
ABSTRACT
In contrast to cytoplasmic localization of spliced mRNAs, many spliced lncRNAs are localized in the nucleus. To investigate the mechanism, we used lncRNA MEG3 as a reporter and mapped a potent nuclear retention element (NRE), deletion of this element led to striking export of MEG3 from the nucleus to the cytoplasm. Insertion of the NRE resulted in nuclear retention of spliced lncRNA as well as spliced mRNA. We further purified RNP assembled on the NRE in vitro and identified the proteins by mass spectrometry. Screen using siRNA revealed depletion of U1 snRNP components SNRPA, SNRNP70 or SNRPD2 caused significant cytoplasmic localization of MEG3 reporter transcripts. Co-knockdown these factors in HFF1 cells resulted in an increased cytoplasmic distribution of endogenous lncRNAs. Together, these data support a model that U1 snRNP components restrain spliced lncRNAs in the nucleus via the interaction with nuclear retention element.
Introduction
Recent progresses with expanded data from next-generation sequencing reveal that most of the genome is transcribed [Citation1]. Among these transcripts, lncRNAs turn out to be a novel class of RNAs that has the largest predicted number although the abundance of most lncRNAs is low compared to mRNAs. Previous studies revealed that lncRNAs played quite diverse roles and multiple functions by interacting with RNAs, DNAs and proteins [Citation2].
LncRNAs are similar to mRNAs as both are transcribed by RNA pol II, capped at the 5ʹ end, most of them have splicing events and polyA tail [Citation3,Citation4]. By definition, lncRNAs do not encode proteins, which is the fundamental difference between lncRNAs and mRNAs. For the localization, all mRNAs are exported to the cytoplasm for translation, whereas lncRNAs can be in the nucleus, cytoplasm or in both compartments. The efficient export of spliced mRNAs to the cytoplasm is apparently due to the recruitment of TREX complex to the 5ʹ end of mRNA during splicing [Citation5], in contrast, many spliced lncRNAs are retained in the nucleus despite splicing events [Citation6].
Several studies have already provided some clues on the nuclear retention of lncRNAs. For example, lncRNA FIRRE contained 156 bp repeating RNA domain that accumulated FIRRE on its target chromosome [Citation7]. Similarly, XIST 5ʹ repeat element helped XIST RNA to localize on the X chromosome [Citation8]. In both cases, depletion of hnRNPU resulted in the de-localization of RNAs from their target sites on the chromosomes [Citation7,Citation9]. Another study suggested Malat1 contained two independent regions that facilitated the localization of Malat1 to nuclear speckles. Knockdown of nuclear speckle-enriched proteins RNPS1, SRm160 and IBP160 showed diffused Malat1 signal in the nucleoplasm [Citation10]. Recently, a short pentamer RNA motif (AGCCC) in mouse lncRNA BORG was shown to be responsible for its nuclear retention [Citation11].
In addition, U1 snRNP component SNRNP70 was shown to be a factor needed for nuclear retention of pre-mRNA [Citation12]. Consensus 5ʹ splice site motif in mature RNAs was reported to facilitate their nuclear retention [Citation13]. Most recently, the strength of 5ʹ splice site was found to be one of the determinants for intron-containing pre-mRNA leakage to the cytoplasm induced by spliceostatin A [Citation14]. Considering the frequent occurrence of potential 5ʹ splice sites also in lncRNAs, it will be interesting to investigate whether U1 snRNP components function in the localization of nuclear lncRNAs. Nevertheless, detailed mechanisms for nuclear retention of spliced lncRNAs are still elusive.
As an attempt to address such mechanism, we used lncRNA MEG3 as a reporter and identified a potent nuclear retention element in the middle of MEG3 RNA sequence, the NRE functioned both in its natural and heterologous contexts. We further showed the NRE recruited U1 snRNP components to restrain the spliced lncRNAs in the nucleus. Our data further support cis-element as a determinant of lncRNA localization and the importance of U1 snRNP components in subcellular distribution of spliced lncRNAs.
Results
MEG3 reporter transcripts localized in the nucleus
To gain deep insight into the mechanism of how some spliced lncRNAs are retained in the nucleus, we constructed MEG3 lncRNA reporter, as the genomic sequence of MEG3 spans about 35kb, MEG3 cDNA was cloned to a vector downstream of an optimized β-globin intron 1 (). As a control, the genomic sequence of a previously reported cytoplasmic lncRNA ANCR was also cloned ().
Figure 1. Subcellular localization of MEG3 reporter transcripts.
A. Schematic of MEG3 genomic structure, MEG3 reporter and ANCR reporter. Introns were showed as a solid line and exons were showed as boxes. For MEG3 gene and ANCR reporter construct, the numbers ontop and beneath indicated the length of each introns and exons, respectively, for MEG3 reporter construct, the number beneath indicated the length of MEG3 cDNA used in this study. B. Western blot to show the efficiency of nucleus/cytoplasm separation. Cyt: cytoplasm; Nuc: nucleus. C. RT-PCR to show that MEG3 reporter transcripts enriched in the nucleus and ANCR reporter transcripts enriched in the cytoplasm. RT: reverse transcriptase. D. RNA-FISH showing nuclear localization of MEG3 and subcellular localization of ANCR after transient transfection.
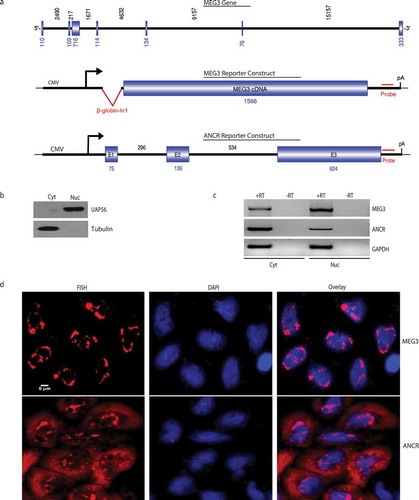
To determine the localization of the reporter transcripts, these reporters were transiently transfected into HeLa cells, RT-PCR was then performed on RNAs purified from the cytoplasmic and nuclear compartments. The cytoplasm/nucleus separation efficiency was showed in using western blot of nuclear marker UAP56 and cytoplasmic marker tubulin. MEG3 and ANCR reporter transcripts were detected in both cytoplasm and nucleus, with apparent enrichment of MEG3 in the nucleus and enrichment of ANCR in the cytoplasm (). The localization of these transcripts were further assayed using RNA-FISH, MEG3 reporter transcripts were observed dominantly in the nucleus, compared to a more even distribution in the cytoplasm and nucleus for ANCR reporter transcripts (). As negative controls, RNA-FISH was also performed on untransfected HeLa cells or cells transfected with empty vector (Suppl Fig. S1). Together, these data suggested the MEG3 reporter we constructed could be used for mechanistic analysis of nuclear retention for spliced lncRNA.
Identification of nuclear retention element in MEG3
Hypotheses of spliced lncRNA retained in the nucleus may include failure to recruit export machinery or nuclear retention by specific retention mechanism. To test the first hypothesis, we purified MEG3 RNP assembled in vivo, and performed mass spectrometry analysis, most of the TREX components were identified such as THOC2, THOC1, THOC3, THOC5, THOC6, ALYREF and UAP56, which suggested the retention of MEG3 in the nucleus was unlikely due to the failure to recruit export machinery.
Next, we further tested the hypothesis that MEG3 might contain nuclear retention element. A series of deletion constructs were made to truncate MEG3 from the 3ʹ end (), transfection of these constructs to HeLa cells followed by RNA-FISH revealed that removal of the 3ʹ end up to 413 nt had no significant impact on the nuclear localization of MEG3 transcripts (), further truncations resulted in partial export of MEG3 transcripts () until the truncation of 769 nt, which led to striking cytoplasmic localization of MEG3 transcripts ().
Figure 2. Mapping nuclear retention element in MEG3.
A. Schematic of MEG3 reporter and RNA-FISH to show subcellular localization of MEG3 transcripts after transient transfection to HeLa cells. B-I. Schematic of MEG3 deletion constructs and RNA-FISH to show subcellular localization of MEG3 transcripts. Dotted line: MEG3 sequence deleted; the numbers beneath indicated the starting and ending position of MEG3 cDNA in the specific construct; dots on top of MEG3 boxes in F&G: two point mutations introduced to eliminate cryptic splicing (See Suppl Fig. S3).
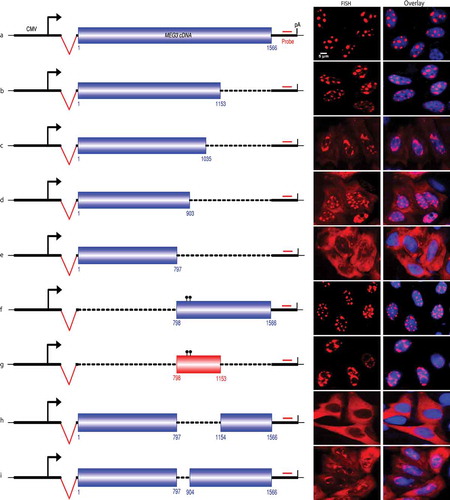
Consistently, removal of the 5ʹ end of MEG3 (1-797nt) showed no impact to the localization of MEG3 transcripts (), further mapping revealed that a 356nt sequence (from 798 to 1153nt) served as nuclear retention element (NRE, ), the transcripts from this construct were typically retained in the nucleus. Whereas deletion of the NRE led to complete cytoplasmic localization (), and partial deletion of NRE resulted in both cytoplasmic and nuclear localization of MEG3 transcripts (). Thus, we concluded that the nuclear localization of spliced lncRNA MEG3 was due to the presence of NRE in the transcripts.
Insertion of MEG3 NRE shifted cytoplasmic lncRNA and mRNA to the nucleus
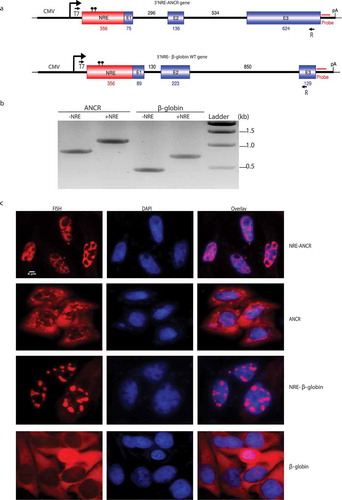
To test whether the MEG3 NRE functions in heterologous context, we inserted it at the 5ʹ end of ANCR or β-globin wild-type constructs (). RT-PCR revealed that the insertion of the NRE had no impact on the splicing of either ANCR or β-globin WT transcripts (). However, as showed by RNA-FISH in , insertion of the NRE to ANCR led to almost complete retention of ANCR transcripts to the nucleus and partial but apparent nuclear retention of spliced β-globin transcripts, suggesting MEG3 NRE is potent to retain not only spliced lncRNA but also spliced mRNA in the nucleus.
Figure 3. Insertion of nuclear retention element enriched cytoplasmic RNA in the nucleus.
A. Schematic of chimeric NRE-ANCR and NRE-β-globin WT constructs. The numbers indicated the length of NRE, each intron and exon. Dots on top of NRE indicated 2-point mutations introduced. T7 and R indicated the positions of primers used for amplification in B. B. RT-PCR showing correct splicing after insertion of NRE. C. RNA-FISH showing subcellular localization of the chimeric transcripts.
Purification of RNP assembled on MEG3 NRE
To investigate which trans-factors bind to MEG3 NRE and mediate the nuclear retention, we first purified RNP assembled in vitro on MEG3 NRE using biotin-streptavidin strategy, NRE transcripts without biotin labelling and the antisense of NRE were used as negative controls. As shown in Suppl Fig. S2A, RNP assembled on NRE showed more protein enrichment after separation on PAGE as compared to RNPs assembled on NRE without biotin labelling or on the antisense NRE. Mass spectrometry analysis revealed major protein components in each RNP, which were listed in Suppl Table S1. Western blot confirmed several proteins enriched in RNP assembled on MEG3 NRE (Suppl Fig. S2B).
Depletion of SNRPA/SNRNP70/SNRPD2 resulted in enhanced cytoplasmic localization of MEG3 reporter transcripts as well as endogenous lncRNAs
To find out which trans-factor mediates the retention, we screened 49 components enriched in MEG3 NRE RNP using siRNA (Suppl Table S1 and S2). Depletion of SNRPA led to cytoplasmic localization of MEG3 in about 50% of cells transfected with MEG3 construct (), whereas depletion of other components screened showed no or minor effect on the nuclear localization of MEG3 transcripts. As SNRPA is a component of U1 snRNP, we further screened other components in U1 snRNP. As showed in , depletion of SNRNP70 or SNRPD2 resulted in cytoplasmic localization of MEG3 in 45% or 70% of transfected cells individually. Consistent with the observation in RNA-FISH, RT-PCR also indicated a significant increase of MEG3 transcripts in the cytoplasm after depletion of SNRPA, SNRNP70 or SNRPD2 ().
Figure 4. Depletion of SNRPA/SNRNP70/SNRPD2 led to cytoplasmic accumulation of MEG3 reporter transcripts and enrichment of endogenous lncRNAs in cytoplasm.
A. Western blot showing knockdown efficiency after treatment of HeLa cells with specific siRNA. B. RNA-FISH showing depletion of SNRPA/SNRNP70/SNRPD2 resulted in shift of MEG3 reporter transcripts from nucleus to cytoplasm. C. The percentage of cells with apparent FISH signal in cytoplasm increased significantly after depletion of SNRPA/SNRNP70/SNRPD2. (n = 3) ***: p < 0.001. D. RT-PCR results showing elevated distribution of MEG3 reporter transcripts in cytoplasm after depletion of SNRPA/SNRNP70/SNRPD2. (n = 3) *: p < 0.05; **: p < 0.01; ***: p < 0.001. E. Western blot showing knockdown efficiency after treatment of HFF1 with siRNAs against SNRPA/SNRNP70/SNRPD2 simultaneously. Si-3KD: triple knockdown of SNRPA, SNRNP70 and SNRPD2. F. RT-PCR showing distribution of endogenous lncRNAs in cytoplasm and nucleus after siRNA treatment of HFF1 cells. C: cytoplasm; N: nucleus; W: whole cell. G. Quantification of lncRNAs localized in the cytoplasm. (n = 3) *: p < 0.05; **: p < 0.01; ***: p < 0.001.
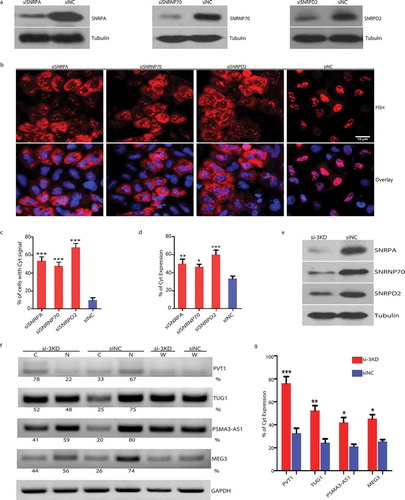
To investigate whether these factors affect the localization of endogenous lncRNAs, we depleted all three proteins in HFF1 cells, the knockdown efficiency was confirmed by western blot (), RT-PCR that is semi-quantitative on RNA purified from cytoplasmic or nuclear compartments revealed that more lncRNAs were detected in the cytoplasm. Specifically, for endogenous MEG3, we observed 44% in the cytoplasm after depletion as compared to 26% in the control (). Similarly, for the other three lncRNAs with dominant nuclear localization, depletion of SNRPA, SNRNP70 and SNRPD2 led to an increase of 33% to 78% (PVT1), 25% to 52% (TUG1) and 20% to 41% (PSMA3-AS1) in the cytoplasm (). These data together supported that U1 snRNP components played a crucial role for restraining spliced lncRNAs in the nucleus.
Discussion
RNA localization is fundamental to its function, it is well known that splicing plays a crucial role in promoting the export of spliced mRNAs from the nucleus to the cytoplasm by recruiting TREX complex [Citation5]. However, although many of the lncRNAs are also spliced, quite a few spliced lncRNAs localize in the nucleus [Citation15,Citation16].
Hypotheses for retention of spliced lncRNA in the nucleus may include failure to recruit export machinery or the presence of nuclear retention element. Using lncRNA MEG3 as a reporter, we showed that it favoured the second hypothesis. Detailed mapping revealed that a 356nt region in the middle of MEG3 served as nuclear retention element, removal of this region led to striking shift of MEG3 localization from the nucleus to the cytoplasm. Two cryptic 3ʹ splice sites in NRE were identified (Suppl Figure 3A, 3B and 3D), the wild type NRE transcripts were accumulated in cytoplasm due to cryptic splicing as revealed by RT-PCR (Suppl Figure 3C). Inactivation of these two sites by point mutations led to nuclear retention of NRE transcripts (Suppl Figure 3B, 3C and 3D). Insertion of the mutated element resulted in dominant nuclear retention of cytoplasmic transcripts including lncRNA ANCR and β-globin WT transcripts, supporting that this element is a potent NRE. Our results are consistent with the previous report that subcellular localization of lncRNAs is mainly determined by primary sequences [Citation17].
Furthermore, a pentamer motif AGCCC was reported to be necessary for the nuclear localization of lncRNA BORG [Citation11]. More recently, it was found that more than 100 unique RNA sequences with C-rich motif could enrich cytosolic transcript in the nucleus using high-throughput screen [Citation18]. Attempts to further narrow down the NRE identified in this study revealed that shorter NREs conferred partial effects. Interestingly, the composition of four nucleotides in the NRE showed increased C (29.5%) and decreased A (18.5%), whether such a change in nucleotide composition is critical for the retention needs further investigation.
Study by Ulitsky et al. reported that short sequence derived from Alu repeats facilitated to retain lncRNAs in the nucleus by interacting with hnRNPK [Citation19]. However, a 107 bp Alu repeat is also present in the nuclear-retained XIST lncRNA at 5235-5341nt, while hnRNPK interaction with XIST is required for XIST silencing effect, such interaction is not necessary for its localization on the chromosome or nuclear retention [Citation20]. In this study, although hnRNPK was also detected in the RNP assembled on NRE, depletion of hnRNPK did not change the nuclear localization of MEG3 reporter transcripts (data not shown). Instead, our study led to the identification of U1 snRNP components including SNRPA, SNRNP70 and SNRPD2 as major trans-factors restraining MEG3 reporter transcripts in the nucleus. Actually, all the three proteins were present in the RNP assembled on NRE as well as the MEG3 RNP assembled in vivo, although only SNRPA was included in the first round of siRNA screen since the other two proteins were also detected in RNPs assembled on antisense NRE. It should be noted that although SNRPA was only identified in NRE RNP in the mass spectrometry analysis, faint bands in the controls were detected when we verified proteins present in the RNPs using western blot, suggesting a non-specific binding to beads. Depletion of these proteins in HFF1 cells resulted in the enriched distribution of endogenous MEG3 as well as several other spliced lncRNAs, thus such mechanism for nuclear retention apparently is not unique to MEG3. It will be interesting to further determine the impact of these factors on nuclear localization of overall-spliced lncRNAs.
During the preparation of this manuscript, a pre-print from the Shen Lab (https://www.biorxiv.org/content/10.1101/310433v1) demonstrated that a U1 motif CAGGUGAGU was vital for chromatin retention of ncRNAs, and several U1 snRNP components including SNRNP70, SNRPA and SNRPC facilitated the retention. These findings are consistent with our results that U1 snRNP components function in lncRNA retention by interacting with the embedded NRE. Bioinformatic analysis using FIMO with the motif from the Shen lab (CAGGTGAGT) or the Lander lab [GGTAAGTA, Citation21] revealed the presence of such motifs in the NRE (CAGGTCAGT, p = 8.66e-05 and GGTCAGTC, p = 8.38e-04, Suppl Fig. S4), which are lacking in the β-globin or ANCR cDNA sequences. These two motifs are overlapping in the MEG3 NRE and supporting the U1 motif facilitates the retention of MEG3 in the nucleus. However, as removal of a shortened NRE containing such motifs resulted in partial export of MEG3 transcripts (), the 356nt MEG3 NRE may harbour additional motifs or structural features needed for its full function. In addition, it should be noted that RNA-FISH performed in this study can only determine the nuclear retention or cytoplasmic accumulation, the resolution is not good enough to conclude whether the transcripts are tethered to chromosome or not.
As U1 snRNP components were part of the spliceosome, we also tested the effect of co-knockdown on splicing efficiency by RT-PCR and assayed the endogenous lncRNA isoforms. At the level of protein depletion in this study, the change on the percentage of spliced transcripts was minor (Suppl Fig. S5A&S5B) and no additional isoforms were detected (Suppl Fig. S5C).
To sum up, our study supports a model of NRE presenting in the lncRNAs recruits nuclear proteins to retain spliced lncRNAs in the nucleus and pinpoints to a critical role of U1 snRNP components in the nuclear localization of spliced lncRNAs.
Materials and methods
Constructs and antibodies
MEG3 reporter was constructed by PCR amplification from HFF1 cDNA and inserted into pcDNA5/FRT/TO vector (Invitrogen) at KpnI/XhoI sites. To create a splicing event, β-globin intron 1 with optimized 5ʹ and 3ʹ splice sites was inserted into HindIII site upstream of MEG3 cDNA. MEG3 truncated constructs were generated by PCR with primers listed in Suppl Table S3, deletion constructs were generated by two-step PCR as described previously [Citation22]. To construct ANCR reporter, nested PCR was performed to amplify ANCR gene in full-length from 293T cells genomic DNA. The amplicon was then cloned in pcDNA5/FRT/TO vector at KpnI/ApaI sites. To generate the chimeric constructs of NRE-ANCR and NRE-β-globin WT, overlap PCR was performed and cloned into the pcDNA3.1 vector at KpnI site (for NRE-ANCR) or KpnI/XbaI sites (for NRE-β-globin WT). All the constructs were verified by DNA sequencing. Primers used for cloning were listed in Suppl Table S3.
Rabbit antibodies against SNRPA (Proteintech, 1:1000), SNRNP70 (Abclonal, 1:3000), SNRPD2 (Proteintech, 1:2000), PTBP1 (Proteintech, 1:2000), ILF3 (Proteintech, 1:5000), ELAVL1 (Proteintech, 1:2000), DHX9 (Proteintech, 1:1000), UAP56 (Gift from Reed lab, Harvard Medical School) and mouse antibody against tubulin (Sigma, 1:10,000) were used in western blot.
Cell culture and transfection
HeLa cells were a kind gift from Reed lab, Harvard Medical School, cultured in DMEM (Life Technologies) supplemented with 10% FBS (Life Technologies). HFF1 cells were purchased from ATCC, cultured in DMEM supplemented with 15% FBS. For transient transfection, 1 µg of plasmid DNA was transfected to cells cultured in 12-well plate at 80% confluency using Lipofectamine 2000 (Invitrogen).
To perform RNAi, cells were seeded in 12-well plate and transfected with siRNAs at 30% confluency using Lipofectamine 3000 (Invitrogen). Non-targeting siRNA was used as a negative control. All the siRNAs were purchased from RiboBio, China. Sequences of siRNAs used were showed in Suppl Table S2.
RNA-FISH
RNA-FISH was performed as previously described [Citation23]. Briefly, after 30 min fixation with 4% paraformaldehyde in 1 × PBS, cells were permeabilised with 1 × PBS/0.1% Triton X-100 for 30 min. Cells were then washed with 1 × SSC/50% formamide twice and incubated at 37°C with vector probes for 16 h. DAPI was used for nuclei staining. Fluorescence was detected using DMI8 microscope (Leica).
Mini nuclear extract preparation
Nucleo-cytoplasmic separation was performed as previous described [Citation24]. In brief, HeLa cells were cultured in 150 mm plates until 90% confluency. Cells were scraped using a cell lifter and transferred into an Eppendorf tube. Cells were then swelled in hypotonic buffer and lysed using a dounce homogeniser. The nuclei portion was extracted by adding low salt and high salt buffers. Nuclear extracts were transferred into Ultra-0.5mL Centrifugal Filters (Amicon) for concentration by centrifuging at 12,000rpm and then dialyzed in Dialysis Buffer (20 mM HEPES, 100 mM KCL, 0.2 mM EDTA, 20% Glycerol) using mini-dialysis Slide-A-Lyzer MINI Dialysis Units (Thermo). The nuclear extract can be used immediately or stored at −80°C.
Purification of RNP assembled in vivo and in vitro
For purification of MEG3 RNP assembled in vivo, MEG3 cDNA was cloned into modified vector pcDNA5 containing MS2 hairpins, this vector was co-transfected with vector encoding OG44 into modified HeLa cells to generate inducible stable cell line. After induction, nuclear extract was prepared and MEG3 RNP assembled in vivo was purified using MS2-MBP affinity purification. The antisense of MEG3 was used as a negative control.
For RNP purification in vitro, PCR products containing T7 promoter and MEG3 NRE or its antisense were used as templates for in vitro transcription. Templates were transcribed by T7 RNA polymerase with or without biotin-UTP at 37°C for 2 h. After removal of template DNAs with DNase I treatment, the transcripts were purified with Mini Quick Spin Column (Roche). For RNP assembly, 1ug in vitro transcribed RNAs labelled with or without biotin was incubated with 200 ul HeLa mini nuclear extract in RNA binding buffer (50mM KCl, 1.5mM MgCl2, 10mM HEPES, 0.5% NP40) and incubated at 30°C for 30 min. For affinity purification, 50ul streptavidin agarose resin (Thermo) was added into the mixture and rotated at room temperature for 50 min. Proteins were eluted with SDS loading buffer after wash.
Mass spectrometry analysis
Mass spectrometry analysis was performed in the core facility of Tsinghua University. Gel bands of proteins with catalase activity were excised for in-gel digestion, and proteins were identified by mass spectrometry as previously described [Citation25]. Briefly, proteins were disulfide reduced with 5 mM dithiothreitol (DTT) and alkylated with 11 mM iodoacetamide. In-gel digestion was performed using sequencing grade-modified trypsin in 50 mM ammonium bicarbonate at 37°C overnight. The peptides were extracted twice with 1% trifluoroacetic acid in 50% acetonitrile aqueous solution for 1 h. The peptide extracts were then centrifuged in a SpeedVac to reduce the volume. Peptides were analyzed with Orbitrap Fusion Lumos Tribrid mass spectrometry from Thermo Scientific.
Supplemental Material
Download Zip (7.3 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108.
- Marchese F, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206–219.
- Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346.
- Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927.
- Cheng H, Dufu K, Lee CS, et al. Human mRNA export machinery recruited to the 5ʹ end of mRNA. Cell. 2006;127:1389–1400.
- Wu H, Yang L, Chen LL. the diversity of long noncoding RNAs and their generation. Trends Genet. 2017;33:540–552.
- Hacisuleyman E, Goff LA, Trapnell C, et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol. 2014;21:198–206.
- Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet. 2002;30:167–174.
- Hasegawa Y, Brockdorff N, Kawano S, et al. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell. 2010;19:469–476.
- Miyagawa R, Tano K, Mizuno R, et al. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. Rna. 2012;18:738–751.
- Zhang B, Gunawardane L, Niazi F, et al. A novel RNA motif mediates the strict nuclear localization of a long noncoding RNA. Mol Cell Biol. 2014;34:2318–2329.
- Takemura R, Takeiwa T, Taniguchi I, et al. Multiple factors in the early splicing complex are involved in the nuclear retention of pre-mRNAs in mammalian cells. Genes Cells. 2011;16:1035–1049.
- Lee ES, Akef A, Mahadevan K, et al. The consensus 5ʹ splice site motif inhibits mRNA nuclear export. PLoS One. 2015;10:e0122743–e0122743.
- Yoshimoto R, Kaida D, Furuno M, et al. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA. 2017;23:47–57.
- Cabili MN, Dunagin MC, McClanahan PD, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015;16:20.
- van Heesch S, van Iterson M, Jacobi J, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15:R6.
- Gudenas BL, Wang L. Prediction of LncRNA subcellular localization with deep learning from sequence features. Sci Rep. 2018;8:16385.
- Shukla CJ, McCorkindale AL, Gerhardinger C, et al. High-throughput identification of RNA nuclear enrichment sequences. Embo J. 2018;37:e98452.
- Lubelsky Y, Ulitsky I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature. 2018;555:107–111.
- Chu C, Zhang QC, Da Rocha ST, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416.
- Engreitz JM, Sirokman K, McDonel P, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell. 2014;159:188–199.
- Lei H, Dias AP, Reed R. Export and stability of naturally intronless mRNAs require specific coding region sequences and the TREX mRNA export complex. Proc Natl Acad Sci U S A. 2011;108:17985–17990.
- Lei H, Zhai B, Yin S, et al. Evidence that a consensus element found in naturally intronless mRNAs promotes mRNA export. Nucleic Acids Res. 2013;41:2517–2525.
- Folco EG, Lei H, Hsu JL, et al. Small-scale nuclear extracts for functional assays of gene-expression machineries. J Vis Exp. 2012;e4140.
- Li X, Tao J, Han J, et al. The gain of hydrogen peroxide resistance benefits growth fitness in mycobacteria under stress. Protein Cell. 2014;5:182–185.