
ABSTRACT
The extra 5ʹ guanine nucleotide (G-1) on tRNAHis is a nearly universal feature that specifies tRNAHis identity. The G-1 residue is either genome encoded or post-transcriptionally added by tRNAHis guanylyltransferase (Thg1). Despite Caenorhabditis elegans being a Thg1-independent organism, its cytoplasmic tRNAHis (CetRNAnHis) retains a genome-encoded G-1. Our study showed that this eukaryote possesses a histidyl-tRNA synthetase (CeHisRS) gene encoding two distinct HisRS isoforms that differ only at their N-termini. Most interestingly, its mitochondrial tRNAHis (CetRNAmHis) lacks G-1, a scenario never observed in any organelle. This tRNA, while lacking the canonical identity element, can still be efficiently aminoacylated in vivo. Even so, addition of G-1 to CetRNAmHis strongly enhanced its aminoacylation efficiency in vitro. Overexpression of CeHisRS successfully bypassed the requirement for yeast THG1 in the presence of CetRNAnHis without G-1. Mutagenesis assays showed that the anticodon takes a primary role in CetRNAHis identity recognition, being comparable to the universal identity element. Consequently, simultaneous introduction of both G-1 and the anticodon of tRNAHis effectively converted a non-cognate tRNA to a tRNAHis-like substrate. Our study suggests that a new balance between identity elements of tRNAHis relieves HisRS from the absolute requirement for G-1.
Introduction
Aminoacyl-tRNA synthetases (aaRSs) belong to an ancient family of enzymes that charge amino acids to their cognate tRNAs and decipher genetic code via base pairing between the anticodon of aminoacyl-tRNA and the codon of mRNA [Citation1]. The accuracy of tRNA aminoacylation is crucial for maintaining the fidelity of translation. AaRSs establish a tRNA’s identity through recognizing certain identity elements present on the tRNA, which could be single nucleotides, base pairs, or post-transcription modifications [Citation2–Citation4]. Identity elements often reside in the acceptor stem or anticodon loop of a tRNA. In eukaryotic cells, the translation process occurs in both the cytoplasm and organelles (e.g. mitochondria and chloroplasts). Hence, there exists at least two distinct sets of aaRSs in each eukaryote [Citation5,Citation6]. For example, humans possess two distinct histidyl-tRNA synthetase (HisRS) genes, one encoding the cytoplasmic enzyme and the other encoding its mitochondrial counterpart [Citation7]. Nevertheless, in some cases, both forms of a given aaRS are encoded by the same gene through alternative transcription or translation, examples of which include genes encoding yeast alanyl-, glycyl-, histidyl-, and valyl-tRNA synthetases [Citation8–Citation11].
In the majority of species, the major identity element of tRNAHis is an extra guanine nucleotide, G-1, at the 5′ end of the acceptor stem, which is positioned across from the discriminator base N73, a minor identity element. The position −1 is normally empty in mature tRNAs, except for tRNAHis. Therefore, the G-1 residue is an exclusive feature to tRNAHis [Citation12]. The essentiality of this residue (and its monophosphate) is evidenced by the widespread existence of two distinct pathways to yield mature tRNAsHis with G-1. In nearly all eubacteria, eukaryotic organelles, and some archaea, this universal identity element is encoded by the tRNAHis genes opposite C73 and is preserved during processing by ribonuclease P [Citation13]. By contrast, In the majority of eukaryotic cytoplasm and some archaea, G-1 is post-transcriptionally added by tRNAHis guanylyltransferase (Thg1) [Citation14], which places G-1 opposite A73 in eukaryotes [Citation15] and opposite C73 in archaea [Citation16]. Thus, in eubacteria, archaea, and eukaryotic organelles, G-1:C73 constitutes an extra base pair in the acceptor stem. However, in eukaryotic cytoplasm, the additional base pair G-1:A73 is actually a non-Watson-Crick mismatch.
Although the G-1 residue is a nearly universal feature for identification of tRNAHis in life, a few exceptions have been identified. These include tRNAsHis recovered from the cytoplasm of Acanthamoeba castellanii [Citation17] and Trypanosoma brucei [Citation18] and some α-proteobacteria such as Caulobacter crescentus [Citation19]. In A. castellanii and T. brucei, G-1 is missing from their cytoplasmic tRNAsHis, and their cytoplasmic HisRSs can efficiently charge yeast tRNAnHis without G-1 [Citation17,Citation18].
In the worm Caenorhabditis elegans, HARS-1 is the only HisRS gene which provides functions in both compartments. Since no THG1 orthologue exists in this eukaryote [Citation18], this raised the question of whether its cytoplasmic and mitochondrial tRNAHis isoacceptors carry the canonical identity element. A recent report indicated that the cytoplasmic tRNAHis of C. elegans (CetRNAnHis) retains a genome-encoded G-1 [Citation18]. In contrast, there is no G residue existing in the corresponding position in the tRNAmHis gene. To gain further insight, we isolated and sequenced the mature CetRNAmHis and studied the tRNAHis identity mechanism in this eukaryote. Our results showed that the mature CetRNAmHis lacks G-1. Despite CetRNAmHis lacking this canonical identity element, it can be efficiently aminoacylated in vivo. Regardless, CeHisRS still preferred CetRNAHis with G-1, but could tolerate CetRNAnHis without G-1. Moreover, the anticodon has also taken a leading role in tRNAHis identity through evolution. The above findings suggest a pioneering mechanism which allows CeHisRS to recognize tRNAHis isoacceptors with or without G-1.
Results
C. elegans HARS-1 is a dual-functional HisRS gene
Sequence homology search indicated that HARS-1 is the only HisRS gene in the nuclear genome of C. elegans. This gene contained 6 exons (exon 1 to exon 6), with alternative intron splicing yielding two mature transcripts, HARS-1a (containing exons 1, 3, 4, 5, and 6) and HARS-1b (containing exons 2, 3, 4, 5, and 6) ()). The polypeptides translated from these two transcripts were essentially identical in sequence except for the N-terminal domain. HARS-1a-translated polypeptide carried an N-terminal domain, which shares significant sequence homology with the WHEP domain, while HARS-1b-translated polypeptide carried an N-terminal domain, which is rich in hydroxylated and positively charged residues but devoid of acidic residues, a feature characteristic of a mitochondrial targeting signal (MTS) ()). Hence, the former (CeHisRSc) is likely to function in the cytosol, and the latter (CeHisRSm) in mitochondria. As the MTS of CeHisRSm is expected to be cleaved off upon being imported into mitochondria, the only difference between CeHisRSc and the processed CeHisRSm is the WHEP domain.
Figure 1. C. elegans HisRS isoforms and tRNAHis isoacceptors.
(a) Generation of two CeHisRS isoforms. CeHisRSc and CeHisRSm possess essentially the same polypeptide sequence except for the N-terminal domain, with WHEP (residues 1 ~ 63) in CeHisRSc and MTS (residues 1 ~ 33) in CeHisRSm. Relative positions of the functional domains in CeHisRS are marked. CD, catalytic domain; ABD, anticodon-binding domain. (b) tRNAHis isoacceptors. Identity elements that are crucial for recognition of SctRNAnHis by ScHisRS are boxed. The defective TψC arm in CetRNAmHis is marked with a dashed circle. The small letters in SctRNAnHis denote the different nucleotides between SctRNAnHis and CetRNAnHis.
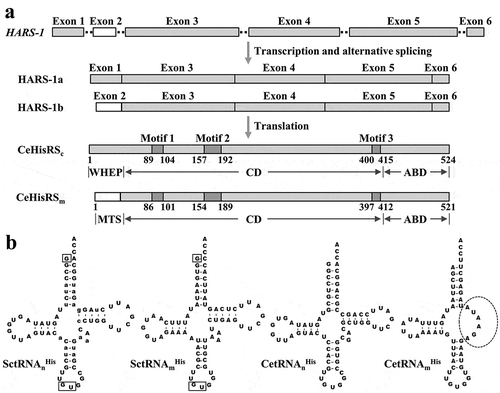
G-1 is a nearly universal feature that specifies tRNAHis identity. For example, the primary identity element of S. cerevisiae tRNAHis (SctRNAHis) is G-1, and deletion of this nucleotide resulted in an unchargeable tRNA mutant [Citation20,Citation21]. ) shows the nuclear- and mitochondrial-encoded tRNAHis isoacceptors. In contrast to the scenario of E. coli tRNAHis, the discriminator base N73 plays almost no role in SctRNAHis identity [Citation20]. On the other hand, the anticodon has become an important identity element of SctRNAHis. Despite CetRNAnHis retaining a structure similar to that of SctRNAnHis, its G-1 is genome encoded, which has so far only been observed in prokaryotes and organelles. Most surprisingly, CetRNAmHis lacks a G-1 nucleotide at the corresponding position in the mitochondrial genome, and contains U73 instead of C73. Moreover, CetRNAmHis lacks a TψC arm, which makes it very different from most tRNAHis species.
The C. elegans mitochondrial tRNAHis lacks the canonical identity element
Because no THG1 orthologue exists in C. elegans and its mitochondrial tRNAHis gene does not contain a G nucleotide at position −1, we wondered whether an editing enzyme or pathway exists to salvage the G-1 nucleotide. To take a closer look, we isolated total RNA from a C. elegans culture, circularized the RNA with T4 RNA ligase, and amplified the acceptor stem sequence of tRNAmHis by RT-PCR using a set of gene-specific primers ()). The PCR products were cloned into pUC18 and ten of the resultant clones were sequenced. ) shows that almost all of the tRNAmHis clones (clones 1 ~ 9) contained a CCA tail (nucleotides 74 ~ 76), suggesting that they represent mature tRNAmHis species. However, none of these clones contained G-1 (clones 1 ~ 10).
Figure 2. Determining the G-1 status of the C. elegans mitochondrial tRNAHis.
(a) Strategy used to determine the G-1 status of CetRNAmHis. (b) Sequencing data. For clarity, the expected position of G-1 is boxed. (c) In vivo aminoacylation assay. The charged and uncharged tRNAs were separated in an acid 10% polyacrylamide-7 M urea gel, blotted onto a positively charged nylon membrane, and hybridized with specific 32P-labled probes. The symbols ‘+’ and ‘–’ denote the treatment with alkaline pH. The arrows mark the positions of charged and uncharged tRNAsHis.
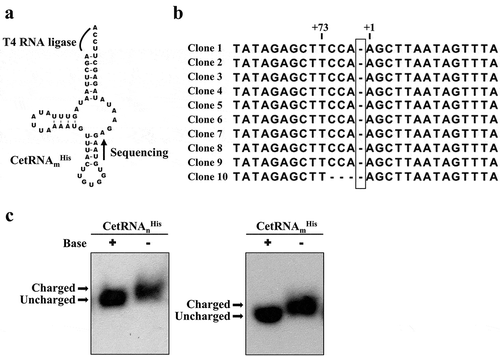
To check whether CetRNAmHis is indeed a natural substrate for CeHisRS, two micrograms of total mitochondrial RNA were isolated from C. elegans under acidic conditions, and a part of the RNA sample was incubated at alkaline pH to hydrolyze aminoacyl-tRNA ester bonds. These RNA samples were electrophoresed through an acid (pH 5.2) 10% polyacrylamide-7 M urea gel and then blotted onto a positively charged nylon membrane. The hybridization was performed with 32P-labeled oligonucleotide probes specific for the cytoplasmic and mitochondrial CetRNAHis isoacceptors, respectively. As expected, CetRNAmHis (without G-1) was efficiently charged in vivo ()). To our knowledge, this is the first example studied to date in which an organellar tRNAHis completely lacks G-1. While this outcome was not totally surprising, it prompted us to ask how CeHisRS can accommodate two such divergent tRNAsHis, one with and the other without the canonical identity element.
C. elegans HiSRs can recognize yeast tRNAhis with G-1:A73 or G-1:C73
Since the native tRNA substrates of CeHisRS are so different in G-1 and N73, we suspected that these two identity elements are not as crucial as previously anticipated. To test this hypothesis, we cloned the genes encoding CeHisRSc and CeHisRSm into a yeast expression vector, and transformed the constructs into a yeast strain deleted of THG1 (encoding Thg1) or HTS1 (encoding HisRS). The rescue activities of these constructs are summarized in . As described earlier, the G-1 residue in SctRNAmHis is genome encoded, while the G-1 residue in SctRNAnHis is post-transcriptionally added. Therefore, no G-1 can be added to SctRNAnHis in the thg1− strain. 5-FOA and YPG plates were respectively used to test the cytoplasmic and mitochondrial HisRS functions of a gene of interest.
Figure 3. Heterologous rescue activity of C. elegans HisRS isoforms.
(a) Summary of the HisRS constructs and their rescue activities. The symbols ‘+’ and ‘–’ respectively denote positive and negative complementation. (b) Complementation of the genetic loss of yeast THG1 or HTS1. (c) Western blotting. The amount of cellular protein extracts loaded into each well is indicated at the bottom of the blot. Numbers 1 ~ 6 (circled) shown in (b) and (c) represent constructs shown in (a).
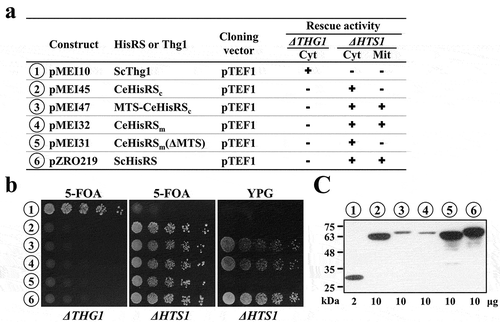
As shown in ), CeHisRSc efficiently restored the growth phenotype of the HTS1 knockout strain on 5-FOA (number 2 in middle panel), and when fused with a yeast MTS, it efficiently rescued the growth defect of the same knockout strain on YPG (number 3 in right panel). Similarly, CeHisRSm with its own MTS efficiently rescued the growth defect of the HTS1 knockout strain on YPG (number 4), and upon deletion of its native MTS, CeHisRSm efficiently restored the growth phenotype of the null allele on 5-FOA (number 5). We also noticed that CeHisRSc fused with a heterologous MTS or CeHisRSm carrying its own MTS could function in both compartments when overexpressed (numbers 3 and 4), possibly due to incomplete mitochondrial targeting of the fusion enzyme, a scenario often seen in aaRS rescue assays [Citation10,Citation22]. Likewise, overexpressed ScHisRS (carrying its own MTS) could rescue the growth defects of the null allele on both 5-FOA and YPG (number 6). Consistent with a previous observation [Citation18], neither CeHisRSc or CeHisRSm were able to complement the genetic loss of THG1 on 5-FOA (left panel), suggesting that CeHisRS cannot charge SctRNAnHis without G-1 (SctRNAnHis(∆G-1)). Together, these results suggest that CeHisRS can charge SctRNAsHis with G-1:A73 or G-1:C73, but cannot charge SctRNAHis without G-1.
We next examined the relative protein expression levels of these constructs in yeast using an anti-His6 tag antibody as a probe. As shown in ), CeHisRSc had a relatively higher expression level when localized in the cytoplasm than in mitochondria (numbers 2 and 3). A similar scenario was observed for CeHisRSm (numbers 4 and 5). The expression levels of CeHisRSc and CeHisRSm(∆MTS) were similar to that of ScHisRS (numbers 2, 5, and 6). Despite the fact that MTS-CeHisRSc and CeHisRSm were expressed at a much lower level than their cytoplasmic counterparts ()), they could provide sufficient rescue activity in both compartments ()).
Cehisrs is more tolerant than ScHisRS towards tRNAHis without G-1
Consistent with a previous observation [Citation18], our study indicated that CeHisRS poorly recognizes SctRNAnHis without G-1 (). We next tested whether CetRNAHis(∆G-1) is a better substrate for CeHisRS in vivo. Pursuant to this objective, constructs that harbor the genes encoding HisRS and tRNAHis(∆G-1) were co-transformed into the yeast THG1 knockout strain and the growth phenotypes of the resultant co-transformants on 5-FOA were examined ()). To express tRNAHis in the THG1 knockout strain, the genes encoding SctRNAHis and CetRNAHis were cloned into pRS313 (a low-copy-number yeast shuttle vector for normal expression of the target gene) and pRS423 (a high-copy-number yeast shuttle vector for overexpression of the target gene).
Figure 4. Complementation of the genetic loss of yeast THG1.
(a) Strategy used to determine the rescue activity of an orthogonal or non-orthogonal pair of HisRS/tRNAHis. (b) Co-expression of ScHisRS and various tRNAsHis(∆G-1). (c) Co-expression of CeHisRS and various tRNAsHis(∆G-1). pRS423 is a high-copy-number yeast vector with a 2μ replication origin, while pRS313 is a low-copy-number yeast vector with a CEN replication origin.
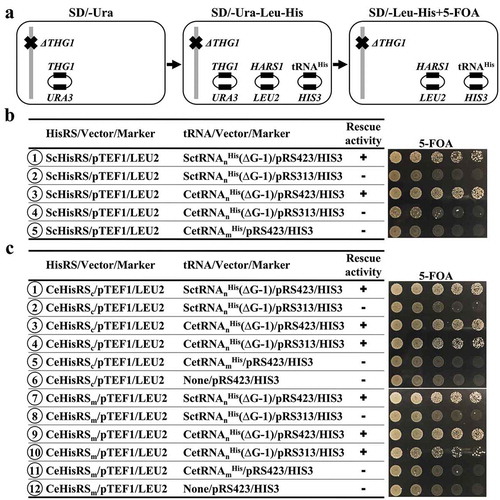
As shown in ), overexpression of both ScHisRS and SctRNAnHis(∆G-1) circumvented the requirement for Thg1 (number 1), a scenario that was reported earlier [Citation23]. Conceivably, overexpression of both the enzyme and substrate enhances the reaction rate to a level that can sustain the growth of the knockout strain. When ScHisRS was overexpressed and SctRNAnHis(∆G-1) was non-overexpressed, no growth was observed (number 2). A similar result was obtained when CetRNAnHis(∆G-1) was used as the substrate (numbers 3 and 4). However, we did notice that non-overexpressed CetRNAnHis(∆G-1), but not SctRNAnHis(∆G-1), conferred a very weak yet distinguishable complementation (numbers 2 and 4). No growth was observed when the tRNA substrate was replaced with CetRNAmHis (lacking G-1) (number 5).
We next tested whether CeHisRS is more tolerant than ScHisRS towards tRNAHis(∆G-1). As shown in ), overexpression of both CeHisRSc and SctRNAnHis(∆G-1) effectively complemented the genetic loss of THG1 (number 1). However, the complementation turned negative if the tRNA substrate was not overexpressed (number 2). In contrast to SctRNAnHis(∆G-1), CetRNAnHis(∆G-1) appeared to be a more favorable substrate for CeHisRSc. Overexpressed CeHisRSc was able to restore the growth phenotype of the null allele even when the substrate CetRNAnHis(∆G-1) was not overexpressed (numbers 3 and 4). No growth was observed when the tRNA substrate was replaced with CetRNAmHis (which lacks both G-1 and a TψC arm) (number 5). Considering the fact that CetRNAmHis requires a species-specific post-transcriptional modification to fold into a proper L-shape structure [Citation24], this outcome was not surprising. Because the mature CeHisRSm is different from CeHisRSc by only an N-terminal WHEP domain, we next tested whether the WHEP domain is required for the rescue activity. As expected, the mature CeHisRSm (or CeHisRSc deleted of WHEP) retained a rescue activity almost indistinguishable from that of CeHisRSc (numbers 7 ~ 12). Hence, the WHEP domain is dispensable for the rescue activity of CeHisRS. Collectively, these data suggest that CeHisRS is more tolerant than ScHisRS towards tRNAHis without G-1, and CeHisRS prefers CetRNAnHis(∆G-1) over SctRNAnHis(∆G-1).
A change in identity element balance enables C. elegans HiSRs to tolerate tRNAHis without G-1
To determine the steady-state kinetic parameters for tRNAHis aminoacylation, His6-tagged CeHisRSc and CeHisRSm (without MTS) were purified to homogeneity through Ni-NTA affinity chromatography. As one of the native tRNA substrates of CeHisRS lacks the canonical identity element G-1, we wondered whether CetRNAHis has gained an additional identity element during evolution. Moreover, we were prompted to ask whether the WHEP domain is really dispensable for tRNAHis histidylation. To this end, various mutations were introduced into CetRNAnHis and the aminoacylation efficiencies of the resultant tRNA mutants were determined.
As shown in , deletion of G-1 from CetRNAnHis reduced the aminoacylation efficiency (kcat/KM) to 5% for CeHisRSc. Unexpectedly, this deletion reduced not only the kcat value (75-fold) but also the KM value (3.5-fold). In contrast, mutation at the discriminator base A73 had only a minor effect on aminoacylation. Mutation of A73 to U, C, or G respectively reduced the aminoacylation efficiency to 94%, 22%, and 11%, suggesting that CeHisRSc slightly prefers A and U at this position. This finding might account for the presence of A73 and U73 on CetRNAnHis and CetRNAmHis, respectively. We next focused on the anticodon GUG (nucleotides 34–36) of CetRNAnHis. As mutations G34U, U35C, and G36C were previously shown to have the strongest effect on histidylation of SctRNAnHis by ScHisRS [Citation20], similar mutations were introduced into CetRNAnHis. G34U, U35C, and G36C respectively reduced the aminoacylation efficiency to 19%, 8% and 2%. Apparently, the importance of specific nucleotides in the anticodon is shifted. ScHisRS relies more on G34 and U35, while CeHisRS relies more on G36.
Table 1. Kinetic parameters for aminoacylation of tRNAnHis by CeHisRSc.
We next tested whether the WHEP domain is actually dispensable for tRNAHis aminoacylation. Note that the ‘processed’ or mature CeHisRSm is essentially equivalent to CeHisRSc minus the WHEP domain. shows that deletion of the WHEP domain from CeHisRSc had a very limited effect on its histidylation activity. The kcat/KM value for histidylation of the WT CetRNAnHis by CeHisRSm was close to that by CeHisRSc (4.23 versus 5.67 μM−1min−1). Moreover, deletion of G-1 and mutation of G36 to C respectively reduced the histidylation efficiency to 9% and2%, while mutation of A73 to U increased the efficiency to 123% (). These results support our hypothesis that the WHEP domain is dispensable for the histidylation activity of CeHisRS.
Table 2. Kinetic parameters for aminoacylation of tRNAnHis by CeHisRSm.
As the mature mitochondrial CetRNAHis (without G-1) can be efficiently aminoacylated in vivo ()), we wondered whether addition of G-1 to this tRNA affects its aminoacylation efficiency. As shown in , addition of G-1 to CetRNAmHis significantly enhanced its aminoacylation efficiency (~8-fold). Thus, CeHisRS prefers CetRNAHis with G-1, regardless of whether it is within the context of cytoplasmic or mitochondrial CetRNAHis.
Figure 5. In vitro aminoacylation assay. The in vitro aminoacylation activity of CeHisRSm (1 μM) was tested using in vitro-transcribed CetRNAs (5 μM) as the substrates. The relative amounts of [14C]Histidine that were incorporated into tRNAmHis were subsequently determined by a liquid scintillation counter. The arrows indicate the tRNA species used. CetRNAmGly served as a negative control.
![Figure 5. In vitro aminoacylation assay. The in vitro aminoacylation activity of CeHisRSm (1 μM) was tested using in vitro-transcribed CetRNAs (5 μM) as the substrates. The relative amounts of [14C]Histidine that were incorporated into tRNAmHis were subsequently determined by a liquid scintillation counter. The arrows indicate the tRNA species used. CetRNAmGly served as a negative control.](/cms/asset/03f2e950-8042-4588-b33f-f1f955d759d0/krnb_a_1626663_f0005_b.gif)
tRNAThr can be converted to a tRNAHis-like substrate by insertion of both G-1 and the anticodon of tRNAHis
So far, we have shown that both G-1 and the anticodon significantly contribute to the specific recognition of CetRNAnHis. To be sure that these two elements are necessary to establish a tRNAHis identity for CeHisRS, we next examined whether a non-cognate tRNA, CetRNAnThr, can be converted to a chargeable tRNA substrate by insertion of one or both of these two elements. A similar strategy has previously been shown in converting SctRNAThr to a tRNAHis-like substrate [Citation25]. As shown in , the WT CetRNAnThr could not be charged by CeHisRSm, but insertion of G-1 or changing the anticodon AGU to GUG partially rescued its aminoacylation efficiency (~0.7% relative to the WT CetRNAnHis). Interestingly, when both of these two elements were introduced into CetRNAnThr, the resultant tRNA became a good substrate for CeHisRSm, with 21% histidylation efficiency relative to the WT CetRNAnHis. Thus, both the G-1 residue and anticodon are necessary to specify a tRNAnHis identity for CeHisRS.
Our rescue assays indicated that CetRNAnHis(∆G-1) is a better substrate than SctRNAnHis(∆G-1) for CeHisRS (). To obtain quantitative data, SctRNAnHis and its ∆G-1 mutant were also tested. As expected, the WT SctRNAnHis was an excellent substrate for CeHisRS, with 55% efficiency relative to the WT CetRNAnHis. However, deletion of G-1 from SctRNAnHis yielded a ∆G-1 mutant unsuitable for histidylation by CeHisRSm; no detectable charging activity was observed for this substrate (). Thus, CeHisRS can tolerate its own tRNAnHis without G-1, but not yeast tRNAnHis without G-1.
The WHEP domain plays little role in the thermal stability and structural flexibility of CeHiSRs
As the only difference between CeHisRSc and the mature CeHisRSm is the WHEP domain, we wondered whether this domain regulates the thermal stability of CeHisRS. To this end, purified CeHisRSc and CeHisRSm proteins were subjected to circular dichroism (CD) spectroscopy at 222 nm between 10°C and 100°C. As shown in , both proteins retained more than 80% of their α-helix structure (compared with the molar ellipticity value determined at 20°C) at temperatures below 40°C, but lost almost half of their α-helix structure when the temperature reached 50°C or higher (,b)). As a result, CeHisRSc and CeHisRSm had a melting temperature of 52°C and 55°C, respectively ()). This result suggests that the WHEP domain does not play a significant role in the thermal stability of CeHisRS.
Figure 6. Thermal stability and structural flexibility of C. elegans HisRS isoforms.
Melting temperatures of (a) CeHisRSc and (b) CeHisRSm were determined at 222 nm using CD spectroscopy between 10℃ and 100℃. (c) Relative CD signal changes of CeHisRSc and CeHisRSm. (d) Limited proteolysis of CeHisRSc and CeHisRSm. The protein amount used was 4 μg per lane. The arrows indicate CeHisRSc and CeHisRSm.
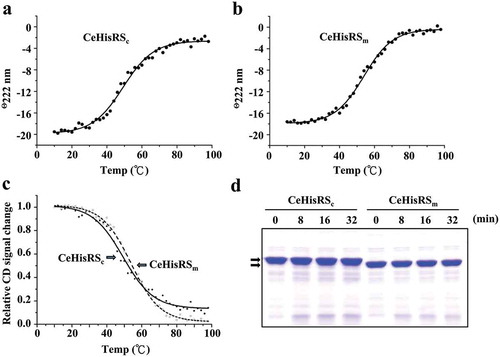
To study the effect of the WHEP domain on the structural flexibility of CeHisRS, limited proteolysis was carried out with CeHisRS/trypsin in a ratio of 10:1. Limited proteolysis is often used to probe the structure and dynamics of proteins. Exposed regions such as loops and flexible regions are more susceptible to the prolific protease. As shown in ), both forms of CeHisRS were rather resistant to trypsin. The relative protein levels of CeHisRSc and CeHisRSm remained almost unchanged in high concentrations of trypsin throughout the time period tested. This result suggests that the WHEP domain does not play a significant role in regulating the dynamic structure of CeHisRS.
Discussion
The G-1 residue is the major identity element for recognition of tRNAHis, regardless of whether it is of eukaryotic or prokaryotic origin [Citation20,Citation26,Citation27]. Deletion of this canonical identity element from E. coli and yeast tRNAsHis respectively reduced the aminoacylation efficiency (kcat/KM) ~250 and ~740-fold [Citation20,Citation27]. Similarly, deletion of G-1 from human tRNAHis led to a mutant tRNAHis unsuitable for histidylation [Citation7]. In contrast, CeHisRS exhibits relatively high tolerance towards CetRNAnHis lacking G-1; deletion of this identity element only reduced the aminoacylation efficiency ~15-fold (,). Hence, overexpression of CeHisRS was able to rescue a yeast THG1 null allele, regardless of whether CetRNAnHis(∆G-1) was also overexpressed (). In contrast, both ScHisRS and SctRNAnHis(∆G-1) had to be overexpressed in order to rescue the THG1 knockout strain () [Citation23]. While the importance of specific nucleotides in the anticodon is shifted between CeHisRS and ScHisRS, the anticodon still plays a significant role in tRNAHis recognition by CeHisRS. As a result, a non-cognate tRNA can be converted to a tRNAHis-like substrate for CeHisRS via simultaneous insertion of both the G-1 residue and anticodon of tRNAHis ().
Consistent with our observations ( and ), a recent study showed that CeHisRS strongly prefers SctRNAnHis with G-1 over SctRNAnHis without G-1, and that overexpression of CeHisRS alone fails to rescue a yeast THG1 knockout strain. However, their study also showed that CeHisRS can still charge SctRNAnHis without G-1, albeit to a low level [Citation18]. In contrast, no charging of this tRNA variant by CeHisRS was detected in our assay. A careful examination of the reaction conditions used by these two studies suggested that the slight difference in aminoacylation activity might result from different concentrations of histidine used in the assays. They used 40 µM of histidine (and labeled 32P at tRNAHis), while we used only 26 µM of histidine (and labeled 14C at histidine). It is possible that CeHisRS possesses a KM value for histidine higher than expected, leading to the slight difference in aminoacylation activity.
Interestingly, our data also indicated that SctRNAnHis without G-1 is a poorer substrate for CeHisRS than CetRNAnHis without G-1 ( and ). A more detailed sequence and structural comparison between these two tRNAsHis indicated that SctRNAnHis is different from CetRNAnHis at 16 nucleotide positions, including 6 in the acceptor stem, 1 at the corner of the D stem and anticodon stem, 4 in the anticodon stem, 2 in the variable loop, and 3 in the TΨC stem (shown in )). Despite the fact that none of these positions have ever been reported to be critical for aminoacylation of tRNAHis by HisRS, it is possible that these nucleotides (or base pairings) play a role in aminoacylation only when the major determinant G-1 is missing. As reported earlier, the nucleotides (or base pairings) in the acceptor stem contribute to the structural flexibility of tRNA [Citation28], while the nucleotides in the variable loop are involved in the overall folding of tRNA [Citation29]. In turn, some of these differences in these two tRNAs may confer a unique conformation or a secondary determinant to CetRNAnHis without G-1. As C. elegans genetically lacks the G-1-addition enzyme, this ability to tolerate a G-1-deficient CetRNAnHis variant might provide certain survival advantages under conditions where the G-1 nucleotide of CetRNAnHis is aberrantly removed during processing by RNase P.
The essentiality of G-1 for tRNAHis identity is evidenced by the prevalence of two distinct mechanisms for acquiring the element, one depending on co-transcriptional incorporation and the other on post-transcriptional addition [Citation3,Citation30,Citation31]. The former pathway occurs in bacteria, eukaryotic organelles, and certain archaea, while the latter pathway occurs in eukarya and some archaea. Nevertheless, certain eukaryotes such as A. castellanii and T. brucei lack a THG1 orthologue [Citation17,Citation18], and thus cannot add G-1 to their cytoplasmic tRNAsHis. As a result, the HisRSc enzymes of these two eukaryotes can efficiently charge G-1-deficient tRNAHis. On the other hand, plant mitochondria appear to retain both pathways for acquisition of the G-1 residue [Citation32]. Unlike other Thg1-dependent eukaryotes, the slime mold Dictyostelium discoideum possesses up to four THG1 homologues. Deletion of the homologue that is responsible for adding G-1 to the mitochondrial tRNAHis resulted in a slow-growth phenotype rather than cell death [Citation33], suggesting a certain level of tolerance towards tRNAmHis without G-1.
Despite the fact that both T. brucei and A. castellanii are Thg1 independent and their cytoplasmic HisRSs are only responsible for recognition of tRNAHis isoacceptors without G-1 in vivo [Citation17,Citation18], these two HisRSs can also efficiently charge tRNAHis with G-1 in vitro. Although C. elegans is also a Thg1-independent eukaryote, its cytoplasmic tRNAHis contains a genome-encoded G-1 residue, which has so far only been found in prokaryotes and organelles. Moreover, its mitochondrial tRNAHis isoacceptor lacks the universal identity element (), a phenomenon never observed in any organelle. To be dual functional and at the same time be able to charge two highly divergent tRNAHis isoacceptors (one with G-1 and the other without), CeHisRS was forced to evolve from a G-1-dependent enzyme to a G-1-preferred enzyme. It remains unclear how the G-1 residue of tRNAnHis can be retained during processing by nuclear RNase P and whether losing the G-1 identity element in tRNAmHis confers any selective advantage to the mitochondrial translational machinery of this organism. In summary, our study provided pioneering insights on how the anticodon might work alongside with G-1 in identity recognition of CetRNAHis.
Materials and methods
Construction of plasmids
Cloning of the gene encoding CeHisRSc or CeHisRSm into pTEF1 (a high-copy-number yeast shuttle vector with a strong TEF1 promoter, a LEU2 maker, and a 2μ replication origin) followed a standard protocol. Briefly, a pair of gene-specific primers were used to amplify the gene via a polymerase chain reaction (PCR) using C. elegans cDNA as the template. The forward primer (with an SpeI site) was located immediately upstream of the open reading frame (ORF), while the reverse primer (with an XhoI site) was located immediately upstream of the stop codon. The amplified DNA fragment was digested with SpeI and XhoI and then cloned into the SpeI/XhoI sites of pTEF1 for complementary assays. To fuse a heterologous MTS to HisRS, a DNA sequence encoding amino acid residues 1 ~ 46 of the mitochondrial precursor form of yeast valyl-tRNA synthetase was amplified by a PCR as an XbaI-SpeI fragment and then inserted into the 5ʹ SpeI site of the target gene.
For protein purification, the target genes were cloned into pET21b (an E. coli expression vector with a T7 promoter) following a similar protocol. The plasmids harboring the target genes were transformed into BL21-CodonPlus(DE3), and His6-tagged proteins were purified to homogeneity through Ni-NTA affinity chromatography as previously described [Citation34]. Western blotting followed a standard protocol and used an HRP-conjugated anti-His6 tag antibody as a probe [Citation35].
Cloning of the SctRNAHis gene into pRS423 (a high-copy-number yeast shuttle vector with a HIS3 marker and a 2μ replication origin) or pRS313 (a low-copy-number yeast shuttle vector with a HIS3 marker and a CEN replication origin) followed a similar protocol, except that the 5ʹ and 3ʹ ends of the sequence respectively extended 345 bp upstream and 375 bp downstream of the gene. To clone and express the genes encoding CetRNAnHis and CetRNAmHis, a ~ 800-bp DNA sequence with the CetRNAnHis or CetRNAmHis gene replacing the SctRNAnHis gene in the aforementioned DNA fragment was in vitro synthesized by a manufacturer (Bio-Basic, Markham ON, Canada). The synthesized DNA sequence was then cloned into pRS423 and pRS313 for expression in yeast. Thus, transcription of CetRNAHis and SctRNAnHis was driven by the same promoter.
Complementation assay for cytoplasmic activity on 5-FOA
Construction of a haploid HTS1 knockout strain (MATα, hts1::kanMX4, his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) was reported earlier [Citation7]. To examine the cytoplasmic HisRS activity of a gene of interest, a test plasmid carrying the target gene and a LEU2 marker was transformed into the haploid knockout strain and the ability of the resulting transformants to grow in the presence of 5-FOA (1 mg/ml) was tested [Citation36]. Starting with a cell density of 1.0 A600, cell cultures were 3-fold serially diluted, and 10-μl aliquots of each dilution were spotted onto the 5-FOA plates. Plates were then incubated at 30°C for 3 days. In the presence of 5-FOA, the transformants evicted the maintenance plasmid (which carried a WT HTS1 gene and a URA3 marker) and therefore failed to grow unless the test plasmid provided a functional cytoplasmic HisRS. To test whether a gene of interest can complement the genetic loss of yeast THG1, a similar protocol was followed using a haploid thg1− strain (MATα, thg1::kanMX4, his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0).
Complementation assay for mitochondrial activity on YPG
To examine the mitochondrial HisRS activity of a gene of interest, a test plasmid (which carried the target gene and a LEU2 marker) and a second maintenance plasmid (which carried a HIS3 marker and an initiator mutant of HTS1 that encodes only cytoplasmic HisRS activity) were co-transformed into the yeast hts1− strain. The first maintenance plasmid (which carried a URA3 marker) was evicted from the co-transformants in the presence of 5-FOA, while the second maintenance plasmid (which carried a HIS3 marker) was retained. Following 5-FOA selection, the survived co-transformants were spotted on yeast extract-peptone-glycerol (YPG) plates, which were then incubated at 30°C for 3 days. Because glycerol is a non-fermentable carbon source, a yeast cell cannot survive in this medium without functional mitochondria. Thus, the co-transformants could not survive on YPG plates unless the test plasmid provided a functional mitochondrial HisRS.
In vitro transcription of tRNAhis
Preparation of the WT and mutant tRNAHis transcripts followed a previously described protocol [Citation27]. Briefly, a synthetic DNA sequence containing both a T7 promoter and a gene encoding tRNAHis (GUG) was cloned into the SmaI site of pUC18 using a set of complementary primers. The transcription template was enriched by PCR amplification of the insert. The in vitro transcription reaction of tRNAHis (with a 5ʹ-GMP) was performed at 37°C for 3 h with 0.3 μM T7 RNA polymerase in 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine, 2 mM of each NTP, and 20 mM GMP. The tRNAHis transcript was purified by a 15% denaturing urea-polyacrylamide gel. After ethanol precipitation and vacuum-drying, the tRNA pellet was dissolved in 1× TE buffer (20 mM Tris-HCl (pH 8.0) and 1 mM EDTA) and then refolded by heating to 80°C and gradually cooled to room temperature after addition of 10 mM MgCl2. ~80% of in vitro-transcribed tRNAHis were active in aminoacylation.
Northern blotting
Total mitochondrial RNA was purified from the mitochondrial fractions of C. elegans by using TRIzol (Invitrogen) as previously described [Citation37]. A portion of the RNA sample was incubated at alkaline pH to hydrolyze aminoacyl-tRNA ester bonds. Two micrograms of total mitochondrial RNA were electrophoresed by an acid 10% polyacrylamide-7 M urea gel in 100 mM sodium acetate-EDTA running buffer (pH 5.2), electroblotted onto a positively charged nylon membrane (GeneScreen Plus, PerkinElmer), cross-linked by UV, and followed by probing with specific 32P-labeled primers: CetRNAnHis (5ʹ-TGCCTGCGGCTGGAATCG) and CetRNAmHis (5ʹ-AAGCTCTATATTCTCTTA), respectively [Citation38].
Aminoacylation assay
Kinetic parameters for histidylation of tRNA were determined by measuring the initial rate of charging over the first 4 min [Citation39]. Initial rates of aminoacylation were determined at 25°C in a buffer containing 50 mM HEPES (pH 7.5), 50 mM KCl, 15 mM MgCl2, 5 mM dithiothreitol, 10 mM ATP, 0.1 mg/ml bovine serum albumin, and 26.25 μM histidine (6.25 μM 14C-histidine; PerkinElmer, Waltham, MA, USA), with tRNAHis concentrations ranging 0.16 ~ 20 μM and HisRS concentrations ranging 50 ~ 1,000 nM. The specific activity of 14C-histidine used was 325 mCi/mmol. Reactions were quenched by spotting 10-μl aliquots of the reaction mixture onto Whatman filters (Maidstone, Kent, UK) presoaked in 5% trichloroacetic acid (TCA) and 2 mM histidine. The filters were washed three times for 15 min each in ice-cold 5% TCA before liquid scintillation counting. The chargeable tRNAHis concentration was determined by aminoacylation reactions to a saturated state using a high concentration of enzyme. The kinetic parameters were derived from Lineweaver-Burk plots. Determination of active protein concentrations by active site titration was as previously described [Citation40]. Data were obtained from three independent experiments and averaged. Error values represent 2× standard deviations.
Miscellaneous methods
Circular dichroism (CD) spectroscopy [Citation41], tRNAHis 5ʹ end sequencing [Citation17], and limited proteolysis [Citation42] followed previously described protocols.
Abbreviations
| aaRS | = | aminoacyl-tRNA synthetase |
| cDNA | = | complementary DNA |
| 5-FOA | = | 5-fluoroorotic acid |
| HisRS | = | histidyl-tRNA synthetase |
| MTS | = | mitochondrial targeting signal |
| ORF | = | open reading frame |
| PCR | = | polymerase chain reaction |
| tRNAnHis | = | nuclear-encoded cytoplasmic tRNAHis |
| tRNAmHis | = | mitochondrial-encoded mitochondrial tRNAHis |
| Thg1 | = | tRNAHis guanylyltransferase |
| WT | = | wild-type |
| YPG | = | yeast extract-peptone-glycerol |
Acknowledgments
We are grateful to Prof. Yi-Chun Wu (NTU) for providing C. elegans cosmids and Chieh-Min Chu for careful proofreading of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Carter CW Jr. Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu Rev Biochem. 1993;62:715–748.
- McClain WH. Transfer RNA identity. Faseb J. 1993;7:72–78.
- Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035.
- Ardell DH. Computational analysis of tRNA identity. FEBS Lett. 2010;584:325–333.
- Burbaum JJ, Schimmel P. Structural relationships and the classification of aminoacyl-tRNA synthetases. J Biol Chem. 1991;266:16965–16968.
- Giege R. The early history of tRNA recognition by aminoacyl-tRNA synthetases. J Biosci. 2006;31:477–488.
- Lee YH, Chang CP, Cheng YJ, et al. Evolutionary gain of highly divergent tRNA specificities by two isoforms of human histidyl-tRNA synthetase. Cell Mol Life Sci. 2017;74:2663–2677.
- Natsoulis G, Hilger F, Fink GR. The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell. 1986;46:235–243.
- Chatton B, Walter P, Ebel JP, et al. The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J Biol Chem. 1988;263:52–57.
- Chang KJ, Wang CC. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J Biol Chem. 2004;279:13778–13785.
- Tang HL, Yeh LS, Chen NK, et al. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J Biol Chem. 2004;279:49656–49663.
- Juhling F, Morl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162.
- Orellana O, Cooley L, Soll D. The additional guanylate at the 5ʹ terminus of Escherichia coli tRNAHis is the result of unusual processing by RNase P. Mol Cell Biol. 1986;6:525–529.
- Cooley L, Appel B, Soll D. Post-transcriptional nucleotide addition is responsible for the formation of the 5ʹ terminus of histidine tRNA. Proc Natl Acad Sci U S A. 1982;79:6475–6479.
- Gu W, Jackman JE, Lohan AJ, et al. tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5ʹ end of tRNAHis. Genes Dev. 2003;17:2889–2901.
- Abad MG, Rao BS, Jackman JE. Template-dependent 3ʹ-5ʹ nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc Natl Acad Sci U S A. 2010;107:674–679.
- Rao BS, Mohammad F, Gray MW, et al. Absence of a universal element for tRNAHis identity in Acanthamoeba castellanii. Nucleic Acids Res. 2013;41:1885–1894.
- Rao BS, Jackman JE. Life without post-transcriptional addition of G-1: two alternatives for tRNAHis identity in Eukarya. RNA. 2015;21:243–253.
- Wang C, Sobral BW, Williams KP. Loss of a universal tRNA feature. J Bacteriol. 2007;189:1954–1962.
- Nameki N, Asahara H, Shimizu M, et al. Identity elements of Saccharomyces cerevisiae tRNA(His). Nucleic Acids Res. 1995;23:389–394.
- Gu W, Hurto RL, Hopper AK, et al. Depletion of Saccharomyces cerevisiae tRNAHis guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m5C. Mol Cell Biol. 2005;25:8191–8201.
- Chiu WC, Chang CP, Wen WL, et al. Schizosaccharomyces pombe possesses two paralogous valyl-tRNA synthetase genes of mitochondrial origin. Mol Biol Evol. 2010;27:1415–1424.
- Preston MA, Phizicky EM. The requirement for the highly conserved G-1 residue of Saccharomyces cerevisiae tRNAHis can be circumvented by overexpression of tRNAHis and its synthetase. RNA. 2010;16:1068–1077.
- Sakurai M, Ohtsuki T, Watanabe K. Modification at position 9 with 1-methyladenosine is crucial for structure and function of nematode mitochondrial tRNAs lacking the entire T-arm. Nucleic Acids Res. 2005;33:1653–1661.
- Su D, Lieberman A, Lang BF, et al. An unusual tRNAThr derived from tRNAHis reassigns in yeast mitochondria the CUN codons to threonine. Nucleic Acids Res. 2011;39:4866–4874.
- Jackman JE, Gott JM, Gray MW. Doing it in reverse: 3ʹ-to-5ʹ polymerization by the Thg1 superfamily. RNA. 2012;18:886–899.
- Himeno H, Hasegawa T, Ueda T, et al. Role of the extra G-C pair at the end of the acceptor stem of tRNA(His) in aminoacylation. Nucleic Acids Res. 1989;17:7855–7863.
- Choi H, Gabriel K, Schneider J, et al. Recognition of acceptor-stem structure of tRNA(Asp) by Escherichia coli aspartyl-tRNA synthetase. RNA. 2003;9:386–393.
- Brennan T, Sundaralingam M. Structlre of transfer RNA molecules containing the long variable loop. Nucleic Acids Res. 1976;3:3235–3250.
- Rudinger J, Felden B, Florentz C, et al. Strategy for RNA recognition by yeast histidyl-tRNA synthetase. Bioorg Med Chem. 1997;5:1001–1009.
- Heinemann IU, Nakamura A, O’Donoghue P, et al. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 2012;40:333–344.
- Placido A, Sieber F, Gobert A, et al. Plant mitochondria use two pathways for the biogenesis of tRNAHis. Nucleic Acids Res. 2010;38:7711–7717.
- Long Y, Abad MG, Olson ED, et al. Identification of distinct biological functions for four 3ʹ-5ʹ RNA polymerases. Nucleic Acids Res. 2016;44:8395–8406.
- Chang CP, Lin G, Chen SJ, et al. Promoting the formation of an active synthetase/tRNA complex by a nonspecific tRNA-binding domain. J Biol Chem. 2008;283:30699–30706.
- Chang KJ, Lin G, Men LC, et al. Redundancy of non-AUG initiators. A clever mechanism to enhance the efficiency of translation in yeast. J Biol Chem. 2006;281:7775–7783.
- Boeke JD, Trueheart J, Natsoulis G, et al. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–175.
- King MP, Attardi G. Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J Biol Chem. 1993;268:10228–10237.
- Janssen BD, Diner EJ, Hayes CS. Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Methods Mol Biol. 2012;905:291–309.
- Francklyn C, Schimmel P (1990) Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc Nat Acad Sci USA, 87, 8655–8659.
- Fersht AR, Ashford JS, Bruton CJ, et al. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry. 1975;14:1–4.
- Chang CY, Chien CI, Chang CP, et al. A WHEP domain regulates the dynamic structure and activity of Caenorhabditis elegans Glycyl-tRNA synthetase. J Biol Chem. 2016;291:16567–16575.
- Ladror US, Egan DA, Snyder SW, et al. Domain structure analysis of elongation factor-3 from Saccharomyces cerevisiae by limited proteolysis and differential scanning calorimetry. Protein Sci. 1998;7:2595–2601.