
ABSTRACT
N6-methyladenosine (m6A) is considered as a reversible RNA modification occurring more frequently on the GAC than AAC context in vivo, which regulates post-transcriptional gene expression in mammalian cells. m6A ‘writers’ METTL3 and METTL14 demonstrate a strong preference for binding AC-containing motifs in living cells. However, this evidence is currently lacking for m6A erasers, leaving the dynamics of the internal m6A modification under debate recently. We analysed three recently published FTO CLIP-seq data sets and two generated in this study, one of the two known m6A ‘erasers’. FTO binding peaks from all cell lines contain RRACH motifs. Only those from K562, 3T3-L1and HeLa cells were enriched in AC-containing motifs, while those from HEK293 were not. The exogenously overexpressed FTO effectively binds to m6A motif-containing RNA sites. FTO overexpression specifically removed m6A modification from GGACU and RRACU motifs in a concentration-dependent manner. These findings underline the dynamics of FTO in target selection, which is predicted to contribute to both the m6A dynamics and the FTO plasticity in biological functions and diseases.
Introduction
In the early 1970s, several groups found that the methylation modification is not restricted to rRNAs and tRNAs, also occurs in polyadenylated cellular [Citation1,Citation2] and viral [Citation3–Citation5] mRNAs. The methylated nucleosides in mRNAs were further determined as the 5ʹ-terminal m7G(5ʹ)ppp(5ʹ)Nm [Citation6–Citation9] and the internal N6-methyl adenosine (m6A) [Citation10–Citation12]. The dimethylated nucleoside, N6, 2ʹ-O-dimethyladenosine (m6Am), and the four common 2ʹ-O-methylribonucleosides (Gm, Am, Um, Cm) at the second position were reported soon after [Citation9]. Then, internal m6A was reported to occur primarily in two sequence contexts, Gm6AC (70%) and Am6AC (30%) in HeLa cell [Citation13], and 60% in Gm6AC in SV40 mRNA [Citation14].
Recently, MeRIP-Seq (methylated RNA immunoprecipitation followed by sequencing) technology, also called m6A-seq, allows the mapping of m6A sites at the whole-transcriptome level [Citation15,Citation16]. MeRIP-seq and a miCLIP-seq (m6A cross-linking) analysis confirmed that m6A modification preferably occurs within the consensus motif RRACH (R = G or A; H = A, C, or U), more in the sequence context of GAC than AAC [Citation15–Citation17]. The currently published MeRIP-seq experiments commonly used a commercial antibody against both the internal m6A and the 5ʹ-terminal m6Am [Citation18–Citation20]. miCLIP recovers mutation and truncations in cDNAs, which allows the mapping of about 18,000 high-confident cross-linking m6A sites among which a few hundred associates with the 5ʹ terminal m6Am [Citation17].
The RNA m6A modification is considered a reversible modification in living cells. The reversible m6A modification dictates the dynamics of m6A modification in transcriptomes, which has been shown to regulate multiple processes of RNA metabolism, including alternative splicing, RNA stability and translation [Citation21–Citation27]. Consistently, m6A modification modulates important biological processes, such as stem cell differentiation, heat shock response, oocyte competence and early zygotic development, neuronal and other development, and innate immunity [Citation24,Citation26,Citation28–Citation31], and is also involved in cancers [Citation32–Citation38] and other diseases [Citation39].
Nevertheless, a number of contradicting results have led to the current debate of the m6A dynamics [Citation40–Citation44]. m6A is added by RNA methyltransferase (‘‘writer’) and removed by demethylases (‘“eraser”’) [Citation23,Citation45]. Photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) analysis have demonstrated that the binding peaks of methyltransferase METTL3 and METTL14 are highly enriched in RRACH motifs, consistent with a strong selection of the sequence context for their writing of m6A [Citation46]. So far, two RNA demethylases, fat mass and obesity-associated protein (FTO) and alkylated DNA repair protein AlkB homolog 5 (ALKBH5), have been identified to remove m6A modification [Citation39,Citation47]. However, neither of them has been shown to selectively bind to the internal m6A motifs. In vitro studies showed that neither FTO nor ALKBH5 shows strict dependence on RRACH motifs in their demethylation activity [Citation48], although the sequence and structure of RNAs affect the demethylation activity of FTO [Citation49]. Three recently published works have performed FTO CLIP-seq experiments; however, none of them reports FTO binding of any m6A motifs [Citation50–Citation52]. Moreover, a recent study showed that FTO could remove the methyl group from the 5ʹ-cap m6Am with a much higher efficiency than that from the internal m6A [Citation53]. Therefore, it remains highly questionable whether FTO recognizes and removes m6A from the internal m6A motifs, which is a key of the m6A reversibility and dynamics.
To investigate the FTO demethylation target sites in cells, we analysed the published FTO CLIP data sets from human HEK293 cell line [Citation50], mouse embryonic fibroblasts (3T3-L1) [Citation51], and FTO eCLIP data set from human erythroleukemia K562 cell line [Citation52]. FTO binding peaks from 3T3-L1 and K562 cells, but not from HEK293 cells, showed the presence of the GAC consensus in the top-enriched motifs. We then performed FTO CLIP-seq in HeLa cells, showing that the GAC consensus was present in the top-represented motifs. The m6A motifs were more enriched in FTO binding peaks upon its overexpression. We further showed that overexpression of FTO robustly removed m6A modification from RRACH motifs. Additionally, the number of transcripts subjected to FTO demethylation were increased with the FTO concentration. The cell type- and concentration-dependence of FTO binding and demethylation selectively from the internal m6A motifs support the m6A dynamics and reversibility.
Results
The m6A context GAC is enriched in FTO binding peaks from K562 and 3T3-L1 but not in HEK293T
To study whether FTO selectively binds to RRACH motif, we firstly analysed three FTO CLIP/eCLIP data sets which were generated by three different labs from HEK293 [Citation50], K562 [Citation52] and 3T3-L1 [Citation51]. The usable reads could estimate library complexity indicative of the binding strength of a RNA binding protein [Citation52]. FTO eCLIP-seq data contained significantly less usable reads than those of PTBP1 (K562), and much less in FTO CLIP-seq data from HEK293 than that from K562 (Figure S1(a)). This data profile could indicate a weak RNA binding affinity of FTO with its targets.
Plot of the distribution of FTO eCLIP/CLIP-seq reads and peaks showed a preferable location in the intronic region (Figure S1(b)), consistent with its reported role in regulating alternative splicing [Citation24,Citation50]. When the regional distribution of CLIP/eCLIP reads was normalized by pre-mRNA length in each genic region, FTO-bound reads were enriched in 5ʹUTR in data sets from HEK293 and K562 ()). Consistent with the reported high efficiency of FTO demethylation of the 5ʹ-end m6Am [Citation53], a sharp enrichment of FTO binding signals around the transcription start site (TSS) was clearly demonstrated by all three replicates of CLIP-seq data from HEK293 cells, while the FTO binding signals were depleted around the stop codon ()). However, the TSS enrichment was not evident in the eCLIP-seq data from K562 cells, an enriched distribution across the entire 5ʹUTR was instead observed. Meanwhile, the depleted FTO binding at the stop codon and the 3ʹ UTR region was seen, but to a much lesser extent than that of HEK293 ()). The distribution profile of FTO CLIP reads from 3T3-L1 around the TSS was between HEK293 and K562 ()).
Figure 1. Transcriptome-wide landscape of FTO binding shows cell type-specificity. Three recently published FTO eCLIP/CLIP-seq data were analysed.
(a) FTO-binding reads were enriched in 5ʹUTR in HEK293 and K562, but not 3T3-L1 cells. CLIP/eCLIP reads mapped to each region of the genome was normalized by the length of the region. There is no replicate and input control for 3T3-L1 cell, a suggested RNA-seq data were analysed as input control. The enrichment of each region was calculated relative to CDS region. Nc_exon, non-coding exon. (b) Enrichment of FTO binding around the transcription start site (TSS) and stop codons was different among HEK293, K562 and 3T3-L1 cells. Normalized FTO binding reads count (RPM) in a ± 1000 bp window around TSS (left) and stop codons (right) for all transcripts. RPM, reads per million. (c) GAC-containing motifs were enriched in K562 and 3T3-L1, but not in HEK293 cells. The 5-nt FTO binding motifs were detected from ABLIRC peaks by HOMER. Those from one sample were presented, and all others were shown in Fig. S2. Motifs include GAC-consensus were marked with a black box. Original usable reads (upper) and randomly selected the same number (102,470) of usable reads (lower) were used for peak calling and the motif analysis, respectively.
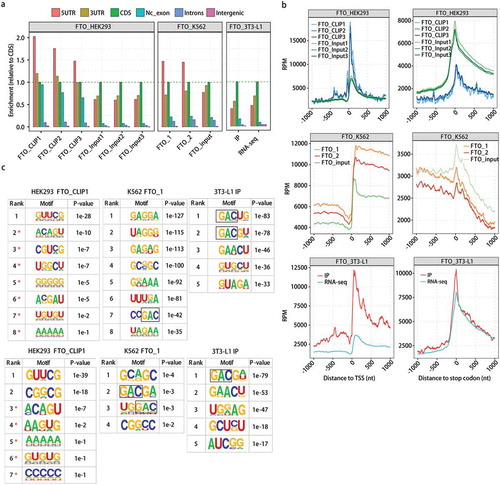
We next identified FTO-binding peaks using three different pipelines, Piranha [Citation54], CIMS [Citation55] and ABLIRC [Citation56], all displaying similar peak distribution in different genomic regions (Figure S1(c)). A de novo motif search of the 5-mer motifs from ABLIRC peaks using the HOMER algorithm revealed that the GAC- and/or GGAC-containing motifs were overrepresented in FTO binding peaks from K562, particularly in 3T3-L1, but not in those from HEK293T () and S2(a)). The search from CIMS peaks resulted in the GGAC-containing overrepresented motif in one of K562 replicates, while no AC-containing motif was resulted from Piranha peaks from either K562 or HEK393 replicates (Figure S2(a)). It should be noted that non-m6A FTO-binding motifs were different among cell lines. For example, GA-rich motifs and UUURA motif were more overrepresented than GAC and GGAC-motifs in K562 cells in both replicates, and GUUCG motif was highly overrepresented in HEK293T cells in three replicates, based on the motif resulted from ABLIRC peaks (Figure S2(a)).
Given that the percentage of usable reads for the HEK293T cells was low, we randomly extracted 102,470 usable reads from each of all samples to call FTO binding peaks and then perform motif analysis. The results showed that GAC and GGAC-motifs were enriched in FTO peaks from K562 and 3T3-L1 cells, but not in those from HEK293T cells (), lower and S2(b)). Therefore, FTO binding of both m6A motifs and non-m6A motifs showed cell type-dependence.
FTO binds to GAC-containing and RRACH motifs in HeLa cells, and the exogenously overexpressed FTO shows increased selectivity
As suggested above, FTO might not strongly associate with the internal m6A GAC motifs in some cell types. Theoretically, the increase of the cellular FTO concentrations would increase the concentration of FTO-target complex sensitive to CLIP-seq capture ()). To test this hypothesis, we overexpressed the Flag-tagged FTO excessively in HeLa cell line where the endogenous FTO expression level was low (Figure S3(a)). Confocal fluorescence microscopy showed that FTO was localized both in nucleus and cytoplasm. When FTO was overexpressed, its localization in the nucleus apparently increased (Figure S3(b)). The methylation level of total RNA and polyadenylated mRNA was then measured by dot blot analysis using antibody against both m6A and m6Am. It has been recently reported that the total amount of internal m6A is at least 10-fold larger than the amount of 5ʹ-end m6Am in mRNAs, and FTO demethylates more m6A than m6Am in HeLa cells [Citation49,Citation51]. FTO overexpression reduced the overall RNA methylation level as predicted, and a much more pronounced reduction was observed for total RNA than purified mRNA (Figure S3(c)). This finding is in good agreement with the recent report that FTO has a higher preference to remove m6A from the primary transcript level [Citation57], and with the early findings of high m6A occurrence in 18S and 28S rRNAs [Citation1], as well as in tRNA [Citation1,Citation19,Citation51].
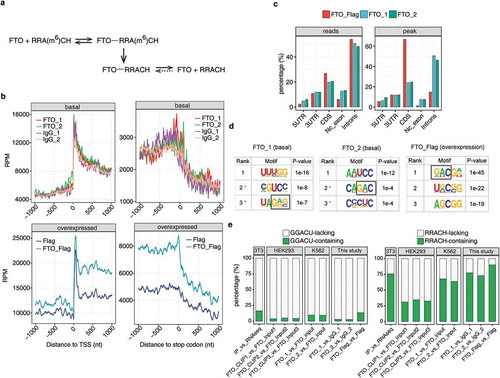
Figure 2. FTO selectively binds to m6A motifs in HeLa cells, and the exogenously overexpressed FTO shows higher selectivity.
(a) A model shows the proposed dynamics of reactions forming FTO-RRACH and FTO-RRA(m6A)CH that could be captured by anti-FTO immunoprecipitation. (b) FTO CLIP reads were not enriched around the TSS or 5ʹUTR region compared to IgG control in normal HeLa cells (upper panel), but were enriched in all transcribed regions when captured the overexpressed Flag-FTO (lower panel). Normalized FTO binding reads count in a ± 1000 bp window around TSS (left) and stop codons (right) for CLIP-seq data. FTO_1 and FTO_2, and IgG_1 and IgG_2, represent two replicated CLIP-seq data obtained from normal HeLa cells using anti-FTO and anti-IgG antibodies, respectively. FTO_Flag represents CLIP-seq data obtained from HeLa cells transfected with plasmid expressing FTO-Flag fusion protein, and Flag represents that from the control HeLa cells transfected with Flag-only plasmid. RPM, reads per million. (c) FTO binding was increased in the protein coding regions upon overexpression. Genomic distribution of FTO binding reads (left) and peaks (right) in HeLa cells. (d) The top FTO binding motifs detected by HOMER. (e) The fractions of FTO binding peaks containing GGACU and RRACH motifs were different among 3T3-L1, HEK293T, K562 and HeLa cells. The overexpressed FTO shows higher fractions of GGACU and RRACH-containing peaks in HeLa cells.
Under this functional context of FTO overexpression, we performed CLIP-seq experiments similar to eCLIP to identify FTO binding targets. To exclude the potential artefacts associated with a specific antibody, we used anti-FTO antibody in normal HeLa cells for two biological replicates and anti-Flag antibody in FTO-overexpressing (FTO-Flag) and control (Flag control) HeLa cells. Please be noted that in the CLIP-seq library construction approach applied here, the 5ʹ adapter was ligated to the 5ʹ-end of RNase-digested RNA after the end repair. This should eliminate the ligation to the 5ʹ-end m6Am with a cap structure; only the FTO-bound m6Am-containing RNA fragments without a 5ʹ cap structure could be recovered together with the internal m6A-containing RNA fragments. Both the FTO and Flag antibodies showed high specificity and efficiency in immunoprecipitation (Figure S3(d,e)). The gel region containing protein–RNA complex above the molecular weight of FTO and FTO-Flag was excised and used for sequencing library preparation (Figure S3(f,g)). A positive control CLIP library of PTBP1 was included. The data were summarized in Table S1.
We observed the similar low fraction of usable reads from FTO CLIP data in HeLa cells, while FTO overexpression increased the usable reads fraction (Figure S4(a)). FTO CLIP tags in one replicate were highly accumulated near the FTO binding sites identified in the other biological replicate (Figure S4(b)), demonstrating the confidence of the RNA binding activity of FTO. We further confirmed the specificity of the CLIP assay by performing UV-RIP-PCR analysis on a panel of anti-FTO enriched RNA fragments (Figure S4(d)).
When compared with the enrichment in IgG controls, FTO CLIP reads did not enrich around the TSS nor 5ʹUTR region in normal HeLa cells () and S4(c)). The overall FTO binding in transcribed regions was increased upon FTO overexpression (), below). At the basal level of FTO expression, the majority of FTO binding peaks (46.43%-50.59%) was located in the intronic regions, similar as those of the previously published data () and S1(c)). When FTO was overexpressed in HeLa cells, the FTO binding peaks preferentially shifted to the protein coding regions ()).
Analysis of the 5-mer motifs showed that the most enriched motif harboured the GAC consensus sequence in both biological replicates of endogenous FTO binding sites () and S4(e)). Upon FTO overexpression, motif enrichment scores increased consistent with the increased concentration of FTO-target RNA complex. Strikingly, the GAC-containing motif ranked as the most overrepresented one, supporting that the exogenously expressed FTO was functional ()). When we increased the motif length during the motif search, the enrichment of GGAC and GGACU in top motifs appeared (Figure S4(f)). We then calculated the fraction of FTO binding peaks containing GGACU motif and RRACH motifs in all the CLIP/eCLIP data. It is shown the peaks containing these m6A motifs were presented in all cell lines, although the frequency differed. FTO overexpression increased the m6A motif frequency in HeLa cells ()). Given that relatively low percentage of usable reads for normal control (Figure 4S(a)), we randomly selected the same small number of usable reads as in ) (lower panel) from each sample to call FTO binding peaks and motif analysis. Similar results were obtained (Figure S4(g)). These results confirmed that FTO binds to the m6A sequence context, and suggested that the binding is sensitive to the FTO concentration.
FTO overexpression effectively removes m6A modification from polyadenylated mRNA/lncRNAs at GGACU motifs in HeLa cells
Given that the overexpressed FTO-Flag bound to GGACU motif in HeLa cells, we then explored whether FTO-Flag could remove m6A from this typical m6A motifs, by performing m6A-seq experiments to capture m6A sites on polyadenylated mRNAs/lncRNAs in control (Flag-control) and FTO-overexpression (FTO-Flag) HeLa cells. The sequence data from two different benches of experiments were shown, with 22.2-fold (H) and 4.3-fold (L) of FTO overexpression by comparing the FPKM values of FTO in the corresponding input controls (Table S2, )). Analysis of the input controls showed that the expression level of key m6A writer and eraser genes was not changed upon FTO overexpression, which eliminated the complication of the result interpretation ()).
Figure 3. m6A demethylation by FTO is RRACU-dependent in HeLa cells.
(a) FTO overexpression did not affect the expression of other m6A writers and erasers in m6A-seq input samples, which were polyadenylated RNAs. FTO-H and FTO-L represent samples from HeLa cells with high-fold (22.2) or low-fold (4.3) FTO overexpression in relative to the Flag-control, respectively. Normalized FPKM values of each gene were presented. (b) GGACU motif was the top first motif in m6A peaks from control HeLa cells, which was disappeared upon FTO overexpression. (c) Relative proportions of m6A peaks with or without RRACU motifs. RRACU-bearing m6A peaks were detected in a higher frequency in Flag-control cells than that in FTO-Flag cells with FTO overexpression (P < 0.05). The P values were determined using Student’s unpaired t test. FTO-H and FTO-L were regarded as replicates. (d) Metagene distribution of m6A-seq reads around the centre of m6A peaks identified in Flag-control cells. m6A peaks were divided into two groups according to the presence or absence of the RRACU motif. (e) Examples of FTO demethylation from mRNA transcripts at the m6A peak containing an RRACU motif (DFFA), but not at the m6A peak lacking an RRACU motif (SFPQ) in HeLa cells. The m6A read density as shown for each gene in reads per million (RPM). Each gene was diagrammed by vertical black bars (exons) and thin horizontal lines (introns). m6A peak regions were shadowed by light green rectangles and arrow. (f) m6A qRT-PCR validation of the RRACU-dependence in FTO demethylation of m6A. The P values were determined using Student’s unpaired t-test. * P< 0.05. ** P< 0.01. *** P< 0.001. Results are shown as mean ± SE.
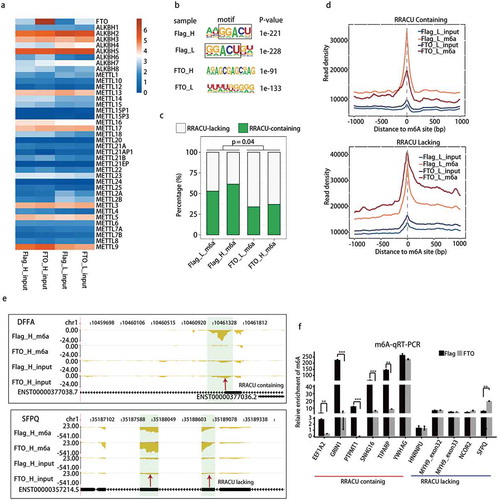
Analysis of the distribution of all mapped reads revealed that 96.5%-99.7% of input and m6A-seq reads of each sample were mapped to nuclear-encoded protein genes, pseudogenes and long non-coding RNAs (Table S3), which supported the purity of mRNAs used in this study. For the following peak calling and motif analysis, only uniquely mapped reads were involved, which removed any remaining complication from tRNAs and rRNAs. We called m6A peaks from m6A-seq data by running ABLIRC pipeline and MACS, followed by a de novo motif search with the HOMER algorithm, using input reads as backgrounds. Motif analysis showed that in control cells (Flag-control), the most significantly enriched motifs in m6A-bound peaks in both replicates were characterized by GGACU consensus sequence, which exactly matched the canonical m6A motif ()). Consistent with the hypothesis that FTO demethylates from its bound m6A motif, GGACU motif disappeared from the top-ranking motifs in FTO-overexpressed cells () and S5(a)).
Calculating the ratio of RRACU-bearing peaks in all m6A peaks confirmed the majority of peaks (52.90%~ 61.36%) detected in Flag-control cells contained RRACU motif, which reduced significantly (33.97%~36.91%) upon FTO overexpression ()). Additionally, the frequency of RRACU at the m6A peak positions in FTO overexpressed cells was substantially lower than that observed at Flag-control cells (Figure S5(b)). These results are all in line with the conclusion that FTO selectively erases the m6A modification from the RRACU motif of polyadenylated mRNAs/lncRNAs, likely in a FTO concentration-sensitive manner.
To further study the FTO selectivity of RRACU motif for its ‘eraser’ function, we separated the m6A peaks into two groups by either containing or lacking a RRACU motif. We then plotted the m6A-seq reads around the centre of these m6A peaks. In accordance with the m6A-demethylation activity of FTO, we observed a substantially decreased amount of m6A-seq reads around the centre of peak containing RRACU motif after FTO overexpression, in both sets of the experiments (), upper and S5(c), left). In contrast, around the centre of peaks lacking the RRACU consensus sequence, the level of m6A-seq reads was not decreased, but somewhat increased upon FTO overexpression (), upper and S5(c), right)
As examples, FTO specifically removed m6A signals from the RRACU-containing peaks in 3ʹ UTRs of the apoptosis-associated gene DFFA [Citation58] and lipid metabolism genes PPARD and VLDLR [Citation59,Citation60] () and S6), which contained the GGACU motif. It is possible that FTO regulates body mass index and obesity, partially by specifically removing the m6A modification from PPARD and VLDLR. We also showed examples whose m6A levels were not decreased by FTO, and these m6A modification occurred at RRACU-lacking peaks. These included m6A clusters on the internal exons of the splicing factor SFPQ ()) and RPL23A (Figure S6) and one cluster in the intronic region of ZNF701 (Figure S6).
We further validated the dependence of RRACU motif for FTO demethylation by RIP-qRT-PCR. Consistent with our findings above, all but one RRACU-bearing m6A sites showed decreased m6A signals upon FTO overexpression, whereas all of those lacking RRACU motif showed unchanged or increased m6A levels upon FTO overexpression ()). Taken together, overexpressed FTO preferentially removed m6A modification from the GGACU motif.
The extent of FTO demethylation from mRNAs/lncRNAs is concentration-dependent
FTO selection of RRACH motif for m6A demethylation in HeLa cells might represent a unique mechanism for FTO specificity in demethylating a population of genes but not the other. We noticed the m6A peak calling approach was not sensitive to the genes with high mRNA level. For example, we found that FTO specifically removed m6A modification from activating transcription factor ATF4 mRNA ()), a recently reported FTO target [Citation61]. One m6A modification being removed was well correlated with the reported alternative translation site controlled by m6A dynamics ()). However, these m6A modifications were not recovered by peak calling pipelines, due to the high input signals.
Figure 4. The extent of FTO demethylation is concentration-dependent.
(a) FTO-mediated demethylation of m6A in gene ATF4 is concentration-dependence. FTO-demethylated regions were marked with black box. (b) The number of genes whose mRNA/lncRNA methylation levels were down-regulated by FTO overexpression were 2.3–3.5 folds of the up-regulated, and increased with FTO levels. The number of up-regulated and down-regulated DMGs was showed in the bar plot. (c) FTO down-methylated genes are significantly overlapped with the m6A peak genes in both high-fold and low-fold FTO overexpression samples. The p values indicated statistical significance of the number of overlapping genes.
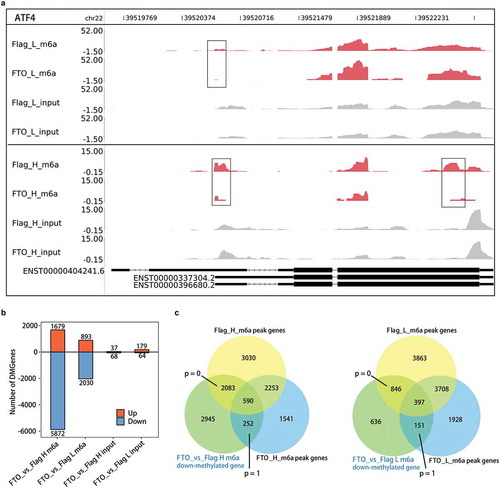
We, therefore, subjected m6A-seq reads to the edgeR software to identify differentially methylated genes (DMG) associated with FTO overexpression. For both benches of overexpression experiments, a much larger number of DMGs were down-methylated compared with those up-methylated. We showed the down-methylated genes were 5,872 and 2030, and the up-methylated genes were 1679 and 893, for the 22.2-fold and 4.3-fold FTO overexpression, respectively ()). As a control, the difference between the corresponding input samples was very small ()). The extent of FTO demethylation of mRNA/lncRNAs was apparently concentration-dependent () and S5(b)).
We further showed that m6A peak-containing genes from the control Flag samples were more significantly overlapped with the FTO down-demethylated genes, than those from samples with FTO overexpression ()). This further supported that the FTO demethylation occurs at the m6A-modified transcripts.
Discussion
The presented results together support a model for the dynamics of FTO in mediating m6A demethylation. FTO binds to GAC-containing and RRACH motifs in mRNAs/lncRNAs typical for m6A modification in a cell type-dependent manner. FTO binds to m6A motifs and removes the m6A modification in a concentration-dependent manner in HeLa cells. This model supports the m6A dynamics and diverse regulatory functions, and provides mechanistic insights into the FTO plasticity in biological functions and its reported involvement in multiple diseases.
This is the first report showing that FTO recognizes and binds to m6A motifs in cells, which is similar to the two m6A ‘writers’ methyltransferases METTL3 and METTL14 [Citation46]. Therefore, in contrast to non-selectively of the demethylation activity of two ‘eraser’ proteins FTO and ALKBH5 in vitro [Citation48], our results demonstrated that the selectively of FTO binding and demethylation from m6A motifs in living cells. In fact, a recent in vitro studies showed both the tertiary structure and sequence of the RNA substrates can affect the catalytic activity of FTO [Citation49]. Our results are consistent with the recent findings of the specific targets of FTO and ALKBH5 involved in diverse biological functions [Citation35,Citation37,Citation61–Citation63].
We have showed that FTO binds mRNA/lncRNA towards GAC-containing and RRACH motifs in multiple cell lines, with its motif selectivity being cell-type dependent. The association between FTO and m6A motifs is enhanced by FTO overexpression, and the overexpressed FTO preferentially removes m6A modification from GGACU and RRACU motifs also in a concentration-dependent manner, at least in HeLa cells. We propose that the concentration-dependence of FTO action shall contribute to the observed m6A/m6Am dynamics and enable a larger regulatory plasticity of m6A/m6Am mark under different physiological and pathological states. Given the structural and functional similarity between FTO and ALKBH5, it could be possible that ALKBH5 also contacts with the internal m6A similarly.
The finding of FTO binding and demethylation from the internal m6A motifs might reconciliate some of the recent conflicted results. For example, Mauer et al. showed that FTO controls mRNA stability in HEK293T cells with its demethylation activity towards m6Am [Citation53]. Results from the other two groups showed a certain contradiction [Citation51,Citation64], which add the debate in this field. In one study, FTO demethylation on internal m6A has a greater effect on mRNA stability than the ones with cap m6Am in the tested cells [Citation51]. In the other study, m6Am promotes the translation of capped mRNAs, but not stabilize A-starting capped mRNAs [Citation64].In the context of the findings presented in this study, the discrepancy might result from the different FTO expression levels and cell lines among these studies. In fact, one of these two studies demonstrates that the efficiency of FTO in demethylating m6A and m6Am is distinct among different cell lines, which depends on the relative levels of m6A/m6Am and the nucleus and cytoplasm distribution of FTO protein [Citation51].
Additionally, our analysis showed that FTO binding was mostly enriched at the TSS region in HEK293 cells, although the CLIP-seq method applied to obtain the data disfavour the recovery of FTO-bound m6Am-containing RNA fragments with an intact 5ʹ cap [Citation50]. This finding is well correlated with the previously reported high preference of FTO for m6Am as a substrate [Citation53], if we assume that at least a fraction of the TSS binding signals of FTO represents its demethylation of m6Am. The more preferred distribution of FTO binding sites in the 5ʹUTR and 3ʹUTR body regions of K562 and HeLa cells is consistent with the reported functions and the m6A profiles regulated by FTO in other leukaemia cells and mouse embryonic fibroblasts [Citation34,Citation37]. We suggest that the cell type-dependent binding of FTO, a potential indicator of its demethylation target selection, should be taken into account in explaining data in the future studies.
In conclusion, we have demonstrated the selectivity of FTO in binding and erasing m6A modification from m6A motifs, which support FTO functions in regulating the dynamics and distribution of m6A/m6Am mark under different physiological and pathological states [Citation26,Citation34,Citation37,Citation50,Citation51,Citation65–Citation68]. We propose that FTO-mRNA/lncRNA association is weak, which leaves rooms for additional cellular factors to corporately work with this eraser to choose among the internal m6A sites and 5ʹ-end m6Am in different transcripts. The dynamics of FTO in target selection is predicted to be sensitive to physiological and pathological changes, and contribute to both m6A dynamics and the FTO plasticity in biological functions and its reported involvement in multiple diseases. Further study of the FTO dynamics and m6A/m6Am demethylation regulation should be critical for m6A biology and the related disease control.
Materials and methods
Cell culture, plasmid and transfection
HeLa cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Hyclone) and 1% penicillin/streptomycin (Hyclone) at 37 °C in a humidified incubator with 5% CO2.
The cDNA of human FTO gene (GenBank: NM_001080432.2) amplified by PCR and subcloned into pCMV-Tag.2B-Flag vector (Stratagene). pCMV-Tag.2B-Flag vector containing FTO gene (FTO-Flag) or not containing the FTO gene (Flag-control) were transfected into HeLa cells using Lipofectamine 2000 (Invitrogen, 11668–027) following the manufacturer’s instructions.
Western blots
Total protein lysate was extracted from FTO overexpression/control cells with RIPA buffer. A total of 40–60 μg protein extracts were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, cat. 1620177, Foster, California, USA). Membranes were blocked with 5% non-fat milk, followed by overnight incubation with primary antibodies against either FTO (CUSABIO, Shanghai) or Flag (Sigma, cat. F7425) overnight at R.T. After the incubation with Rhodamine (TRTTC) goat anti-rabbit IgG (Abclonal, cat. A5040) (1: 10,000) for 1 h at R.T, the signals were detected with Clarity Max™ Western ECL Substrate (Bio-Rad, cat. 1705062).
Immunofluorescence analysis
After washed with PBS, HeLa was fixed in 4% formaldehyde. Cells were blocked in 5% BSA in PBS with 0.2% Triton X-100 for 10 min. Cells were incubated with antibody against FTO (MBL, RN121PW) overnight at 4°C. After washing with PBS, cells were incubated with an anti-rabbit IgG (Abclonal, cat. A5040) (1:100) in the dark at R.T for 1 h. DAPI staining for DNA was performed. Images were obtained using a confocal LSM780 microscope (Carl Zeiss, Germany).
CLIP-seq
HeLa cells (~106) cultured to a confluence of 70–80% with or without FTO-Flag or Flag transfection were subjected to UV cross-linking treatment on ice for 400 mJ/cm2. The cross-linked cells were then lysed and treated with RQ1 RNase-Free DNase (Progema, cat. M6101) to prevent DNA contamination, followed by partial digestion with MNase (Thermo, cat. EN0181) to further release the FTO-protected RNA fragments in a FTO-RNA complex form. FTO–RNA complex was immunoprecipitated by incubating with DynaBeads protein A/G conjugated with anti-FTO antibody (MBL, cat. RN121PW, Japan), IgG (Millipore, cat. 12–370, USA) or anti-Flag antibody (sigma, cat. F7425, USA) at 4°C for 2 h. Efficiency of the immunoprecipitation was shown in Figure S3. RNA was dephosphorylated at 3ʹ end and phosphorylated at 5ʹ end. The protein–RNA complex was separated by 4–12% NuPAGE Bis-Tris gel (Nvirogen, cat. NP0321 BOX) and the region of the membrane 30 kDa above the protein size was excised (Figure S3(e)). Protein was digested by proteinase K and RNA was isolated to prepare the sequencing library using Balancer sm/miRNA Library Preparation Kit according to the manufacturer’s instructions (Gnomegen, cat. K02420). The cDNA libraries were subsequently quantified and sequenced on the Illumina Nextseq platform. Image processing and base-calling were performed by the Illumina pipeline.
Analysis of CLIP-seq data
The clean reads were generated from raw sequencing reads after removing adaptor sequences and low-quality sequences, which were mapped to human GRCh38 genome using Tophat (v2.1.1). Reads aligned to more than one genome location were discarded. ‘Usable’ reads were defined as reads that uniquely mapped to the genome and remained after discarding PCR duplicates.
FTO CLIP-seq peaks were identified by running Piranha [Citation54], CIMS [Citation55] and ABLIRC [Citation56]. The target genes of FTO were finally determined by analysing the genomic locations of all the FTO binding peaks. A de novo motif search with the HOMER algorithm [Citation69] was performed to identify FTO binding sites.
UV-RIP-qPCR
HeLa cells (~106) were UV cross-linked, which were operated similarly as CLIP-seq described above to obtain the cross-linked protein-RNA complex right after immunoprecipitation, without further treatment. MNase was not used to further fragment RNA associated with FTO. Then, RNA was purified from the protein-RNA complex by proteinase K treatment, phenol/chloroform extraction and precipitated with ethanol. The isolated FTO-associated RNA was reverse transcribed using random primer, and analysed by quantitative, strand-specific RT-PCR using the Real-time detection system using a QuantStudio 6 Flex System (ABI). Gene-specific PCR primer pairs were presented in Table S4.
m6A dot blot assay
Total RNA was extracted using TRIzol Reagent (Ambion) following the manufacturer’s instructions, and mRNA was then purified by GenElute™ mRNA Miniprep Kit (sigma, MRN10). The m6A-dot-blot was performed on the Bio-Dot®Microfiltration Apparatus (170–6545, GE Healthcare) using Amersham Hybond-N+ membrane (GE Amersham, RPN303B) in two-fold dilutions. After UV cross-linking, the blotted membrane was washed by 1× PBST buffer, blocked with 5% of the non-fat milk, and incubated with primary rabbit anti-m6A antibody (sysy, cat. 202003) overnight at 4°C. After incubated with Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (DakoCytomation, p0448) secondary antibody, the membrane was visualized by supersignal westPico ECL substrate box (Thermo Pierce, 34087).
m6A immunoprecipitation and m6A-seq
The polyadenylated mRNAs isolated from total RNA using GenElute™ mRNA Miniprep Kit (sigma, MRN10) were fragmented into 100 nt length by using RNA Fragmentation buffer (0.1 M Tris-HCL PH 7.0, 0.1 M ZnCl2). Then, 50 and 100 ng fragmented mRNAs were incubated for 2 h at 4°C with 12.5 μg of anti-m6A antibody (sysy, 202003) in IP buffer (0.05 M Tris-HCL pH 7.4, 0.375 M NaCl, 0.5% Igepal CA-630). The mixture was then subjected to immunoprecipitation by incubation with Pierce™ ChIP-grade Protein A/G Magnetic Beads (Thermo, 26162) at 4°C for 2 h. After sufficient washing, m6A antibody-bound RNA was eluted from the beads with Elution buffer (1× IP buffer, 7mM m6A, RNase inhibitor), and then ethanol-precipitated. The eluted RNA was resuspended in H2O and used to generate the cDNA library according to RNA-Seq Library Preparation Kit for Transcriptome Discovery–Illumina Compatible, which was then sequenced using the HiSeq 2000 system (Illumina) according to the manufacturer’s instructions.
Analysis of m6A_seq data
For m6A-Seq data, adaptors and low-quality bases were trimmed from raw sequencing reads using CutAdapt, and reads less than 16nt were discarded. After quality control and data filtering, reads were aligned to the reference genome GRCh38 by TopHat2 (v2.1.1). To assess the m6A level of each gene, only reads unambiguously aligned were preserved to calculate reads number and RPKM value (RPKM represents reads per kilobase and per million). To identify the m6A regions (statistically significant m6A peaks), we called m6A peaks from m6A-seq data by running ABLIRC pipeline [Citation56].
m6A-qRT-PCR
m6A immunoprecipitation was performed as above. The ethanol-precipitated m6A antibody-bound RNA fragments were reverse transcribed using random primer, and then subjected to quantitative, strand-specific RT-PCR sing the Real-time detection system using a QuantStudio 6 Flex System (ABI). Gene-specific PCR primer pairs were presented in Table S5.
Differentially demethylated genes (DMG)
Differentially demethylated genes between the paired groups were analysed by using edgeR in R packages4. For each gene, significance p-value was obtained based on the model of the negative binomial distribution. Fold changes of gene expression were also estimated within the edgeR statistical package. The criterion for DMG has been set as fold change >2 or <0.5 and P < 0.01.
Statistical analysis
For the comparison of proportions of m6A peaks with or without the RRACU motif and the qPCR results, statistical analyses were carried out using Student’ s t-test. The results of qPCR are shown as the mean ± SE. Statistical analysis was performed using R (v3.1.3).
Data availability
All CLIP-seq and m6A-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE101955. PTB CLIP data are available at NCBI with BioProject ID: PRJNA377229 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA377229). A link to a UCSC genome browser session displaying the uploaded sequence tracks has been created (https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=erhuoyi&hgS_otherUserSessionName = fto).
Supplemental Material
Download Zip (13.6 MB)Acknowledgments
We are grateful to Yaqiang Xue, and Yali Ye (ABLife) for their contributions to the experimental parts, and to Yaxun Wei (ABLife) in helping some graphics. This work was supported by the grants from National Key R&D Program of China (2018YFD0500402), National Natural Science Foundation of China (31660641), Guangxi Natural Science Foundation (2017GXNSFAA198139), Guangxi Hundred-Talent Program, State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources (SKLCUSA-a201808), and Scientific Research Foundation of Guangxi University (XTZ130719). This study is also supported by grant from ABLife Inc. (ABL2013-07002).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975.
- Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42.
- Furuichi Y. “Methylation-coupled” transcription by virus-associated transcriptase of cytoplasmic polyhedrosis virus containing double-stranded RNA. Nucleic Acids Res. 1974;1:809–822.
- Rhodes DP, Moyer SA, Banerjee AK. In vitro synthesis of methylated messenger RNA by the virion-associated RNA polymerase of vesicular stomatitis virus. Cell. 1974;3:327–333.
- Shatkin AJ. Methylated messenger RNA synthesis in vitro by purified reovirus. Proc Natl Acad Sci U S A. 1974;71:3204–3207.
- Abraham G, Rhodes DP, Banerjee AK. The 5ʹ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell. 1975;5:51–58.
- Furuichi Y, Miura K-I. A blocked structure at the 5ʹ terminus of mRNA from cytoplasmic polyhedrosis virus. Nature. 1975;253:374.
- Furuichi Y, Morgan M, Muthukrishnan S, et al. Reovirus messenger RNA contains a methylated, blocked 5ʹ-terminal structure: m-7G(5ʹ)ppp(5ʹ)G-MpCp. Proc Natl Acad Sci U S A. 1975;72:362–366.
- Wei CM, Gershowitz A, Moss B. 5ʹ-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15:397–401.
- Adams JM, Cory S. Modified nucleosides and bizarre 5ʹ-termini in mouse myeloma mRNA. Nature. 1975;255:28–33.
- Furuichi Y, Morgan M, Shatkin AJ, et al. Methylated, blocked 5 termini in HeLa cell mRNA. Proc Natl Acad Sci U S A. 1975;72:1904–1908.
- Wei C, Gershowitz A, Moss B. N6, O2ʹ-dimethyladenosine a novel methylated ribonucleoside next to the 5ʹ terminal of animal cell and virus mRNAs. Nature. 1975;257:251–253.
- Wei CM, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16:1672–1676.
- Canaani D, Kahana C, Lavi S, et al. Identification and mapping of N6-methyladenosine containing sequences in simian virus 40 RNA. Nucleic Acids Res. 1979;6:2879–2899.
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3ʹ UTRs and near stop codons. Cell. 2012;149:1635–1646.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206.
- Linder B, Grozhik AV, Olarerin-George AO, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772.
- Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887.
- Munns TW, Liszewski MK, Sims HF. Characterization of antibodies specific for N6-methyladenosine and for 7-methylguanosine. Biochemistry. 1977;16:2163–2168.
- Bringmann P, Luhrmann R. Antibodies specific for N6-methyladenosine react with intact snRNPs U2 and U4/U6. FEBS Lett. 1987;213:309–315.
- Fu Y, Jia G, Pang X, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798.
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2013;505:117–120.
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326.
- Zhao X, Yang Y, Sun BF, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419.
- Meyer Kate D, Patil Deepak P, Zhou J, et al. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010.
- Zhou J, Wan J, Gao X, et al. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594.
- Yang Y, Fan X, Mao M, et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017;27:626–641.
- Zheng Q, Hou J, Zhou Y, et al. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol. 2017;18:1094–1103.
- Chen T, Hao YJ, Zhang Y, et al. m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301.
- Haussmann IU, Bodi Z, Sanchez-Moran E, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304.
- Ivanova I, Much C, Di Giacomo M, et al. The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell. 2017;67:1–9.
- Liu J, Eckert MA, Harada BT, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074–1083.
- Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270.
- Li Z, Weng H, Su R, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–141.
- Cui Q, Shi H, Ye P, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634.
- Barbieri I, Tzelepis K, Pandolfini L, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552:126–131.
- Su R, Dong L, Li C, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:1–16.
- Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–E56.
- Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
- Zhao BS, Nachtergaele S, Roundtree IA, et al. Our views of dynamic N6-methyladenosine RNA methylation. Rna. 2018;24:268–272.
- Mauer J, Jaffrey SR. FTO, m6Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett. 2018;592:2012–2022.
- Darnell RB, Ke S, Darnell JE. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics”. Rna. 2018;24:262–267.
- Rosa-Mercado NA, Withers JB, Steitz JA. Settling the m6A debate: methylation of mature mRNA is not dynamic but accelerates turnover. Genes Dev. 2017;31:957–958.
- Meyer KD, Jaffrey SR. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–342.
- Fu Y, Dominissini D, Rechavi G, et al. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15:293–306.
- Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95.
- Huang Y, Yan J, Li Q, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–384.
- Zou S, Toh JD, Wong KH, et al. N6-Methyladenosine: a conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci Rep. 2016;6:25677.
- Zhang X, Wei LH, Wang Y, et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc Natl Acad Sci U S A. 2019;116:2919–2924.
- Bartosovic M, Molares HC, Gregorova P, et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3ʹ-end processing. Nucleic Acids Res. 2017;45:11356–11370.
- Wei J, Liu F, Lu Z, et al. Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71:973–985.
- Van Nostrand EL, Pratt GA, Shishkin AA, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods. 2016;13:508–514.
- Mauer J, Luo X, Blanjoie A, et al. Reversible methylation of m6Am in the 5ʹ cap controls mRNA stability. Nature. 2017;541:371–375.
- Uren PJ, Bahrami-Samani E, Burns SC, et al. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012;28:3013–3020.
- Moore MJ, Zhang C, Gantman EC, et al. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc. 2014;9:263–293.
- Xia H, Chen D, Wu Q, et al. CELF1 preferentially binds to exon-intron boundary and regulates alternative splicing in HeLa cells. Biochim Biophys Acta. 2017;1860:911–921.
- Ke S, Pandya-Jones A, Saito Y, et al. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31:990–1006.
- Liu X, Zou H, Slaughter C, et al. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184.
- Seedorf U, Aberle J. Emerging roles of PPARδ in metabolism. Biochim Biophys Acta. 2007;1771:1125–1131.
- Tacken PJ, Hofker MH, Havekes LM, et al. Living up to a name: the role of the VLDL receptor in lipid metabolism. Curr Opin Lipidol. 2001;12:275–279.
- Zhou J, Wan J, Shu XE, et al. N6-methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell. 2018;69:1–12.
- Zhang S, Zhao BS, Zhou A, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.
- Zhang S-Y, Zhang S-W, Liu L, et al. m6A-driver: identifying context-specific mRNA m6A methylation-driven gene interaction networks. PLoS Comp Biol. 2016;12:e1005287.
- Akichika S, Hirano S, Shichino Y, et al. Cap-specific terminal N6-methylation of RNA by an RNA polymerase II–associated methyltransferase. Science. 2019;363:eaav0080.
- Shun Z, Zhou‐Lan B, Di X, et al. FTO regulates the chemo‐radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–597.
- Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent m6A regulates cardiac function during remodeling and repair. Circulation. 2019;139:518–532.
- Engel M, Eggert C, Kaplick PM, et al. The role of m6A/m-RNA methylation in stress response regulation. Neuron. 2018;99:389–403.
- Xiang Y, Laurent B, Hsu CH, et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576.
- Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589.