
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Coxsackievirus B3 is an enterovirus, with positive-sense single-stranded RNA genome containing ‘Internal Ribosome Entry Site’ (IRES) in the 5ʹUTR. Once sufficient viral proteins are synthesized in the cell from the input RNA, viral template switches from translation to replication to synthesize negative-strand RNA. Inhibition of translation is a key step in regulating this switch as the positive-strand RNA template should be free of ribosomes to enable polymerase movement. In this study, we show how a host protein hnRNP C1/C2 inhibits viral RNA translation. hnRNP C1/C2 interacts with stem-loop V in the IRES and displaces poly-pyrimidine tract binding protein, a positive regulator of translation. We further demonstrate that hnRNP C1/C2 induces translation to replication switch, independently from the already known role of the ternary complex (PCBP2-3CD-cloverleaf RNA). These results suggest a novel function of hnRNP C1/C2 in template switching of positive-strand from translation to replication by a new mechanism. Using mathematical modelling, we show that the differential affinity of hnRNP C1/C2 for positive and negative-strand RNAs guides the final ± RNA ratio, providing first insight in the regulation of the positive to negative-strand RNA ratio in enteroviruses.
Introduction
Enteroviruses have positive sense single-stranded RNA genome (ssRNA) and their life cycle begins upon the entry of ssRNA genome into the cytoplasm. Viral RNA undergoes IRES-mediated translation in the cytoplasm and viral proteins are synthesized [Citation1]. The genomic RNA that undergoes translation is also utilized as a template for replication to produce negative-strand RNA. As both the processes cannot occur simultaneously on the same template, the genomic RNA undergoes a switch from translation to replication [Citation2]. The newly synthesized negative-strand RNA is now used as a template to synthesize more positive-strand RNA. Positive-strand synthesis is much more efficient than negative-strand synthesis which leads to far greater amounts of positive-strand RNAs as compared to negative-strand RNA. In fact, the average ratio of positive to negative-strand RNA is 20:1 [Citation3]. This is partly because positive-strand RNA has two functions, translation and replication while negative-strand RNA undergoes only replication. The RNP complex assembled at the UTRs of the genomic RNAs could be the deciding factor in determining the fate of the template for translation or replication. This was observed in the case of Hepatitis C virus (HCV) where, host RNA binding proteins, HuR and La, interact with 3ʹUTR causing the recruitment of the NS5b polymerase leading to template switching from translation to replication [Citation4].
Molecular mechanisms that regulate the switching of positive-strand RNA from translation to replication are poorly understood in Enteroviruses. For this process to be successful two conditions must be satisfied; 1) assembly of replication complex to initiate polymerase movement from 3ʹ end and 2) repression of translation to make the template free of translating ribosomes. Earlier report suggests that recruitment of viral protein 3CD and host protein PCBP-2 at the cloverleaf RNA forms a ternary complex, which induces translation to replication switch in poliovirus [Citation2]. Disrupting the ternary complex by mutation in cloverleaf RNA that prevents 3CD interaction leads to complete abrogation in replication [Citation2]. Earlier reports that have studied the mechanism of translation to replication switch in enteroviruses have been restricted to cleavage of IRES trans acting factors (ITAFs), that positively regulate viral RNA translation. For example, host protein PCBP-2 is also known to interact with the IRES and positively regulate IRES-mediated translation [Citation5]. It has been shown that cleavage of PCBP-2 by viral protease 3C abrogates PCBP-2-IRES interaction and favours interaction of PCBP-2 with cloverleaf RNA causing translation to replication switch [Citation6]. Similarly, host factor PTB, that positively regulates IRES-mediated translation in enteroviruses, is also known to be cleaved by 3C protease and this could also contribute in translation to replication switch [Citation7,Citation8]. Cleavage of IRES ITAFs by viral proteases is an interesting strategy to control the translation to replication switch. However, the cleavage of these proteins begins only at 4-h post-infection and major proportion of the protein remains intact during virus life cycle [Citation6,Citation7]. Also, negative strands have been detected as earlier as 2-h post-infection, indicating that template switching to replication occurs at much earlier time points before the cleavage of ITAFs [Citation9]. This suggests that an alternative mechanism of translation to replication switch could exist during viral life cycle.
Host protein hnRNP C1/C2 is a well-characterized protein with respect to its role in poliovirus life cycle. It is known that hnRNP C1/C2 interacts with the genomic ends of negative-strand RNA and encourages positive-strand synthesis [Citation9,Citation10]. Knockdown of hnRNP C1/C2 in cells leads to delayed kinetics of poliovirus replication [Citation9,Citation11]. In the same study, it was also mentioned that hnRNP C1/C2 interacted with the 5ʹUTR of poliovirus positive-strand RNA but with a lower affinity, but the consequence of this interaction was not studied [Citation10]. Here we report that hnRNP C1/C2 interaction with 5ʹUTR of positive-strand RNA leads to inhibition of viral RNA translation. Affinity of hnRNP C1/C2 for positive-strand RNA was weaker than negative-strand RNA. The binding site of hnRNP C1/C2 was mapped to the basal region of stem-loop V where PTB is known to interact. In our experiments, hnRNP C1/C2 displaced PTB at IRES, providing the mechanism of translation repression caused by hnRNP C1/C2. We also find that hnRNP C1/C2 could induce translation to replication switch and this was independent of the assembly of the ternary complex. These results suggest a novel role of hnRNP C1/C2 in operating the translation to replication switch, before the cleavage of ITAFs. Incorporating the interactions of hnRNP C1/C2 and PTB with viral RNAs, a mathematical model of intracellular viral replication and translation was built. Simulations show that the relative affinity of hnRNP C1/C2 for positive and negative-strand RNAs guides the final ± RNA ratio. The differential interaction of hnRNP C1/C2 with viral RNAs further buffers the ± RNA ratio from perturbations in levels of host factors.
Results
hnRNP C1/C2 interacts with 5ʹUTR in positive-strand RNA
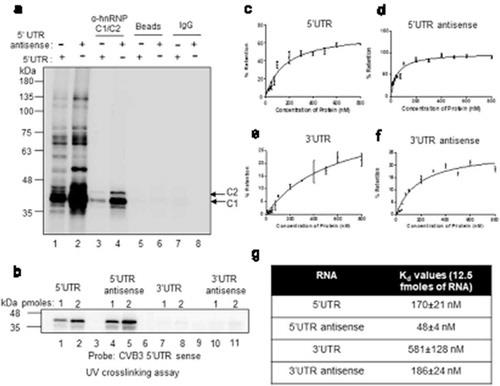
hnRNP C1/C2 is known to interact with both the genomic ends of negative-strand RNA and also with 5ʹUTR of positive-strand RNA of poliovirus [Citation9,Citation10]. Here we have investigated the binding of hnRNP C1/C2 with CVB3 positive negative-strand RNAs. CVB3 5ʹUTR and 3ʹUTR RNAs were used to study positive-strand RNA interaction and antisense corresponding to the 5ʹUTR and 3ʹUTR RNAs were used to study negative-strand RNA interaction. First, UV-crosslinking assay was carried out with radiolabelled probes corresponding to the 5ʹUTR and antisense RNA of 5ʹUTR using S10 extracts prepared from HeLa S3 cells, followed by immuno-precipitation by anti-hnRNP C1/C2 antibody. Non-specific antibody was used as control. Consistent with previous reports, interaction of hnRNP C1/C2 was observed with both the probes ()). Recombinant hnRNP C1 protein was also able to interact with 5ʹUTR of positive-strand RNA and 5ʹUTR antisense RNA as observed by direct UV-crosslinking experiment ()). To further confirm these results, competition UV-crosslinking experiments using recombinant hnRNP C1 were performed either with radiolabelled 5ʹUTR sense (Figure S1A) or radiolabelled 5ʹUTR antisense RNAs (Figure S1B). Various competitor RNAs were used, which include, un-labelled 5ʹUTR RNA, 5ʹUTR antisense RNA, 3ʹUTR RNA and 3ʹUTR antisense RNA (Figure S1A and S1B). These experiments also suggested the interaction of hnRNP C1 with 5ʹUTR in positive strand and both genomic ends of negative-strand RNAs but not with 3ʹUTR of positive strand. To measure the affinity of hnRNP C1 for these RNAs, filter binding experiments were performed with all above-mentioned RNAs (–)) and the results are summarized in ). hnRNP C1 displayed strongest interaction with the 5ʹUTR antisense followed by 5ʹUTR sense and 3ʹUTR antisense. A very weak or negligible interaction of hnRNP C1 protein with 3ʹUTR sense was observed with very high kD value ()). Interaction of hnRNP C1/C2 with both positive and negative-strand RNAs during intracellular viral life cycle was confirmed by performing RNA immuno-precipitation using hnRNP C1/C2 antibody (Figure S1C). These results show that hnRNP C1/C2 protein interacts with both genomic ends of negative-strand RNA but interacts only with 5ʹUTR of positive-strand RNA.
Figure 1. Host protein hnRNP C1/C2 interacts with both positive and negative-strand RNA. (a) UV crosslinking carried out with cytoplasmic S10 extracts and radiolabelled 5ʹUTR (lane 1) or radiolabelled antisense 5ʹUTR (lane 2) followed by immuno-precipitation by using hnRNP C1/C2 antibody (lane 3 and 4) or IgG antibody (lane 7 and 8) as control. Pulldown carried out without antibody is indicated as beads alone (lane 5 and 6). For each reaction, 30 µg of S10 extract was used. (b) Direct UV-crosslinking assay performed by using recombinant hnRNP C1 protein and different riboprobes, 5ʹUTR sense (lane 1 and 2), 5ʹUTR antisense (lane 3 and 4), 3ʹUTR sense (lanes 5and 6) and 3ʹUTR antisense (lanes 6 and 7). For each RNAs, 1 hundred thousand counts of radiolabelled RNA probe were used for UV-crosslinking along with indicated amounts of protein. (c–f) Filter binding assays carried out with radiolabelled 5ʹUTR sense (c), 5ʹUTR antisense (d), 3ʹUTR sense (e) and 3ʹUTR antisense (f) RNAs. In all the case, 12500 counts per minute (CPM) of RNA was used per reaction and RNA-protein interactions were carried out at indicated amounts (x-axis) of recombinant hnRNP C1 protein. The dissociation constant (Kd) values are represented in table (g). Error bars indicated standard deviation from three independent experiments.
hnRNP C1/C2 acts as a repressor of viral RNA translation
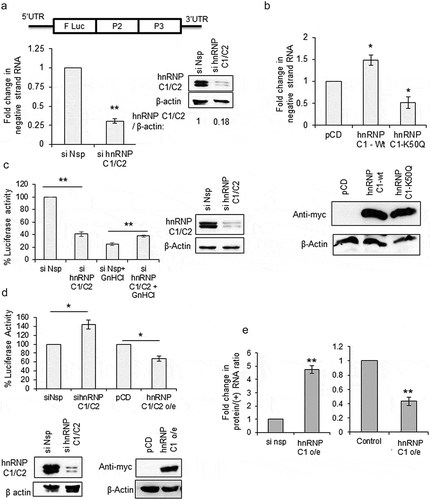
Earlier reports on poliovirus suggest that, hnRNP C1/C2 protein stimulates positive-strand synthesis step of viral RNA replication by interacting with the negative-strand RNA [Citation9,Citation10]. However, the consequence of hnRNP C1/C2 positive-strand RNA interaction was not known. We have utilized CVB3 replicon RNA to study the consequence of hnRNP C1/C2-5ʹUTR interaction on viral RNA translation and replication. In the replicon RNA the structural proteins P1 is replaced by Firefly Luciferase (F luc) gene and the efficiency of viral life cycle can be measured quantitatively by measuring F luc activity and also by estimating negative-strand RNA by RT-qPCR ()) [Citation12]. Initially, the effect of knockdown of hnRNP C1/C2 on CVB3 replication was studied. CVB3 replicon RNA was transfected in cells treated with either si-hnRNP C1/C2 or si-Nsp. At 8-h post-transfection, negative-strand RNA levels were measured by RT-qPCR. Significant reduction in negative-strand RNA levels was observed in knockdown cells as compared to control cells ()). Similarly, upon overexpression of wild-type hnRNP C1 protein, replication was found to be increased ()). However, overexpression of mutant hnRNP C1-K50Q protein, known to possess compromised RNA binding activity [Citation9,Citation10,Citation13], acted as dominant negative suppressing the replication ()). These results are consistent with earlier results reported using poliovirus, where silencing of hnRNP C1/C2 led to reduction in virus RNA as well as plaque forming units (PFUs) [Citation9]. Next, the same experiment was repeated along with 4 mM GnHCl, an inhibitor of enterovirus replication, and the luciferase activity was measured. As expected, knockdown of hnRNP C1/C2 led to reduced luciferase activity, however the luciferase activity was increased upon partial silencing of hnRNP C1/C2 in presence of GnHCl ()). In the presence of GnHCl, luciferase activity is the measure of only the translation as replication is inhibited. This suggests that although hnRNP C1/C2 is a positive regulator for replication of virus, it is a repressor of translation. The same experiment was repeated without GnHCl and cells were harvested at 1.5-h post transfection of replicon RNA, as by this time only translation occurs in cells. Knockdown of hnRNP C1/C2 led to increased luciferase activity and overexpression of hnRNP C1 led to reduced luciferase activity suggesting repressive role of hnRNP C1/C2 in viral RNA translation ()). Knockdown of hnRNP C1/C2 did not affect the cap-dependent translation (Figure S2B). In another experiment, the ratio of protein (as measured by luciferase activity) and positive-strand RNA (as determined by RT-qPCR) was plotted to uncouple translation and replication. This ratio indicates the amount of protein produced per positive-strand RNA template. Upon partial silencing of hnRNP C1/C2 about five-fold increase in viral RNA translation was observed and overexpression of hnRNP C1 protein reduced viral protein synthesis by 50% ()), suggesting hnRNP C1/C2 as inhibitor of viral RNA translation ()). Supplementation of hnRNP C1 recombinant protein during in vitro translation of replicon RNA carried out in rabbit reticulocyte lysate supplemented with HeLa S10 extract, a method previously described in the literature to study IRES-mediated translation of enteroviruses [Citation14–Citation17], also resulted in reduction in translation (Figure S2A). Recombinant PTB was used as positive control. These results suggest that hnRNP C1/C2 has opposite effects on viral RNA replication and translation. It stimulates viral RNA replication while it represses viral RNA translation.
Figure 2. hnRNP C1/C2 is inhibitor of viral RNA translation. (a) Effect of partial knockdown of hnRNP C1/C2 on CVB3 replication. Schematic of CVB3 sub-genomic Replicon RNA used in the current study. Structural proteins are replaced by Firefly Luciferase gene. Graph shows the negative-strand RNA levels as determined by qPCR. Western blot indicates the knockdown of hnRNP C1/C2 upon siRNA treatment. (b) Effect of over-expression wild type and RNA binding mutant hnRNP C1 protein on CVB3 RNA replication. Negative-strand RNA level as measured by qPCR is indicated in the graph. Western blot indicates the overexpression of myc-tagged hnRNP C1 (c) Effect of partial knockdown of hnRNP C1/C2 on CVB3 life cycle in presence or absence of replication inhibitor GnHCl. Cells were lysed after 8-h post-transfection of CVB3 replicon RNA and processed for measuring luciferase activity. Fold change in luciferase activity is indicated in the graph. Western blot indicates the knockdown of hnRNP C1/C2 upon siRNA treatment. (d) Effect of silencing or overexpression of hnRNP C1/C2 on CVB3 RNA translation. Cells were harvested at 1.5-h post-transfection of CVB3 replicon RNA and processed for measuring luciferase activity. Fold change in luciferase activity upon silencing or overexpression of hnRNP C1/C2 are indicated. Western blot indicates the knockdown of hnRNP C1/C2 upon siRNA treatment and the overexpression of myc-tagged hnRNP C1. (e) To uncouple replication and translation without using inhibitor, translation per positive-strand RNA template was derived by calculating protein (luciferase activity)/positive strand RNA (as estimated by qPCR) ratios. Fold change in protein/(+) strand RNA is indicated. * indicates P < 0.05 and ** indicates P <0.01 throughout. Two-tailed Students t-test was used for statistical analysis.
hnRNP C1/C2 displaces PTB, an ITAF, at CVB3 IRES
To determine the binding sites of hnRNP C1/C2 on CVB3 5ʹUTR, UV-crosslinking assay was performed with radiolabelled CVB3 5ʹUTR and recombinant hnRNP C1 protein in the presence of individual stem-loop RNAs derived from CVB3 5ʹUTR ()). Strongest competition was observed by stem-loop V RNA (), lane numbers 8 and 9) indicating the presence of hnRNP C1/C2 binding site. hnRNP C1/C2 binding sequence is known from the SELEX experiments [Citation13,Citation18] and a similar sequence was found to be present in the stem-loop V ()). Stem-loop V in CVB3 IRES is followed by poly-pyrimidine tract where 40S subunit of ribosome is proposed to land [Citation19]. Also, C-terminal eIF4G1 and an important ITAF, PTB interacts with this region and promotes viral RNA translation [Citation20]. Interestingly, nucleotides interacting with the PTB on stem-loop V were found to be overlapping with the predicted hnRNP C1/C2 binding site [Citation20] ()). To study whether binding of hnRNP C1/C2 with stem-loop V interferes with the interaction of PTB with 5ʹUTR, UV-crosslinking experiments were performed with S10 extracts supplemented with increasing concentration of recombinant hnRNP C1. A band corresponding to molecular weight of 63 kDa, previously characterized as PTB in our laboratory [Citation8], was found to be competed out with the addition of increasing concentration of recombinant hnRNP C1 protein ()). The intensities of several other bands (indicated by * in )) remained unchanged, indicating a specific competition between proteins. In a reverse experiment, by increasing the PTB concentration, hnRNP C1/C2 protein was observed to be competed out ()). Competition UV-crosslinking experiments using recombinant hnRNP C1 and PTB proteins confirmed the competitive interaction between these two proteins on 5ʹUTR (Figure S3A). To study whether these competitive interactions occur in cells during CVB3 life cycle, RNA immuno-precipitation (RNA IP) experiments were performed from CVB3 replicon harbouring cells. hnRNP C1 protein was overexpressed in cells followed by immuno-precipitation of PTB and the PTB associated positive-strand RNA was estimated by qPCR. Positive-strand RNA association with PTB was significantly reduced, upon overexpression of hnRNP C1 ()). Similarly, upon overexpression of PTB, association of positive-strand RNA with hnRNP C1/C2 was significantly reduced ()). Western blots corresponding to the IP experiments (, )) indicating the overexpression of PTB and hnRNP C1 are presented in figure S3C. Affinity of PTB with stem-loop V was measured by filter binding experiments and it was observed that PTB interacts with much stronger affinity (kD = 9.76 nM of PTB to stem-loop V as compared to kD = 167 nM of hnRNP C1 to full 5ʹUTR) as compared to hnRNP C1 (Figure S3B). These results suggest that hnRNP C1/C2 binds to stem-loop V and competes with PTB, which is a stimulator of viral RNA translation, providing the mechanism of translation repression caused by hnRNP C1/C2. Of note, hnRNP C1/C2 binding sequence appears to be conserved across different viruses in picronaviridae family containing type I IRES (Figure S3D).
Figure 3. hnRNP C1/C2 displaces PTB at 5ʹUTR. (a) UV crosslinking carried out with recombinant hnRNP C1 protein and radiolabelled 5ʹUTR RNA in the presence of unlabelled RNAs corresponding to the different stem loops of 5ʹUTR. (B) Schematic of stem-loop V and potential binding site of hnRNP C1 on stem-loop V is indicated with respect to the known binding site from SELEX experiments [Citation17]. Grey-shaded region indicated PTB binding site as per earlier report [Citation19]. (c) UV-crosslinking assay with radiolabelled 5ʹUTR and cytoplasmic S10 extracts in the presence of increasing concentration of rhnRNP C1. + = 1 pmole, ++ = 2 pmole, +++ = 3 pmole of protein. Densitometry values representing the PTB band intensity are indicated under each lane. * mark beside the gel image indicates the proteins that did not change upon increasing concentration of hnRNP C1. (d) UV-crosslinking assay with radiolabelled 5ʹUTR and cytoplasmic S10 extracts in the presence of increasing concentration of rPTB. Densitometry values representing the hnRNP C1 band intensity are indicated under each lane. + = 1 pmole, ++ = 2 pmole, +++ = 3 pmole of protein. (e) RNA immuno-precipitation experiment carried out with anti-PTB antibody upon overexpression of hnRNP C1 or vector control (pCDNA3). Cells were transfected with plasmid expressing hnRNP C1 or pCDNA3 vector followed by transfection of CVB3 replicon RNA. This was followed by processing cells for RNA immuno-precipitation with anti-PTB antibody at 8-h post-CVB3 replicon RNA transfection. Fold change in positive-strand RNA associated with PTB upon hnRNP C1 overexpression as compared to pCDNA3 vector is indicated. (f) RNA immuno-precipitation experiment carried out with anti-hnRNP C1/C2 antibody upon overexpression of PTB. Cells were transfected with plasmid expressing PTB or pCDNA3 vector followed by transfection of CVB3 replicon RNA. This was followed by processing cells for RNA immuno-precipitation with anti-hnRNP C1/C2 antibody at 8-h post-CVB3 replicon RNA transfection. Fold change in positive-strand RNA associated with hnRNP C1/C2 upon PTB overexpression as compared to pCDNA3 vector is indicated. ** indicates P <0.01. Two-tailed Students t-test was used for statistical analysis.
![Figure 3. hnRNP C1/C2 displaces PTB at 5ʹUTR. (a) UV crosslinking carried out with recombinant hnRNP C1 protein and radiolabelled 5ʹUTR RNA in the presence of unlabelled RNAs corresponding to the different stem loops of 5ʹUTR. (B) Schematic of stem-loop V and potential binding site of hnRNP C1 on stem-loop V is indicated with respect to the known binding site from SELEX experiments [Citation17]. Grey-shaded region indicated PTB binding site as per earlier report [Citation19]. (c) UV-crosslinking assay with radiolabelled 5ʹUTR and cytoplasmic S10 extracts in the presence of increasing concentration of rhnRNP C1. + = 1 pmole, ++ = 2 pmole, +++ = 3 pmole of protein. Densitometry values representing the PTB band intensity are indicated under each lane. * mark beside the gel image indicates the proteins that did not change upon increasing concentration of hnRNP C1. (d) UV-crosslinking assay with radiolabelled 5ʹUTR and cytoplasmic S10 extracts in the presence of increasing concentration of rPTB. Densitometry values representing the hnRNP C1 band intensity are indicated under each lane. + = 1 pmole, ++ = 2 pmole, +++ = 3 pmole of protein. (e) RNA immuno-precipitation experiment carried out with anti-PTB antibody upon overexpression of hnRNP C1 or vector control (pCDNA3). Cells were transfected with plasmid expressing hnRNP C1 or pCDNA3 vector followed by transfection of CVB3 replicon RNA. This was followed by processing cells for RNA immuno-precipitation with anti-PTB antibody at 8-h post-CVB3 replicon RNA transfection. Fold change in positive-strand RNA associated with PTB upon hnRNP C1 overexpression as compared to pCDNA3 vector is indicated. (f) RNA immuno-precipitation experiment carried out with anti-hnRNP C1/C2 antibody upon overexpression of PTB. Cells were transfected with plasmid expressing PTB or pCDNA3 vector followed by transfection of CVB3 replicon RNA. This was followed by processing cells for RNA immuno-precipitation with anti-hnRNP C1/C2 antibody at 8-h post-CVB3 replicon RNA transfection. Fold change in positive-strand RNA associated with hnRNP C1/C2 upon PTB overexpression as compared to pCDNA3 vector is indicated. ** indicates P <0.01. Two-tailed Students t-test was used for statistical analysis.](/cms/asset/1fed1d7e-258b-499d-8f76-1b5a043a34e8/krnb_a_1629208_f0003_b.gif)
hnRNP C1/C2 regulates translation to replication switch in CVB3
Our results indicate that hnRNPC1/C2 is a repressor of CVB3 RNA translation but it is a positive regulator for replication of RNA. This motivated us to study whether hnRNP C1/C2 is involved in regulating translation to replication switch in CVB3, where both these events of translation repression and replication initiation must co-ordinate. To study the role of hnRNP C1/C2 in regulating translation to replication switch, puromycin drug (known to release the ribosomes from the template) was used to induce translation to replication switch. We hypothesized that hnRNP C1/C2 overexpression might induce translation to replication switch similar to puromycin treatment. Cycloheximide drug treatment, known to freeze ribosomes on the template RNA, was used as control. Puromycin/cycloheximide treatment was given at 6-h post-transfection of replicon RNA and cells were harvested for analysis of translation and replication at 2 h and 4-h post puromycin/cycloheximide treatment ()). Reduction in translation was observed after 2 h and 4-h post puromycin treatment (Figure S4A). Increase in negative-strand level was apparent post puromycin treatment ()), indicating successful induction of translation to replication switch. Interestingly, overexpression of hnRNP C1 protein induced translation to replication switch, similar to the puromycin treatment ()). As expected, cycloheximide treatment did not result in any stimulation in replication ()). A representative western blot depicting the hnRNP C1 overexpression is presented in figure S4B. These results suggest that hnRNP C1/C2 regulate translation to replication switch by causing translation repression.
Figure 4. hnRNP C1/C2 regulates translation to replication switch. (a) Schematic of experimental design to determine the role of hnRNP C1/C2 in regulating translation to replication switch. 24 hprior to transfection of CVB3 replicon RNA, hnRNP C1 overexpression construct was transfected in cells and pCDNA3 vector was transfected as control. Six-hourpost-transfection of CVB3 replicon RNA, puromycin treatment was started and cells were harvest at 2 h and 4-h post puromycin treatment for protein and RNA analysis. (b and c) Negative-strand RNA levels are indicated by line graph. At each time point, pCDNA3 vector alone is used as control and fold change in negative-strand RNA levels is indicated. Concentration of puromycin drug was 100 µg/mL and concentration of cycloheximide drug was 40 µg/mL. Error bars represent standard deviation from three different experiments. (d) Schematic of cloverleaf RNA present in 5ʹUTR. Both wild type and LD.73 mutant, which has the insertion of 4 nucleotides at 72nd position, cloverleaf RNAs are shown. Mutation was incorporated in the CVB3 replicon RNA. (e and f) Effect of partial silencing or overexpression of hnRNP C1/C2 on translation of wild type and LD.73 mutant replicon RNA. Twenty-four hour post-transfection of siRNAs or overexpression plasmid, wild type or LD.73 replicon RNAs were transfected into cells and harvested 1.5-h post-transfection, before the replication begins. Western blots indicate the hnRNP C1/C2 levels or the overexpressed myc-tagged hnRNP C1 protein as compared to β-actin levels. * indicates P< 0.05 and ** indicates P <0.01. Two-tailed student’s t-test was used for statistical analysis throughout.
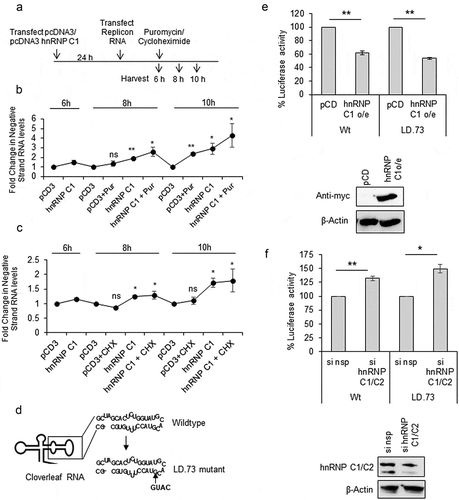
Interaction of viral protein 3CD with cloverleaf RNA along with host protein PCBP2 forms a ternary complex that inhibits viral RNA translation and this is a key step in initiating negative-strand synthesis [Citation2]. Interestingly, 3CD protein is also known to interact with hnRNP C1/C2 [Citation10]. Hence, we asked whether recruitment of hnRNP C1/C2 at IRES is dependent on the assembly of ternary complex at the cloverleaf RNA. For this purpose, 3CD binding site was mutated by inserting 4 nucleotides in ‘d’ arm of cloverleaf region in CVB3 replicon RNA ()). This mutation is already well characterized in the literature (called LD.73 mutant) and was shown to be defective in replication but more efficient in translation as compared to wild-type [Citation2]. In CVB3 replicon RNA system also, we found that the translation was enhanced in the LD.73 mutant as compared to wildtype (Figure S5A). LD.73 mutant was defective in replication as treatment of GnHCl, a replication inhibitor, showed negligible change in luciferase activity as compared to untreated cells, while drastic reduction in luciferase activity was observed in wild-type RNA (Figure S5B). Next, the effect of silencing or over-expression of hnRNP C1/C2 on the efficiency of translation in wild type and LD.73 mutant replicon RNAs was investigated. Wild type and LD.73 mutant CVB3 replicon RNAs were transfected in HeLa cells and at 1.5-h post-transfection, cells were processed for measuring luciferase activity. 1.5-h time point was chosen as per method described earlier [Citation2], as by this time replication does not occur and luciferase activity reflects translation only. As observed earlier, overexpression of hnRNP C1 resulted in suppression of translation and partial silencing of hnRNP C1/C2 resulted in enhanced translation of wild-type RNA (, )). Similar effect was also observed in LD.73 mutant suggesting that translation regulation of hnRNP C1/C2 is independent of 3CD binding to cloverleaf RNA (, )).
hnRNP C1/C2 mediated regulation of positive-strand to negative-strand RNA ratio
In an earlier report, positive and negative-strand RNAs were estimated in poliovirus-infected single cells and the average of the positive to negative-strand RNA ratio (±) was determined to be 20 ± 3:1 across 150 cells [Citation3]. This data suggests that there might be a mechanism by which ± RNA ratio is regulated around 20:1. We hypothesized that regulation of replication of both positive and negative-strand RNA by hnRNP C1/C2 and its differential affinity to both the RNAs might have a role in regulating ± ratio. Interaction of hnRNP C1/C2 with positive-strand RNA leads to synthesis of negative-strand RNA and hence leads to the availability of high-affinity binding sites for hnRNP C1/C2 on the negative strand RNA. This would lead to preferential interaction of hnRNP C1/C2 with negative-strand RNA and initiate positive-strand synthesis. Since positive-strand RNA synthesis is more efficient, number of positive-strand RNAs increase in the cell and this leads to synthesis of larger number of low-affinity binding sites for hnRNP C1/C2. Now, a large number of positive-strand RNAs lead to preferential interaction of hnRNP C1/C2 with positive-strand RNAs and synthesis of negative-strand RNA, and so on ()). Such a system could act as a buffer to prevent the ± RNA ratio to reach extreme ends and control it within an ideal range. To test this hypothesis, we built a mathematical model of intracellular viral replication and translation, based on the viral RNA – host protein interactions identified experimentally (see supplementary methods). Based on these interactions, ordinary differential equations describing the time-evolution of viral RNA and protein concentrations were obtained. Model predictions were able to describe our experimental observations as well as the trends in the intracellular viral life cycle reported previously (Figure S6 and Figure S7A).
Figure 5. Regulation of positive to negative-strand RNA ratio. (a) Schematic of bidirectional regulation of RNA replication by hnRNP C1/C2. 1) Interaction of hnRNP C1/C2 leads to negative-strand synthesis. 2) Negative strand has high-affinity binding sites for hnRNP C1/C2 as (indicated by +++) which leads to preferential interaction of hnRNP C1/C2 with negative-strand RNA. 3) hnRNP C1/C2-negative-strand RNA complex leads to synthesis of positive strands. 4) Since positive-strand synthesis is more efficient, they are more in number and this leads to hnRNP C1/C2 positive-strand RNA interaction. 5) More negative-strand RNA is synthesized as a consequence of step 4. (b) Model predictions (Supplementary Methods) of ± RNA ratios during the entire intracellular CVB3 life cycle. Orange, blue and black lines represent different kD (+)/kD (-) ratios. kD (+)/kD (-) of 4:1 represents the physiological state. (c) ± RNA ratios at various affinities of PTB to positive-strand RNA. Orange line represents the physiological affinity of PTB of 10 nM. (d) ± RNA ratios at different concentrations of PTB. E) ± RNA ratios at different concentrations of hnRNP C1/C2.
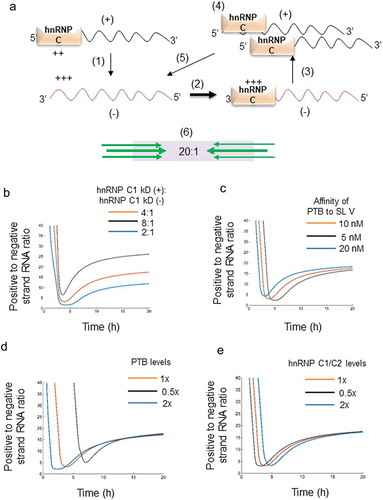
Using this model, we tested the consequence of changing the relative affinity of hnRNP C1/C2 to positive and negative-strand RNA. In our filter binding experiments ()), we had observed that the affinity of hnRNP C1/C2 is around 4 times stronger for negative-strand RNA (kD (-)) than for positive-strand RNA (kD (+)). We simulated the model at this physiological ratio of affinities and at hypothetical affinity ratios of 8:1 and 2:1 [(kD(+)/kD(-)]. We observed that changes in the affinity ratio resulted in large changes in the final positive to negative strand RNA ratio ()). At the same time, changing the affinity of PTB to the positive strand altered the initial dynamics (t < 4 h) but not the final (t = 8 h) ratio between positive and negative-strand RNA ()). The final ± RNA ratio was unchanged even upon varying the initial concentrations of PTB and hnRNP C1/C2 (, )). In our model, replication saturates toward the end of the life cycle (Figure S7A) consistent with earlier experimental reports [Citation9,Citation21]. This saturation, in our model, arises when viral RNA levels rise and all hnRNP C1/C2 molecules become bound to the viral RNAs (Figure S7B). The distribution of hnRNP C1/C2 between positive and negative-strand viral RNAs is dependent on its relative affinity to the positive and negative strands (Figure S7B). The replication rates of the positive and negative strands and hence their relative prevalence are thus determined by the ratio of the affinity of hnRNP C1/C2 to the two strands. To test the robustness of the model, we have performed formal sensitivity analysis by obtaining Partial Rank Correlation Coefficients (PRCC) for various parameters (Figure S7C), to ensure that our results are not artefacts of particular values chosen but are a basic property of the system (see supplementary methods).
The independence of the final ± RNA ratio from factors such as the affinity of PTB for viral RNA, and the abundance of PTB and hnRNP C1/C2 proteins is made explicit by analytical calculations from the mathematical model. We show that the final ratio is
where and
are the concentrations of positive and negative-strand viral RNA, is respectively,
and
are rate constants of the production of positive and negative strands,
and
characterize the binding of the 3D polymerase to the two strands, and
and
are the affinities of hnRNP C1/C2 for the two strands, respectively (See supplementary information). The ratio between
and
is approximately 4:1 (). If the protein 3D has no preference for positive or negative strands, it follows that the ratio of replication rates,
, which is unknown, must be approximately 100:1 for the final positive and negative-strand viral RNA ratio to be is the observed 20:1 [Citation3].
Disrupting hnRNP C1/C2 interaction with CVB3 RNAs in strand-specific manner
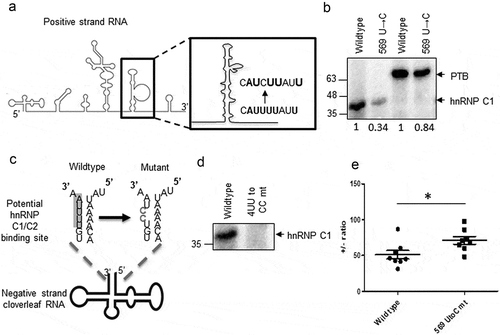
To validate the predictions from the mathematical model, mutations were generated to disrupt hnRNP C1/C2 interaction in a strand-specific manner and ± RNA ratio was estimated. To disrupt hnRNP C1/C2-positive-strand RNA interaction, U to C mutation was generated at 569 position in CVB3 IRES (schematic in )). UV-crosslinking experiment was performed using recombinant hnRNP C1 protein with radiolabelled wild type and mutant CVB3 5ʹUTR. Interaction of hnRNP C1 was found to be reduced in the 569 U →C mutant (), lane 1 and 2). PTB is also known to interact with the same region and this mutation led to marginal reduction in PTB interaction as well (), lane 3 and 4). This result confirms the binding site of hnRNP C1 to basal region of stem-loop V. To disrupt the hnRNP C1/C2-negative-strand RNA interaction, two U residues at position 4 and 5 from 3ʹend in the negative-strand RNA were mutated to C residues (4UU to CC) ()). This site was predicted to be the hnRNP C1/C2 binding site in an earlier report [Citation10]. Recombinant hnRNP C1 protein was UV cross-linked with wild type or 4UU to CC mutant radiolabelled antisense cloverleaf RNA. Complete loss of hnRNP C1 interaction with negative-strand RNA in 4UU to CC mutant was observed as compared to wild type ()). This result validates the hnRNP C1 binding site in the negative-strand RNA. Next, life cycle of wild-type CVB3 replicon RNA and replicon RNA harbouring above-mentioned mutants was compared by measuring luciferase activity in a time-dependent manner by a method as described by Vogt and Andino [Citation22]. LD.73 mutant which is known to be replication deficient was used as control. Interestingly, 4UU to CC mutant showed complete inhibition in replication similar to LD.73 mutant and wild type with GnHCl control (Figure S8). However, 569 U to C mutant replicated efficiently almost equivalent to wild type. Of note, 569 U to C mutation did not completely abrogate hnRNP C1 interaction. Consistent modest effects of hnRNP C1/C2-positive-strand RNA interaction on CVB3 translation and its lower affinity with positive-strand RNA co-relate well with the data represented in ). Since 4UU to CC mutant was unable to replicate, ± RNA ratio in this mutant could not be determined. Compared to wild-type replicon RNA, 569 U →C mutant RNA showed increased ± RNA ratio, consistent with the predictions from the model ()). Of note, the ± RNA ratios are much higher as compared to the published report with virus infection as this experiment was carried out using CVB3 replicon RNA [Citation9]. Another study used RNAase protection assay to estimate the positive and negative-strand RNA in poliovirus also found ± RNA ratio in similar range (40:1 to 70:1) [Citation23]. These experiments provide experimental support to the predictions from mathematical model providing first insights into the regulation of positive to negative-strand RNA ratio in enteroviruses.
Figure 6. Strand-specific mutations to disrupt hnRNP C1/C2 interaction with viral RNAs. (a) Schematic of U to C mutation generated at 569 position in 5ʹUTR. (b) UV-crosslinking assay was carried out using wild type or mutant radiolabelled stem-loop V with rhnRNP C1 or rPTB proteins. PTB and hnRNP C1 bands are indicated by arrows. Densitometry values of band intensities are indicated under the lanes. (c) Schematic of UU to CC mutation generated at fourth and fifth nucleotide position (from 3ʹend) in the negative-strand RNA. (d) UV-crosslinking gel image representing the interaction of recombinant hnRNP C1 to wild type and 4UU to CC mutant negative-strand RNAs. 1–100 nucleotide antisense RNA probe was used for this crosslinking. (e) Positive to negative-strand RNA ratio at 8-h post-transfection of wild type or 569U to C mutant CVB3 replicon RNA. * indicates P< 0.05.
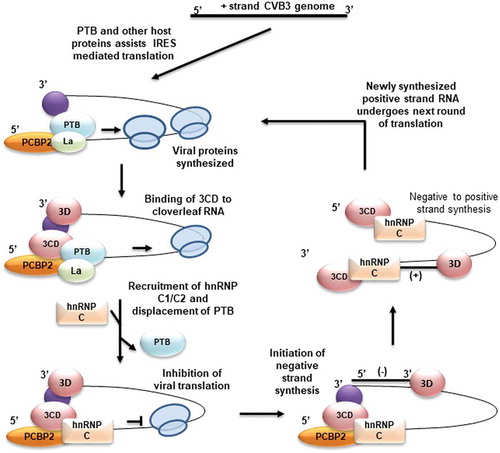
With all these experimental observations we propose a model in which hnRNP C1/C2 plays a role in removing the ribosomes from the positive-strand RNA template by repressing translation initiation. This leads to reduced ribosome flux on the template and indirectly improves the efficiency of the ternary replication complex in negative-strand synthesis (). These results provide novel mechanism of orchestrating viral RNA translation and replication by host proteins hnRNP C1/C2 and PTB.
Figure 7. Model for hnRNP C1/C2 mediated regulation of translation to replication switch. Virus life cycle begins in the cell by IRES-mediated translation of positive-strand RNA with the help of canonical translation initiation factors and IRES trans-acting factors. Once sufficient viral proteins are produced, a ternary complex of cloverleaf RNA, viral protein 3CD and PCBP-2 assemble at the 5ʹend of positive-strand RNA. The activity of the replication complex further indirectly stimulated by displacement of PTB at IRES at stem-loop V and inhibition of translation, as reduced ribosome flux can facilitate polymerase movement. Synthesis of negative-strand RNA leads to the generation of high-affinity binding sites on negative-strand RNA and preferential interaction of hnRNP C1/C2 with negative-strand RNA leads to positive-strand synthesis.
Discussion
In this study, we show that a host protein hnRNP C1/C2 acts as a repressor of translation and this facilitates negative-strand synthesis. This is particularly interesting as hnRNP C1/C2 is known to be a positive regulator of viral RNA replication and here we have observed inhibitory function of hnRNP C1/C2 in viral RNA translation. Earlier study in poliovirus suggested that hnRNP C1/C2 does not play any role in translation using in vitro replication/translation system [Citation9]. In the current study, cell culture-based CVB3 replicon RNA system was used to study the role of hnRNP C1/C2 in viral RNA translation. By uncoupling translation and replication by various methods, we demonstrate that hnRNP C1/C2 acts as a repressor of viral RNA translation in cells. In vitro translation system of rabbit reticulocyte lysate supplemented with HeLa S10 extracts was also utilized and repressive role of hnRNP C1/C2 in translation was observed.
Further, the mechanism of translation repression by hnRNP C1/C2 was also studied. The competitive interactions between PTB, a positive regulator of translation, and hnRNP C1/C2 at CVB3 IRES decides the fate of the template. Binding site of hnRNP C1/C2 is present in the basal region of stem-loop V and mutation at 569 U to C nucleotide position reduced hnRNP C1/C2 interaction with stem-loop V. Since the interaction sites of both PTB and hnRNP C1/C2 are overlapping, 569 U to C mutant also reduced interaction of PTB with stem-loop V. Of note, affinity of PTB seems to be very high as compared to hnRNP C1/C2 (Figure S3B). Hence, the magnitude of hnRNP C1/C2 mediated translation repression is low which is also observed in our experiments. The low affinity of hnRNP C1/C2 could be countered by its very high abundance in a cell relative to PTB [Citation24]. Disrupting the hnRNP C1/C2 interaction with negative-strand RNA completely inhibits viral RNA replication. These results are consistent with an earlier study performed with cloverleaf RNA where mutation in ‘a’ arm of cloverleaf RNA abrogated replication of virus [Citation22]. Our results provide the molecular basis for this as a mutation in the stem-loop ‘a’, of cloverleaf RNA abrogates hnRNP C1/C2 binding in negative strand and hence inhibit positive-strand synthesis.
Various host proteins have been reported to be a negative regulator of viral RNA translation for type 1 IRES. For example, FBP2 and AUF1, are both suppressors of viral RNA translation [Citation25,Citation26]. However, upon knockdown of these proteins, viral lifecycle was found to be more efficient, suggesting that these proteins act more as antivirals and do not favour translation to replication switch. In contrast to this, upon knockdown of hnRNP C1/C2, viral life cycle is abrogated as observed from our experiments and also well characterized in literature [Citation9–Citation11]. This dual role of hnRNP C1/C2 in inhibiting translation and promoting replication makes it an ideal candidate that can function in regulating translation to replication switch. Repression of translation by hnRNP C1/C2 lead to template switching from translation to replication, similar to puromycin drug treatment. This property of hnRNP C1/C2 was independent of the assembly of ternary complex at the cloverleaf RNA, as observed by experiments using LD.73 mutant replicon RNA. This indicates that the interaction of hnRNP C1/C2 with 5ʹUTR in positive-strand RNA causes translation repression and hence reduces ribosome flux on the template. This leads to increase in the efficiency of replication complex indirectly as template in free of obstructing ribosomes favouring polymerase movement.
One of the first reports that studied translation to replication switch suggests the role of host protein PCBP-2 and viral protein 3CD interaction with cloverleaf RNA. Although this explains the mechanism of replication initiation at positive-strand RNA, how translation is inhibited to favour polymerase movement was not clear [Citation2]. Our results suggest that hnRNP C1/C2 functions independently of the ternary complex in template switching, by acting as a repressor of translation. We propose that the efficiency of the ternary complex to induce translation to replication switch is enhanced due to translation repression by hnRNP C1/C2.
Another study suggested that cleavage of host protein PCBP-2 by viral proteinases might regulate translation to replication switch [Citation6]. Several other proteins like PTB, PABP and La are also cleaved by viral proteinases and probably these might also regulate the translation to replication switch [Citation7,Citation27]. Such a mechanism, in which cleavage of an ITAF stimulates replication is also known in case of Hepatitis C virus [Citation28]. Mechanisms of template switching that are based on cleavage of ITAFs could operate during later time points when sufficient amounts of viral proteases accumulate. As evidenced in earlier reports, cleavage of ITAFs begins only after 3–4-h post-infection and intact proteins were observed even at the end of viral life cycle. However, negative strands have been detected as early as 2-h post-infection, suggesting an alternate mechanism of translation to replication switch exist [Citation9]. Displacement of PTB by hnRNP C1/C2, could be an alternate strategy operating at earlier time points of infection, as this mechanism is independent of viral proteases activity.
Affinity of hnRNP C1/C2 for negative-strand RNA is stronger as compared to positive-strand RNA, but the number of positive-strand RNAs are higher as compared to negative strand. This leads to competitive interaction of hnRNP C1/C2 with both RNAs. Such an affinity-based competitive interaction of hnRNP C1/C2 with both strands could optimize translation and replication rates to yield a successful life cycle. Insights from our mathematical model support the hypothesis that bidirectional role of hnRNP C1/C in replication and its differential affinity-based interaction leads to buffering of positive to negative-strand RNA ratio. However, these predictions from the model remain to be validated experimentally. Generating mutants with different affinities of hnRNP C1/C2 could be used to measure the ratio, but unfortunately such a mutant as generated in this study does not replicate at all. In an earlier study where ± RNA ratio was measured in single cells, there were few cells having extremely high ratios (± = 150) and few with very low ratios (± = 5) [Citation29]. It is likely that in these cells, the intracellular host protein abundances are such that relative affinities of hnRNP C1/C2 (and not the abundance) are altered leading to eccentric ± RNA ratios. Using a mutant CVB3 replicon RNA that shows reduced affinity of hnRNP C1/C2 with positive-strand RNA, we were able to demonstrate that indeed the ± RNA ratio was increased in the mutant. These results provide first insights into regulation of positive and negative-strand RNA ratio in enteroviruses.
Taken together our results suggests a novel role of hnRNP C1/C2 in repressing viral RNA translation and regulating template switching from translation to replication. Upon the release of viral genomic RNA in cell, it undergoes IRES-mediated translation with the help of ITAFs including PTB. Repression of translation is achieved by displacement of PTB from stem-loop V by hnRNP C1/C2. This indirectly stimulates 3CD and PCBP-2 mediated negative-strand synthesis, as now the template is free of ribosomes and viral polymerase movement is favoured. Negative strands contain high-affinity binding site for hnRNP C1/C2 which further leads to recruitment of hnRNP C1/C2 and positive-strand synthesis (). This study suggests an alternative mechanism of translation to replication switch involving competitive interaction of host proteins PTB and hnRNP C1/C2 with CVB3 IRES.
Material and methods
Plasmids and RNAs
CVB3 replicon RNA, described in Lanke et al., 2009 [Citation12], was prepared from pRibCB3-T7-luc vector (kind gift from Prof. Frank van Kuppeveld). One microgram of RNA was used for transfection in one well of 24-well plate. Transfection was carried out by using lipofectamine 2000 (Invitrogen). pCD-CVB3-5ʹUTR construct was generated from pRibCB3-T7-luc. pCD-5ʹUTR construct was linearized with XbaI enzyme and used to prepare 5ʹUTR sense RNAs by in vitro transcription carried out using T7 RNA polymerase. The same plasmid was linearized by HindIII and used as template to synthesize 5ʹUTR anti-sense RNA using SP6 RNA polymerase in in vitro transcription. Similarly, 3ʹUTR sense and 3ʹUTR antisense RNAs were prepared using pCD-3ʹUTR construct. The same radiolabelled RNAs were also used in filter binding experiment. pCDNA 3.1-myc-hnRNPC1 construct was generated and used to overexpress hnRNP C1/C2 protein in cells. pCDNA 3.1-myc-PTB was used to overexpress PTB. pET-28a-hnRNP C1 construct was generated from pQE30-hnRNP C1 (kind gift from Prof. Bert Semler) and was used to express hnRNP C1 in bacteria followed by purification using Ni-NTA beads (Qiagen). pET-28a-PTB plasmid was used to express PTB protein in bacteria and purified as described earlier [Citation8,Citation30].
Cell lines, transfections and luciferase assay
HeLa cells from ATCC (kind gift from Dr. Deepak Saini) maintained in DMEM (Sigma) containing 10% serum (GIBCO, Invitrogen) were used for all experiments. CVB3 Replicon RNA was transfected in cells maintained in OptiMEM (Invitrogen) using Lipofectamine-2000 (Invitrogen). Four hours post-transfection, OptiMEM was replaced by DMEM containing 10% serum. Cells were harvested at different time points post-transfection for luciferase assay in 1x Passive Lysis Buffer (Promega) or in trizol for RNA isolation. For partial silencing experiments, cells were transfected with sihnRNP C1/C2 (Dharmacon – M-011869–01-0005), non-specific siRNA (Dharmacon) or lipofectamine alone, 24 h prior to transfection of CVB3 replicon RNA. Luciferase assays were performed using luciferase assay system (Promega) as per manufacturer’s protocol.
Western blot analysis
Cells were lysed in RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1mM sodium orthovanadate and mammalian protease inhibitor. The protein concentration was measured by using Bradford reagent (Bio-Rad) and equal amounts of protein were resolved on SDS-PAGE. Separated proteins were transferred on to nitrocellulose membrane (Pall Biosciences) and analysed by using mouse monoclonal anti-hnRNP C1/C2 (abcam), mouse monoclonal anti-PTB antibody (Calnexin), monoclonal anti-β-Actin antibody (Sigma), followed by HRP conjugated secondary antibody (goat raised anti-rabbit and goat raised anti-mouse, Sigma). Protein antibody complexes were analysed by chemiluminescence using Immobilon western systems (EMD Millipore).
UV-induced cross-linking of RNA-protein complexes and immuno-precipitation assay
To carry out UVcrosslinking experiments, HeLa S10 extracts containing the cytoplasmic proteins used. HeLa S10 extracts were prepared exactly as described in earlier [Citation31]. Purified recombinant protein or HeLa S10 extracts were incubated with the α-32P labelled RNA probes in 1x RNA binding buffer [25 mM HEPES (pH 7.6), 1.25 mM ATP and 2 mM MgCl2, 19% glycerol, 0.5 mM EDTA, 1.25 mM ATP, 2 mM GTP and 10 ug yeast tRNA (Sigma)] and the RNA-protein complexes were cross-linked using UV light of 254 nm wavelength. This was followed by RNase digestion and protected complexes were resolved by SDS PAGE and visualized by phosphor imaging. In experiments involving competition with unlabelled RNAs, indicated amounts of molar excess of unlabelled RNAs were added to reaction mixture.
For immuno-precipitation assays, UV-crosslinked complexes were diluted in IP buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.0, 0.5% NP-40, 1mM DTT, 100 U/ml RNase inhibitor) up to 500 µl volume and incubated with protein G agarose beads that were previously saturated with anti-hnRNP C1/C2 antibody (abcam-ab10294) or IgG antibody for 4 h. The beads were washed three times with IP buffer followed by re-suspension of beads in SDS buffer. This mixture was boiled for 5 min followed by gel electrophoresis. The gel was analysed by autoradiography.
In vitro transcription and translation
To prepare CVB3 sub-genomic replicon RNA, pRib-T7-Luc DNA was linearized with salI enzyme and in vitro transcription reaction was carried out using T7 polymerase (Fermentas) as per manufacturer’s protocol. For UVcrosslinking assays, probes were prepared by including α-32P uridine triphosphate (BRIT) in the in vitro transcription reactions carried out by T7 or SP6 RNA polymerase as per manufacturer’s protocol. The 5ʹUTR sense RNA probe consists of 1–741 nucleotides of the CVB3 genome and 5ʹUTR antisense consist of the same nucleotides but in reverse orientation. 3ʹUTR sense probe corresponds to nucleotide 7300–7399 nucleotide region in the genome and antisense 3ʹUTR probe corresponds to the same region but in reverse orientation.
RNAs corresponding to different stem-loops of 5ʹUTR were prepared as described previously [Citation30]. SL-I corresponds to 1–100 nucleotides, SL-II+III corresponds to 101–239 nucleotides, SL-IV corresponds to 240–447 nucleotides, SL-V corresponds to 448–582 nucleotides and SL-VI+VII corresponds to 583 to 754 nucleotides in the 5ʹUTR.
For in vitro translation reaction, 1 µg of CVB3 replicon RNA was added to Rabbit Reticulocyte Lysate (Promega) supplemented with 10% (v/v) of S10 extracts and amino acids. Reaction mixture was incubated at 30ºC for 90 min. Luciferase activity was measured after the reaction using luciferase assay system (Promega).
RNA isolation and qPCR
Total RNA was isolated from cells using trizol reagent (Sigma) as per manufacturer’s protocol. This RNA was further used for cDNA synthesis using gene-specific reverse primers and Revertaid enzyme (Fermentas). In all experiments, strand-specific reverse transcription was carried out to detect positive or negative-strand RNA specifically. For detection of negative-strand RNA, CVB3 forward primer was used in reverse transcription step and for detection of positive-strand RNA, reverse primer was used. Primer sequences: CVB3-F: 5ʹ GAATGCGGCTAATCCTAACTGC 3ʹ; CVB3-R: 5ʹ GCTCTATTAGTCACCGGATGGC 3ʹ. qPCR was carried out using DyNAmo qPCR kit as per manufacturer’s protocol in which 2 µl of cDNA was added per reaction.
RNA immuno-precipitation
Cells previously transfected with CVB3 replicon RNA were lysed in IP buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.0, 0.5% NP-40, 1mM DTT, 100 U/ml RNase inhibitor) at 8-h post-transfection. Equal amount of lysate was incubated with protein G beads previously saturated with anti-hnRNP C1/C2 antibody or IgG antibody for 4 h at 4°C. RNP complexes were washed with IP buffer three times and the beads were re-suspended in IP buffer and 10% aliquot was used analysis of proteins by western blot, while the remaining was treated with 30 µg proteinase K in presence of 0.1% SDS at 50°C for 30 min. Trizol was added to these reactions and were processed for RNA isolation as per manufacturer’s protocol. This RNA was used for semi-quantitative PCR or qPCR analysis.
Filter binding experiments
For filter binding experiments, radiolabelled CVB3 5ʹUTR or radiolabelled stem-loop V was incubated with increasing concentrations of recombinant hnRNP C1 protein or PTB protein in RNA binding buffer (5mM HEPES pH 7.6, 25 mM KCl, 2 mM MgCl2, 3.8% glycerol, 2 mM DTT, and 0.1 mM EDTA) for 15 min at 30°C. The reaction mixtures were spotted on nitrocellulose filters (Millipore) previously equilibrated with RNA binding buffer and were vacuum dried followed by two washes with RNA binding buffer and one wash with 100% ethanol. The dried filter papers were then used to measure the retained counts by using a scintillation counter. The measures counts were plotted against the concentration of the proteins and kD value was derived from the graph.
Mathematical modelling of CVB3 life cycle based on RNA protein interactions
Mathematical model was built to understand the role of hnRNP C1/C2 in the regulation of positive to negative-strand RNA ratio. The details of mathematical modelling are presented in supplementary methods.
Supplemental Material
Download Zip (4.3 MB)Acknowledgments
Prof. Frank van Kuppeveld and Nora Chapman are acknowledged for various constructs. Prof. Bert Semler is acknowledged for providing pQE30-hnRNP C1 construct. Dr. Deepak Saini, Indian Institute of Science, is acknowledged for providing the HeLa cell line. DST Fund for Improvement of Science and Technology Infrastructure (FIST) level II infrastructure and University Grants Commission Centre of Advanced Studies support to the department is acknowledged. PD and HR are supported by the research fellowship from the Council of Scientific and Industrial Research (CSIR). BG is supported by DS Kothari fellowship from UGC, Government of India. SD acknowledges JC Bose fellowship from Department of Science and Technology (DST), Govt. of India.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary materials
Supplemental materials data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism [10.1038/nrmicro1284]. Nat Rev Microbiol. 2005 Oct;3(10):765–776. print.
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998 Aug 1;12(15):2293–2304.
- Schulte MB, Andino R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J Virol. 2014 Mar 19. DOI:10.1128/jvi.03539-13.
- Shwetha S, Kumar A, Mullick R, et al. HuR displaces polypyrimidine tract binding protein to facilitate la binding to the 3′ untranslated region and enhances hepatitis C virus replication. J Virol. 2015 Nov 15;89(22):11356–11371.
- Blyn LB, Swiderek KM, Richards O, et al. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5ʹ noncoding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc Natl Acad Sci U S A. 1996;93(20):11115–11120. PubMed PMID: PMC38293.
- Chase AJ, Daijogo S, Semler BL. Inhibition of poliovirus-induced cleavage of cellular protein PCBP2 reduces the levels of viral RNA replication. J Virol. 2014 Mar 15;88(6):3192–3201.
- Back SH, Kim YK, Kim WJ, et al. Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3C(pro). J Virol. [Received 2002 Jul 2 Accepted 2002 Dec 4]; 76(5):2529–2542. PubMed PMID: PMC135932
- Verma B, Bhattacharyya S, Das S. Polypyrimidine tract-binding protein interacts with coxsackievirus B3 RNA and influences its translation. J Gen Virol. 2010 May 1;91(5):1245–1255.
- Ertel KJ, Brunner JE, Semler BL. Mechanistic consequences of hnRNP C binding to both RNA termini of poliovirus negative-strand RNA intermediates. J Virol. 2010 May 1;84(9):4229–4242.
- Brunner JE, Nguyen JHC, Roehl HH, et al. Functional interaction of heterogeneous nuclear ribonucleoprotein C with poliovirus RNA synthesis initiation complexes. J Virol. 2005 Mar 15;79(6):3254–3266.
- Brunner JE, Ertel KJ, Rozovics JM, et al. Delayed kinetics of poliovirus RNA synthesis in a human cell line with reduced levels of hnRNP C proteins. Virology. 2010 May 10;400(2):240–247.
- Lanke KHW, van der Schaar HM, Belov GA, et al. GBF1, a guanine nucleotide exchange factor for arf, is crucial for coxsackievirus B3 RNA replication. J Virol. 2009 Nov 15;83(22):11940–11949.
- Cieniková Z, Damberger FF, Hall J, et al. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J Am Chem Soc. 2014 Oct 15;136(41):14536–14544.
- Sweeney TR, Abaeva IS, Pestova TV, et al. The mechanism of translation initiation on type 1 picornavirus IRESs. Embo J. 2014 Dec 16. [Received June 28 Revised Aug 18];33(1):76–92.
- Jünemann C, Song Y, Bassili G, et al. Picornavirus internal ribosome entry site elements can stimulate translation of upstream genes. J Biol Chem. 2007 Jan 5;282(1):132–141.
- de Breyne S, Yu Y, Pestova TV, et al. Factor requirements for translation initiation on the Simian picornavirus internal ribosomal entry site. RNA. 2008 Feb 1;14(2):367–380.
- Brown BA, Ehrenfeld E. Translation of poliovirus RNA in vitro: changes in cleavage pattern and initiation sites by ribosomal salt wash. Virology. 1979 Sep;97(2):396–405. PubMed PMID: 224589.
- Soltaninassab SR, McAfee JG, Shahied-Milam L, et al. Oligonucleotide binding specificities of the hnRNP C protein tetramer. Nucleic Acids Res. 1998 July 1;26(14):3410–3417.
- Ochs K, Zeller A, Saleh L, et al. Impaired binding of standard initiation factors mediates poliovirus translation attenuation. J Virol. 2003 Jan 1;77(1):115–122.
- Kafasla P, Morgner N, Robinson CV, et al. Polypyrimidine tract‐binding protein stimulates the poliovirus IRES by modulating eIF4G binding. Embo J. 2010 Nov 03;29(21):3710–3722.
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol. 1991;65(6): 3384–3387. PubMed PMID: PMC241002.
- Vogt DA, Andino R. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010;6(6):e1000936.
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol. 1991 Jun;65(6):3384–3387. PubMed PMID: 1851886; PubMed Central PMCID: PMCPMC241002.
- Beck M, Schmidt A, Malmstroem J, et al. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7(1). DOI:10.1038/msb.2011.82.
- Cathcart AL, Rozovics JM, Semler BL. Cellular mRNA decay protein AUF1 negatively regulates enterovirus and human rhinovirus infections. J Virol. 2013 Oct 1;87(19):10423–10434.
- Lin J-Y, Li M-L, Shih S-R. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009 Jan 1;37(1):47–59.
- Bonderoff JM, LaRey JL, Lloyd RE. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J Virol. 2008 July 16. [Received 2008 Jan 2 Accepted 2008 Jul 10]; 82(19):9389–9399. PubMed PMID: PMC2546981.
- Ray U, Das S. Interplay between NS3 protease and human La protein regulates translation-replication switch of Hepatitis C virus [Article]. Sci Rep. 2011 June 14 online;1(1). doi:10.1038/srep00001.
- Schulte MB, Andino R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J Virol. 2014 June;88(11):6205–6212. PubMed PMID: 24648454; PubMed Central PMCID: PMCPMC4093869.
- Dave P, George B, Sharma DK, et al. Polypyrimidine tract-binding protein (PTB) and PTB-associated splicing factor in CVB3 infection: an ITAF for an ITAF. Nucleic Acids Res. 2017;45(15):9068–9084.
- Walter BL, Parsley TB, Ehrenfeld E, et al. Distinct Poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J Virol. 2002 Dec 1;76(23):12008–12022.