA and the 5ʹ-end of the target RNA due to the truncation of the spacer.
Previously, we identified a trinucleotide sequence in the crRNA that is crucial for RNA interference and is positioned at nucleotides 28–30 of the spacer [Citation21], we also found that Cmr1α is important for efficient NA interference activities of Cmr-α complex as an integral activator [Citation59]. Thus, we analysed the RNA cleavage activity of Cmr-α and Cmr-αΔ1 (purified from S. islandicus in which the cmr1α gene was deleted) effector complexes using either the wild-type or 28–30 nt mutated SS1-46 target RNA. As shown in , trinucleotide mutation at 28–30 nt substantially reduced the RNA interference of the Cmr-α complex and completely abolished RNA interference of Cmr-αΔ1. Given closer analysis we found that the cutting pattern of mixed complexes to SS1-46 target RNA resulted in the simultaneous enrichment of the third and fourth cutting products, while the cut pattern was of a linear accumulation of four products for the 28–30 nt mutated target RNA (). This suggested that the mismatch at 28–30 nt of crRNA with target RNA (shown in in blue) eliminated the RNA cleavage activity of the 40 nt crRNA complex.
We next examined the binding of target RNA to each purified Cmr-α effector complex. The target RNA was radio-labelled and mixed with the incremental amounts of Cmr-α effector complex indicated in each experiment and incubated in cleavage buffer with or without EDTA for 15 min at 70°C. If the target RNA could strongly bind the Cmr-α effector complex, the resulting target RNA-Cmr-α complex could be detected by electrophoretic mobility shift assay using non-denatured PAGE since it migrates slower due to its large molecular weight. Only Cmr-α-40 strongly bound to the target RNA, forming multiple bands of high molecular weights (). Cmr-α-28 formed a slight aggregation, while the binding shift of Cmr-α-20 and Cmr-α-14 was much weaker. These Cmr-α complexes were also investigated for their RNA-activated DNA cleavage using 5ʹ-labelled S10 ssDNA, which does not show any sequence similarity with the SS1 spacer (Table S2), and the SS1-46 was used as activator RNA. We found that while Cmr-α-14 showed inconsiderable DNA cleavage activity, the remaining four effector complexes, Cmr-α-28 and Cmr-α-20 showed diminished ssDNA degradation compared with Cmr-α-76 and Cmr-α-40 (Figure S3B). Taken together, NA cleavage data correlated with binding affinity, as spacers were shortened, the 3ʹ-end of crRNA that includes the repeat sequence () was gradually not complementary to the target RNA, which greatly reduced the stability of the tertiary complex of Cmr-α further influencing the target cleavage.
Target RNA binding and cleavage by Cmr-α complex
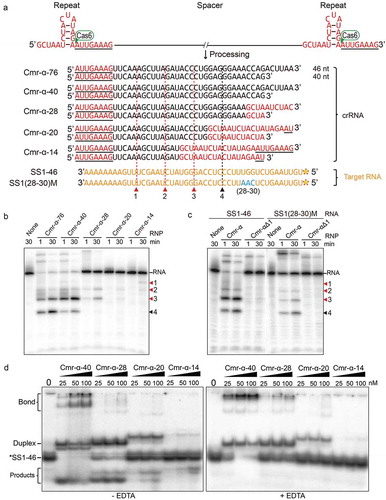
It has been shown that RNA substrates containing non, complementary or mismatch flanking sequence with the 8 nt tag at the 5ʹ-end of crRNA can be cleaved effectively by all reported Type III effector complexes [Citation27,Citation30,Citation60,Citation61]. To further explore insights into the effect of base pairing between target RNA and crRNA for target RNA binding and cleavage, we designed a set of truncations of SS1 target RNA. SS1/40 is a 40 nt RNA substrate that covers the whole guide sequence in both crRNAs, while SS1/32 is only complementary to the 32 nt guide RNA sequence exclusive of the 8 nt tag in the 40 nt crRNA, which would distinguish the two sizes of Cmr-α complexes and characterize the 40 nt one more directly. SS1/24 and SS1/16–1 are 8 nt truncated from the 5ʹ-end of SS1/32 successively, whereas SS1/16–2 is the 5ʹ-end half of the SS1/32 ().
Figure 3. Base pairing at the 3ʹ-end of crRNA with target RNA is important for RNA cleavage and binding of the Cmr-α complex in vitro.
(a) Schematic representation of two species of Cmr-α complexes (with 40/46 nt crRNAs) and RNA substrates (including 40 nt SS1 target RNA and different truncations) used in the cleavage assay. Red (for RNP complexes with 40/46 nt crRNAs) and black (only for RNP complexes with 46 nt crRNAs) triangles show the cleavage sites detected below. (b) RNA cleavage assay of 5ʹ-end labelled and 3ʹ-end labelled substrates (shown in A) by Cmr-α complexes for the indicated time points. (c) Target RNA binding assay of Cmr-α complexes from 0 nM to 100 nM with different lengths of target RNA, reactions were incubated with the same conditions as in (B).
All the RNA substrates were radiolabeled at 5ʹ-ends and 3ʹ-ends, respectively, then 25 nM of each was incubated with 50 nM Cmr-α complexes for 1 min and 30 min at 70 ℃. The products were subsequently analysed by denaturing PAGE. We found that the truncations did not affect the cleavage sites located at fixed positions on target RNAs. As shown in , for 5ʹ-labelled targets, compared with SS1/40 target RNA, cleavage at site 4 in SS1/32 was not obvious, suggesting that the 46 nt crRNA complexes cannot degrade the SS1/32 RNA lacking pairing with the 3ʹ-end of crRNA. For SS1/24 and SS1/16–1, they were resistant to degradation compared with SS1/32 (which were not cut after 1 min) perhaps due to fact the 40 nt crRNA complexes were also deactivated when the 5ʹ-end of target RNA was missing. The remaining SS1/16–2 target RNA also was not cleaved at site 4 presumably due to the same reason as for SS1/32, while site 3 was only cut weakly after 30 min probably due to the loose construction of Cmr-α RNP::target RNA tertiary complexes at site 3 that only one base pair on left. For 3ʹ-labelled targets, we found that the major product of cleavage of SS1/32 target RNA corresponds to cleavage at site 2 and 3 even at the earliest time points, while the generation of products from cutting at site 1 (closest to the crRNA tag) lags behind obviously, and longer products turned into smaller ones as incubating time were increased. The results suggest that the Cmr-α complex cuts target RNAs initially at the cleavage site in the middle of the crRNA and the cleavage at site 1 appears to be last and delayed.
To investigate the binding affinity of Cmr-α complexes to truncated target RNAs, 25 nM of each RNA substrate was incubated with Cmr-α complexes at the indicated concentrations for 15 min at 70°C (), then were analysed by non-denatured PAGE. We found that SS1/40 and SS1/32 could be bound by Cmr-α-40 RNP complex efficiently. Remarkably, the 3ʹ-end truncated substrate SS1/16–2 formed stable RNP::target RNA tertiary complexes with 25 nM RNP similar to SS1/32, while SS1/24 and SS1/16–1 could not, suggesting that the base pairing at the 3ʹ-end of crRNA with target RNA is required for target RNA binding of the Cmr-α effector complex.
A hexanucleotide capture motif important for immune defence of the Cmr-α system was located at the 3ʹ-end of crRNA
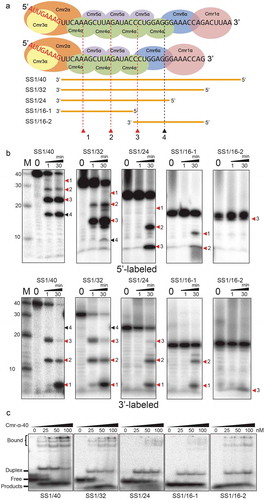
To yield an insight into the capture motif for the efficient defence against invaders by the Cmr-α system, we firstly tested the importance of sequence complementarity between crRNA and target RNA on RNA interference. A number of pSS1 plasmids carrying mutated 32 nt SS1 spacers were introduced to S. islandicus ΔβE233 by transformation and studied for RNA interference activity by measuring the specific activity of β-glycosidase in the transformants. As shown in , the introduced quadruple mutations to the first half of the SS1 spacer (positions 1–16) showed no significant difference with the wild-type spacer. A series of subtle mutations were generated in the second half of the spacer, and we found that five coherent dual mutations, i.e. 25/26, 26/27, 27/28, 28/29 and 29/30 reduced targeting activity by ~65%, ~47%, ~35%, ~50% and ~66%, respectively. This interesting result suggested that the important sequence motif of crRNA was located within 25–30 nt. In view of the fact that two ends of this hexanucleotide showed greater influence on RNA interference, we generated another dual mutation including G25T and C30A. Notably, this dual mutation reduced gene silencing by more than 90%.
Figure 4. Identification of the capture motif in crRNA for RNA interference by the Cmr-α system.
A schematic representation of the Cmr-α complex with 40 nt SS1 crRNA and target RNA (yellow) is shown on the top of the left panel. SS1 spacer and its mutants are annotated according to the positions mutated. The mutated bases are shown in the left panel. The right panel illustrates relative RNA targeting activity of strains with pSS1 [Citation32] (wide-type 32 nt spacer, defined as 100%) and mutated plasmids relative to the former. A dual mutation with greatly reduced gene silencing activity is highlighted in blue. Three independent transformants were analysed for each construct with error bars indicating standard deviations.
![Figure 4. Identification of the capture motif in crRNA for RNA interference by the Cmr-α system.A schematic representation of the Cmr-α complex with 40 nt SS1 crRNA and target RNA (yellow) is shown on the top of the left panel. SS1 spacer and its mutants are annotated according to the positions mutated. The mutated bases are shown in the left panel. The right panel illustrates relative RNA targeting activity of strains with pSS1 [Citation32] (wide-type 32 nt spacer, defined as 100%) and mutated plasmids relative to the former. A dual mutation with greatly reduced gene silencing activity is highlighted in blue. Three independent transformants were analysed for each construct with error bars indicating standard deviations.](/cms/asset/8de26353-1161-4397-8bba-7e149f3b657f/krnb_a_1618693_f0004_oc.jpg)
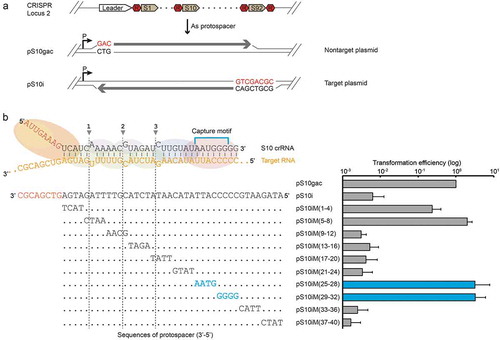
To investigate this further, the transcription-dependent immune defence of Cmr-α system was evaluated by an invader plasmid assay. An invader plasmid carrying an artificial protospacer matching spacer 10 of CRISPR locus 2 (pS10i) and its quadruple mutations were employed together with non-targeted plasmid (pS10gac), which carried an inverted protospacer, as a control plasmid (). While the pS10i was susceptible to Type III-B interference, pS10gac was not targeted by both Type I-A and III-B interference since a GAC sequence replaced the CCN PAM sequence (for Type I interference activity) and the yielding transcripts were non-complementary to crRNAs [Citation23]. As shown in , intriguingly, in comparison with RNA interference activity, not only the tag-distal mismatches (nt 25–32) significantly increased the transformation efficiency but also another tag-proximal important region located at 1–8 nt in crRNA. Indeed, the obvious effect of the tag-proximal region was also observed for other Type III systems [Citation26,Citation36,Citation37,Citation62]. Taken together, these results established that the capture motif (nt 25–30) located at the tag-distal region of crRNA is important for immune defence by the Cmr-α system.
Figure 5. Identification of the capture motif in crRNA for invader plasmid interference of the Cmr-α system.
(a) A scheme of CRISPR locus 2 showing the spacer 10 that was targeted, where R and S denote repeat and spacer, respectively. Two plasmid constructs are presented showing the protospacer orientation and its adjacent sequence. pS10i – an invader plasmid carrying a target sequence of spacer 10 in CRISPR locus 2 in S. islandicus REY15A. (b) A schematic representation of the Cmr-α complex with 40 nt S10 crRNA and target RNA (yellow) is shown on the top of the left panel. Protospacer S10i and its mutants are annotated according to the positions mutated. The mutated bases are shown in the left panel. The right panel illustrates the in vivo DNA interference activity assayed in S. islandicus by transformation with different invader plasmids. Mutated regions consistent with the established capture motif are highlighted in blue. Three independent transformations were performed and average relative transformation efficiencies are given with standard deviations. The transformation efficiency of the reference plasmid pS10gac was taken as 100%.
Discussion
Type III CRISPR-Cas systems employ a sophisticated interference mechanism to degrade the invading genetic elements. Although today there is a unified simple model that seems to apply to all Type III subtypes, many more details are still not clear.
RNP assembly and crRNA maturation of the type III CRISPR-Cas system
Here, we firstly explored the specific characteristics of crRNA maturation in the Type III-B Cmr-α system by using varying length of spacer in S. islandicus REY15A. We found that the crRNA generated from all constructs remained 40 nt and 46 nt in length, even an artificial mini-CRISPR plasmid harbouring only upstream repeat elements followed by a 40 nt spacer also produced mature crRNAs similar to wild-type (Figure S3A). We hypothesized that Cmr-α complex formation is launched from the 5ʹ primary processing site to produce two species of crRNA with a constant length and independent on whether there is a repeat sequence at the 3ʹ end. Interestingly, previous studies on other organisms have shown that mature crRNAs are 31–67 nucleotides in length for Type III-A Csm complexes in S. epidermidis [Citation63], Csm systems of S. thermophilus can also assemble into an RNP filament that contains a long 72 nt crRNA [Citation61], the most abundant crRNAs varied in length from 35 to 53 nt in Csm complexes of T. thermophilus [Citation64], and Cmr RNP complexes can be reconstituted by recombinant Cmr proteins with a 67 nt crRNA which can achieve additional target RNA cleavages elongating the guide sequence in P. furiosus [Citation55]. Csm3 and Cmr4-like subunits of Type III-A Csm complexes in S. epidermidis were confirmed as a ruler that determines the mature crRNA length with multiple copies [Citation65], whereas the determinant of copies of Csm3/Cmr4 is still unknown, especially for the Type III-B system with two constant molecules of complex in our study. Furthermore, although the deletion of SeCsm5 (homologue of Cmr1) caused severe maturation defects [Citation66], our results showed the other five Cmr-α proteins can be purified as a small complex harbouring degenerated crRNA in the absence of Cmr1α suggesting that Cmr1α is not responsible for crRNA maturation [Citation59]. Thus, it remains to be determined what is the regulatory mechanism to determine the constant size of Cmr-α complexes in S.islandicus REY15A.
Capture motif located at the tag-distal region of crRNA is required by Cmr1
Our results showed that the mismatch between the 3ʹ-end of crRNA and the 5ʹ-end of target RNA which was attributed to the spacer truncation and supplement of the downstream repeat element into mature crRNA, significantly reduced both RNA and ssDNA cleavage activity ( and S3B), which is consistent with the results from the target RNA binding assay ()). Moreover, truncation analysis of target RNA also showed that when the target RNA was truncated to 24 nt, there was an abrupt drop in both binding affinity and cleavage activity compared to SS1/32 which is complementary to the 32 nt guide RNA sequence (). Interestingly, the truncated target RNA SS1/16–2 showed stronger binding shifts than SS1/24 and SS1/16–1 in the binding affinity assay (, )), reinforcing the inference that the 3ʹ-end of crRNA in the Cmr-α effector complex is responsible for capture of target RNA [Citation55]. It was also previously shown that mismatch between the tag-distal (nt 18–19) of crRNA and target RNA sequence suppresses the ssDNA cleavage activity of a Type III-A complex [Citation26].
Previously, the rear-end subunits Cmr1 and Cmr6 of the Cmr complex have been predicted to facilitate target RNA capture [Citation55]. Furthermore, PfCmr1 has a nucleotide-binding pocket and cross-links with the 3ʹ region of the crRNA [Citation54,Citation57], and Cmr1 is absent from the complex without crRNA, but all six Cmr proteins purified as a complex in the presence of crRNA [Citation27]. More straightforward, the chimeric Cmr complex without Cmr1 (PfCmr2-Cmr3 and AfCmr4-Cmr5-Cmr6, PDB: 3X1L) carried a 32 nt crRNA containing 8 nt tag and 24 nt guide sequence [Citation67], prompting the proposal that the capture motif (nt 25–30) is the location of the Cmr1 binding region. Combined with our earlier study, which found that the complex without Cmr1α showed the weakest binding affinity and cleavage activity to target RNA [Citation59], we further clearly confirmed that Cmr1 plays a key role in target capture which enables base pairing between the capture motif at the 3ʹ-end of crRNA with target RNA of the Cmr-α complex.
Control of DNase and cOA synthetase activity in type III CRISPR-Cas systems
Cognate target RNAs activate both DNase and cOA synthetase activity of Cas10 in Type III CRISPR-Cas systems and their inactivation is thought to occur upon RNA cleavage and product release [Citation22,Citation25–Citation28,Citation36]. The single-molecule fluorescence microscopy assay investigated that Cas10 is locked in a static configuration on self CRISPR transcripts carrying 3ʹ-flanking sequence (anti-tag) that base pairing with the crRNA 5ʹ-tag, but samples a large conformational space when binds to non-self RNA containing a scrambled 3ʹ-flanking sequence that is non-complementary to the crRNA 5ʹ-tag [Citation37]. The presence of ssDNA and ATP both further enrich the specific Cas10 conformations at the active configuration [Citation37]. Notably, the cryo-EM structural study revealed that the complementary 3ʹ anti-tag of target RNA forms four base pairs with nucleotides −2 to −5 within the 5ʹ-tag of crRNA, and these four nucleotides of target RNA do not interact with the adjacent disordered fragments of Csm1 (Cas10) subunit. In contrast, the interaction between the non-complementary 3ʹ anti-tag region of target RNA and Csm1 Linker is important for both target RNA-activated DNA cleavage and cOA synthesis [Citation38]. Moreover, the mutagenesis assays on the Csm1 Linker showed that DNase and cOA synthetase activities are required by different residues [Citation38]. Taken together, although both DNase and cOA synthetase activities depend on a non-complementary 3ʹ flanking sequence but have little interdependency, consistent with previous study that ssDNA and ATP are two types of substrates of HD and PALM domains of Cas10 [Citation33,Citation34]. If it is the same principle in vivo, the DNase and cOA synthetase have to be carefully regulated to ensure efficient antiviral defence and avoid off-targets from killing the infected cell since activated Cas10 and Csm6/Csx1 function on non-specific substrate DNA and RNA.
Our findings indicate that there is another region of target RNA associated with the tag-proximal of the crRNA important for invader plasmid defence, which has also has been observed in Type III-A systems [Citation37,Citation62]. We reasoned that it would be due to the mismatch at the tag-proximal region of crRNA that influences the active conformational change of Cas10. In this regard, there appears to be a double-check for specific invader defence of Type III-B systems: one is the capture motif for efficient target binding, another one is a relatively stable base-pairing between the tag-proximal of crRNA and target RNA like a ‘fail-safe’ that enabling allostery of Cas10, the premise of both is the non-complementary 3ʹ flanking sequence of target RNA with the 5ʹ-tag of crRNA. Furthermore, the observed delayed cleavage at the tag-proximal site could reflect a requirement of the Cmr complex to help maintain the active state of Cas10 against invaders. On the other hand, Cas10 could be deactivated rapidly as target RNA is cleaved and products dissociate [Citation36]. Moreover, a ring nuclease can degrade cOA and switch off the antiviral state as an important determinant [Citation39].
So far, seed sequences have been found that function by a similar principle to facilitate the recognition of targets for several RNA-based regulatory systems, including eukaryotic microRNAs (miRNAs) [Citation41,Citation68], prokaryotic small RNAs (sRNAs) [Citation69,Citation70] and CRISPR RNAs (crRNAs) [Citation43–Citation49]. For example, the binding between Cas9:gRNA and target DNA exhibited a R-Loop (termed the ‘zipped conformation’) expanding from 3ʹ-end to 5ʹ-end of crRNA [Citation47]. Similar to target recognition by other CRISPR-Cas systems, they share the following characteristics: (i) biochemistry assays in vitro showed mutation in the seed region did not exhibit significant target binding () and the seed region of the target exhibited higher affinity to the RNP complex ()); (ii) loss of function assay in vivo showed mutagenesis at the seed region of crRNA or corresponding protospacer significantly silenced the system (, ). In this respect, we hypothesized that the target recognition and capture of the Cmr-α complex begins in a zipper slider conformation of Cmr1α:crRNA:target then zipping up to tag-distal of crRNA.
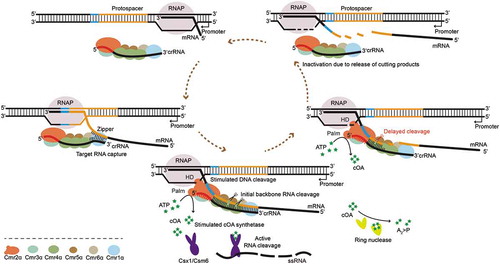
In conclusion, a clearer model has emerged from previous studies and our findings presented here (), in detail, Cmr1α mediates the Cmr-α complex to capture the pre-transcript through base pairing with the capture motif at the 3ʹ-end of crRNA, forming a ‘zipper’ slider. With the passage of the ‘zipper’, Cmr4α cleaves the target mRNA stepwise on four sites. Meanwhile, the mismatch between the 5ʹ-tag of crRNA and 3ʹ-flanking sequence of target RNA activates the DNase and cOA synthetase activity respectively from the HD domain and Palm domain of Cmr2α in the Cmr-α complex. After backbone cleavage of target mRNA, cleavage products are released from the Cmr-α RNP complex. Meanwhile, a ring nuclease degrades cOA and switches off the antiviral state of the RNP complex through another pathway. Then, the Cmr-α RNP complex can start the next round of activity. In addition, non-target RNAs with non or complementary 3ʹ-flanking sequences proceed with the similar processing steps, but without the activated DNase and cOA synthetase activity of Cmr2α which could avoid self-immunity. We require more experiments such as the crystal structure analysis of Type III-B RNP complex to test this model.
Figure 6. A model for target RNA capture mediating immune defence of the Type III-B CRISPR–Cas system in S. islandicus.
(i) Once RNA polymerase transcribes through the protospacer (yellow), the Cmr1α leads the Cmr-α complex to capture the nascent transcript through base pairing with the seed sequence at the 3ʹ-end of crRNA, forming a ‘zipper’ slider. (ii) With the passage of this ‘zipper’, Cmr4α cleaves the target mRNA stepwise on four sites. Meanwhile, the mismatch between the 5ʹ-tag (red) of crRNA and 3ʹ-flanking sequence (blue) of the protospacer region in the transcript activates the DNase and cOA synthetase activity respectively from the HD domain and Palm domain of Cmr2α in the Cmr-α complex. The tag-proximal cleavage site of the Cmr-α RNP complex to target RNA is delayed in action, which probably gives Cmr2α more time to function. (iii) After backbone cleavage of target mRNA, cleavage products are released from the Cmr-α RNP complex. Then, the Cmr-α RNP complex can start the next round of activity. Meanwhile, a ring nuclease degrades cOA and switches off the antiviral state of the RNP complex through another pathway. Non-target RNAs with a non or complementary 3ʹ-flanking sequence to the 8 nt 5ʹ-tag of crRNA proceed through the similar processing steps but without the activated DNase and cOA synthetase activity of Cmr2α.
Material and methods
Strains, growth conditions and transformation of Sulfolobus
Genetic hosts used in this work are listed in Table S1. S. islandicus E233, E233S1, ∆βE233, MF1 and ∆β∆1α were reported previously [Citation30,Citation59,Citation71,Citation72]. Sulfolobus strains were grown at 78°C in an SCV medium (0.2% sucrose, 0.2% casamino acids plus 1% vitamin solution) or SCVy (SCV + 0.0025% yeast extract) and uracil was supplemented to 20 μg/mL if required. Sulfolobus competent cells were prepared and transformed by electroporation as previously described [Citation71].
Construction of plasmids
The reference plasmid pSeSD, invader plasmid pS10gac and pS10i were reported previously [Citation23,Citation73]. pS10iM(N) plasmids were constructed like pS10i as described earlier, carrying mutations on the protospacer (Table S2). All plasmids carrying artificial mini-CRISPR with varying length of spacers were constructed individually by cloning a single spacer (matching the protospacer SS1 in the lacS gene) into pSe-Rp, a Sulfolobus CRISPR-cloning vector [Citation21]. Spacer fragments were generated by annealing of the corresponding complementary oligonucleotides and inserted into pSe-Rp at the BspMI sites, yielding pSS1(N) (Table S2). The 10× His-tagged cmr6α including arabinose promoter and terminator was inserted into pSS1(N) at SalI and SmalI sites, resulting in pAC(N)-cmr6α-10His. The pR40-cmr6α-10His plasmid was constructed through replacing the Repeat-SS1-Repeat fragment in pSS1 [Citation40]-cmr6α-10His with the Repeat-SS1 fragment. All the oligonucleotides (Table S3) were synthesized from Tsingke (Wuhan, China) and the sequences of all plasmid constructs were verified by DNA sequencing (Tsingke, Wuhan, China).
Determination of β- glycosidase activity
To determine relative β-glycosidase activity of each transformant carrying artificial mini-CRISPR plasmids, the S. islandicus strains were grown in SCV medium, and cells were collected for preparing cellular extracts when the A600 (absorbance at 600 nm) of the culture reached 0.2–0.4. Protein content in the cellular extracts was determined using the Bradford Protein Assay Kit (Beyotime) whereas the β-glycosidase activity was determined using the ONPG (ρ-nitrophenyl-β-D-galact- opyranoside) method as described previously [Citation74].
Evaluation of RNA interference efficiency by qRT-PCR
To evaluate the RNA interference efficiency, qRT-PCR was performed as described previously [Citation59]. Briefly, total RNA was extracted from three transformants of each mutant harbouring pSS1(N) and pSeSD (A600 = 0.3) using the TRIZOL® reagent (Invitrogen) according to the manufacturer’s protocol. Then, 2 μL of DNase-treated RNA (DNaseI, Thermo Scientific) was incubated with Revert Aid Reverse Transcriptase (Thermo Scientific) for first-strand cDNA synthesis using a lacS-RT-R primer (Table S3), which annealed downstream of the lacS gene. Two primer sets Qtar (amplifying the target region) and Qref (an internal control) were used for amplification using iTaqTM Universal SYBR® Green Supermix (Bio-Rad) and a CFX384 Real-Time System (Bio-Rad) following the PCR conditions: denaturing at 95°C for 5 min, 40 cycles of 95°C 15 s, 55°C 15 s and 72°C 20 s. The amount of uncut mRNA is expressed as a ratio of the target mRNA (lacS) levels in transformants of pSS1(N) versus pSeSD. Data analysis was performed using the comparative Ct value method.
Purification of Cmr-α effector complex and extraction of crRNA
All the S. islandicus strains carrying artificial mini-CRISPR locus and 10× His-tagged cmr6α expression plasmid were grown in SCVy medium, and cells were collected from at least 6 L of culture for each strain by centrifugation when A600 of the culture reached 0.7–0.8. Then, the Cmr-α effector complexes containing Cmr6α with a 10× His tag to the C-terminus were purified as described previously [Citation59]. crRNAs were extracted from purified Cmr-α effector complexes using TRIZOL® reagent (Invitrogen) following the protocol described previously [Citation30]. Briefly, 100 µL of purified Cmr-α complexes were mixed with 600 µL Trizol and 300 µL chloroform, and after incubation at room temperature for 5 min, the mixture was centrifuged at 12,000 rpm for 15 min. The upper phase was transferred into a new tube and precipitated by one volume of isopropanol. RNA precipitates were washed with 1 mL of pre-chilled 75% ethanol. After air-drying for 30 min at room temperature, RNA pellets were resuspended in 15 µL DEPC-H2O.
Labelling of DNA and RNA
An aliquot of the purified crRNA was first 5ʹ labelled by 32P-γ-ATP (PerkinElmer) using T4 polynucleotide kinase (Thermo Fisher Scientific), and then separated on a large denatured polyacrylamide gel. DNA and RNA substrates used in cleavage assays were 5ʹ labelled with 32P-γ-ATP using T4 polynucleotide kinase and 3ʹ labelled with [32P]pCp (PerkinElmer) using T4 RNA ligase (Invitrogen). All nucleic acids were purified by recovering the corresponding bands from a denaturing polyacrylamide gel.
RNA and ssDNA cleavage assays
RNA and DNA cleavage assays were conducted as described previously [Citation30]. Fifty nanomolars purified Cmr-α effector complexes and 25 nM 32P-5ʹ-labelled (or 32P-3ʹ-labelled) synthetic target RNA were mixed in cleavage buffer (50 mM Tris-Cl pH 7.6, 10 mM MgCl2, 5 mM DTT) and incubated at 70°C for the indicated time period. To test the RNA activated DNA cleavage activity of Cmr-α effector complexes, 25 nM ssDNA substrates (a synthetic single strand DNA fragment S10) were incubated together with 50 nM different Cmr-α complexes and 200 nM unlabelled activator RNA. All reactions were stopped by adding 2× RNA loading dye (New England Biolabs) then heated at 95°C for 5 min and cooling on ice before loading onto a 18% denaturing polyacrylamide gel (18% polyacrylamide, 40% urea, 0.5× TBE). Radioactive signals were visualized by phosphor imaging using a Typhoon FLA 7000 (GE Healthcare).
Target RNA binding assay
For the binding assay, 25 nM of 32P-5ʹ-labelled RNA was incubated with the amount of Cmr-α complex indicated in each assay in cleavage buffer (50 mM Tris-Cl pH 7.6, 10 mM MgCl2, 5 mM DTT) with or without 10 mM EDTA. After 15 min incubation at 70°C, the samples were loaded on a 10% nondenaturing polyacrylamide gel (10% polyacrylamide, 0.5× TBE).
Supplemental Material
Download MS Word (3.3 MB)Acknowledgments
We thank the Danish Archaea Centre in Copenhagen University for its help in biochemical experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170.
- Barrangou R, Marraffini LA. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54:234–244.
- van der Oost J, Westra ER, Jackson RN, et al. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–492.
- Mohanraju P, Makarova KS, Zetsche B, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147.
- Makarova KS, Zhang F, Koonin EV. SnapShot: class 1 CRISPR-Cas systems. Cell. 2017;168(946–946):e941.
- Makarova KS, Zhang F, Koonin EV. SnapShot: class 2 CRISPR-Cas systems. Cell. 2017;168(328–328):e321.
- Rollie C, Schneider S, Brinkmann AS, et al. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife. 2015;4:e08716.
- Arslan Z, Hermanns V, Wurm R, et al. Detection and characterization of spacer integration intermediates in type I-E CRISPR-Cas system. Nucleic Acids Res. 2014;42:7884–7893.
- Silas S, Mohr G, Sidote DJ, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351:aad4234.
- Xiao Y, Ng S, Nam KH, et al. How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature. 2017;550:137–141.
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297.
- Charpentier E, Richter H, van der Oost J, et al. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39:428–441.
- Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964.
- Plagens A, Richter H, Charpentier E, et al. DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol Rev. 2015;39:442–463.
- Anders C, Niewoehner O, Duerst A, et al. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573.
- Westra ER, Semenova E, Datsenko KA, et al. Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013;9:e1003742.
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740.
- Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771.
- Zhang J, Rouillon C, Kerou M, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell. 2012;45:303–313.
- Zebec Z, Manica A, Zhang J, et al. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014;42:5280–5288.
- Peng W, Feng M, Feng X, et al. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015;43:406–417.
- Zhang J, Graham S, Tello A, et al. Multiple nucleic acid cleavage modes in divergent type III CRISPR systems. Nucleic Acids Res. 2016;44:1789–1799.
- Deng L, Garrett RA, Shah SA, et al. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol. 2013;87:1088–1099.
- Goldberg GW, Jiang W, Bikard D, et al. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014;514:633–637.
- Samai P, Pyenson N, Jiang W, et al. Co-transcriptional DNA and RNA cleavage during Type III CRISPR-Cas immunity. Cell. 2015;161:1164–1174.
- Kazlauskiene M, Tamulaitis G, Kostiuk G, et al. Spatiotemporal control of Type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol Cell. 2016;62:295–306.
- Estrella MA, Kuo FT, Bailey S. RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 2016;30:460–470.
- Elmore JR, Sheppard NF, Ramia N, et al. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016;30:447–459.
- Liu TY, Iavarone AT, Doudna JA. RNA and DNA targeting by a reconstituted thermus thermophilus Type III-A CRISPR-Cas system. PLoS One. 2017;12:e0170552.
- Han W, Li Y, Deng L, et al. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017;45:1983–1993.
- Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571.
- Jackson RN, Wiedenheft B. A conserved structural chassis for mounting versatile CRISPR RNA-guided immune responses. Mol Cell. 2015;58:722–728.
- Kazlauskiene M, Kostiuk G, Venclovas C, et al. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017;357:605–609.
- Niewoehner O, Garcia-Doval C, Rostol JT, et al. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017;548:543–548.
- Han W, Stella S, Zhang Y, et al. A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 2018.
- Rouillon C, Athukoralage JS, Graham S, et al. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. Elife. 2018;7:e36734.
- Wang L, Mo CY, Wasserman MR, et al. Dynamics of Cas10 govern discrimination between self and non-self in Type III CRISPR-Cas immunity. Mol Cell. 2019;73(278–290):e4.
- You L, Ma J, Wang J, et al. Structure studies of the CRISPR-Csm complex reveal mechanism of co-transcriptional interference. Cell. 2019;176(239–253):e16.
- Athukoralage JS, Rouillon C, Graham S, et al. Ring nucleases deactivate type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature. 2018;562:277–280.
- Gorski SA, Vogel J, Doudna JA. RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol. 2017;18:215–228.
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233.
- Kunne T, Swarts DC, Brouns SJ. Planting the seed: target recognition of short guide RNAs. Trends Microbiol. 2014;22:74–83.
- Semenova E, Jore MM, Datsenko KA, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A. 2011;108:10098–10103.
- Wiedenheft B, van Duijn E, Bultema JB, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci U S A. 2011;108:10092–10097.
- Sternberg SH, Redding S, Jinek M, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67.
- Blosser TR, Loeff L, Westra ER, et al. Two distinct DNA binding modes guide dual roles of a CRISPR-Cas protein complex. Mol Cell. 2015;58:60–70.
- Lim Y, Bak SY, Sung K, et al. Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat Commun. 2016;7:13350.
- Swarts DC, van der Oost J, Jinek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol Cell. 2017;66(221–233):e224.
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.
- Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573.
- Liu L, Li X, Wang J, et al. Two distant catalytic sites are responsible for C2c2 RNase activities. Cell. 2017;168(121–134):e112.
- Smargon AA, Cox DB, Pyzocha NK, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65(618–630):e617.
- Maniv I, Jiang W, Bikard D, et al. Impact of different target sequences on Type III CRISPR-Cas immunity. J Bacteriol. 2016;198:941–950.
- Benda C, Ebert J, Scheltema RA, et al. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol Cell. 2014;56:43–54.
- Hale CR, Cocozaki A, Li H, et al. Target RNA capture and cleavage by the Cmr type III-B CRISPR-Cas effector complex. Genes Dev. 2014;28:2432–2443.
- Gudbergsdottir S, Deng L, Chen Z, et al. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol. 2011;79:35–49.
- Spilman M, Cocozaki A, Hale C, et al. Structure of an RNA silencing complex of the CRISPR-Cas immune system. Mol Cell. 2013;52:146–152.
- Staals RHJ, Agari Y, Maki-Yonekura S, et al. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of thermus thermophilus. Mol Cell. 2013;52:135–145.
- Li Y, Zhang Y, Lin J, et al. Cmr1 enables efficient RNA and DNA interference of a III-B CRISPR-Cas system by binding to target RNA and crRNA. Nucleic Acids Res. 2017;45:11305–11314.
- Hale CR, Zhao P, Olson S, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956.
- Tamulaitis G, Kazlauskiene M, Manakova E, et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell. 2014;56:506–517.
- Pyenson NC, Gayvert K, Varble A, et al. Broad targeting specificity during bacterial Type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe. 2017;22:343–353.e3.
- Hatoum-Aslan A, Maniv I, Marraffini LA. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci U S A. 2011;108:21218–21222.
- Staals RH, Zhu Y, Taylor DW, et al. RNA targeting by the type III-A CRISPR-Cas Csm complex of thermus thermophilus. Mol Cell. 2014;56:518–530.
- Hatoum-Aslan A, Samai P, Maniv I, et al. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J Biol Chem. 2013;288:27888–27897.
- Walker FC, Chou-Zheng L, Dunkle JA, et al. Molecular determinants for CRISPR RNA maturation in the Cas10-Csm complex and roles for non-Cas nucleases. Nucleic Acids Res. 2017;45:2112–2123.
- Osawa T, Inanaga H, Sato C, et al. Crystal structure of the CRISPR-Cas RNA silencing Cmr complex bound to a target analog. Mol Cell. 2015;58:418–430.
- Brennecke J, Stark A, Russell RB, et al. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85.
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891.
- Bandyra KJ, Said N, Pfeiffer V, et al. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell. 2012;47:943–953.
- Deng L, Zhu H, Chen Z, et al. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles. 2009;13:735–746.
- Li Y, Pan S, Zhang Y, et al. Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res. 2016;44:e34.
- Peng N, Deng L, Mei Y, et al. A synthetic arabinose-inducible promoter confers high levels of recombinant protein expression in hyperthermophilic archaeon Sulfolobus islandicus. Appl Environ Microbiol. 2012;78:5630–5637.
- Peng N, Xia Q, Chen Z, et al. An upstream activation element exerting differential transcriptional activation on an archaeal promoter. Mol Microbiol. 2009;74:928–939.