
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Structural models of large and dynamic molecular complexes are appearing in increasing numbers, in large part because of recent technical advances in cryo-electron microscopy. However, the inherent complexity of such biological assemblies comprising dozens of moving parts often limits the resolution of structural models and leaves the puzzle as to how each functional configuration transitions to the next. Orthogonal biochemical information is crucial to understanding the molecular interactions that drive those rearrangements. We present a two-step method for chemical probing detected by tandem mass-spectrometry to globally assess the reactivity of lysine residues within purified macromolecular complexes. Because lysine side chains often balance the negative charge of RNA in ribonucleoprotein complexes, the method is especially useful for detecting changes in protein-RNA interactions. By probing the E. coli 30S ribosome subunit, we established that the reactivity pattern of lysine residues quantitatively reflects structure models derived from X-ray crystallography. We also used the strategy to assess differences in three conformations of purified human spliceosomes in the context of recent cryo-electron microscopy models. Our results demonstrate that the probing method yields powerful biochemical information that helps contextualize architectural rearrangements of intermediate resolution structures of macromolecular complexes, often solved in multiple conformations.
Introduction
Cellular macromolecular complexes, including ribonucleoproteins (RNPs) like the ribosome, the spliceosome and telomerase, contain many moving parts, which makes it challenging to establish the mechanisms that control their molecular motions. Recent cryo-electron microscopy (cryo-EM) technological advances are now yielding the first models of complicated macromolecular assemblies previously refractory to structure analysis. Many of these assemblies, such as the spliceosome, telomerase and transcription initiation complexes [Citation1–Citation20], have also been captured in different functional conformations. While such structures provide important clues to the molecular mechanisms of these cellular machines, the intermediate resolution range (3–10 Å) of the cryo-EM maps limits the interpretation of interaction details. Furthermore, cryo-EM models, like X-ray crystallographic structures, are derived from stable conformations that often represent only one of many intermediate states within a multi-step process and/or assembly pathway. Additional means of measuring conformational changes of complexes remains essential to wholly describing how such complexes assemble and function.
Partnered with chemical modification, tandem mass-spectrometry (MS/MS) has emerged as a critical tool for gleaning structural information from macromolecular complexes. The presence and position of chemical crosslinks between molecules can be derived from MS/MS peptide sequencing to help pinpoint protein interactions [Citation21]. MS/MS analysis can also be used to quantify hydrogen/deuterium exchange to estimate solvent accessibility and flexibility of protein regions [Citation21]. Similarly, chemical probing (covalent labeling) with reagents that non-specifically modify protein side chains or that selectively target specific amino acids can be evaluated to determine the likelihood of a residue’s exposure to solvent vs. its participation in interactions within a macromolecule [Citation22,Citation23]. Several reagents are available for amino acid probing with well-defined chemistries [Citation22]. For RNPs in particular, the reactivity of lysine side chains to an acetylation reagent is especially informative, because it reflects both solvent accessibility and availability of the lone pair electron of the uncharged ε-nitrogen as a nucleophile. When positively charged, the primary amine of lysine side chains often assists to counteract and position the negatively charged phosphate backbone of nucleic acids, which should render the residue less reactive [Citation24].
Amino acid probing is commonly carried out with well-behaved individual proteins that can be purified in high (> nanomolar) concentrations, and with the goal of examining surface topology and protein/ligand interactions [Citation22]. Extending chemical probing methods to multiple conformations of larger and dynamic assemblies, including RNPs that are often only available at low picomolar amounts, brings a unique set of challenges. One challenge relates to comparing MS/MS data of multiple conformations of the same complex when non-overlapping peptide are generated after chemical probing. To solve the problem, we developed a chemical probing strategy that includes two lysine acetylation steps, and results in comparable peptides from any protein in a sample. In this study, we detail the two-step acetylation protocol and describe its application to two different RNP complexes: 30S ribosome subunit from E. coli and spliceosomes assembled in nuclear extract from human cells. Our results indicate that lysine reactivity is indeed reflective of structure and can be used to characterize conformational changes in macromolecular complexes. In this respect, when probing data is combined with mid-range resolution structural models we found lysines reactivity can guide detailed interpretation of localized conformational change. Although we have focused on RNPs, the method is compatible with single proteins and protein complexes.
Results
Two-step probing methodology and lysine acetylation assignment
Lysine side chains are good targets for chemical probing because they are reactive to acetylation reagents under mild conditions that maintain molecular interactions. The modification state of an individual lysine residue can then be determined by LC-MS/MS of peptides derived from proteins in the macromolecular complex that was subjected to chemical probing. A caveat to such analysis is that peptides for mass spectrometry evaluation are routinely generated by tryptic digestion, which cleaves after unmodified lysine and arginine residues, but not after acetyl-lysine. Assessing changes in lysine modification states between multiple RNP conformations is problematic because differential acetylation patterns will generate different tryptic peptides that are not directly comparable by MS/MS due to varying retention times and ionization profiles. To overcome this challenge, we developed a two-stage lysine acetylation protocol in which the native RNP is first probed with sulfo-NHS, and then denatured by SDS-PAGE before probing a second time with deuterated acetic anhydride ()). Because every lysine present in the protein should be modified in either the first or second step, the strategy will yield identical tryptic peptides. The 3 mass-unit difference between acetyl-lysine and deutero-acetyl-lysine allows us to discriminate between reactivity in the RNP vs. the denatured state ()).
Figure 1. Two-step acetylation protocol. (a) Cartoon diagram of an assembled RNP (70S ribosome) being acetylated, denatured, then deutero-acetylated. (b) Cartoon diagram showing a charged lysine interacting with the negative backbone of RNA and a solvent exposed uncharged lysine. The charged lysine is resistant to the first acetylation step and will be modified during the second acetylation with a deuter-acetyl (red bold acetyl group). (c) Left: Sample MS spectra containing two doubly charged peptides of m/z 498.796 and m/z 500.277 corresponding to the peptide VGFGYKAR from E. coli protein RS5. In the m/z 498.796 version, the lysine is modified with an acetyl group, whereas in the m/z 500.277 version, the lysine is modified with a deuteron-acetyl group, leading to a 3 Dalton mass difference. Right: Theoretical modeling of the predicted isotope pattern for the lighter version using MS-Isotope [Citation25] indicates a fourth isotope peak would contribute 5% peak intensity to the first isotope of the deuterated monoisotopic peak signal, and would have an insignificant effect on overall quantification
![Figure 1. Two-step acetylation protocol. (a) Cartoon diagram of an assembled RNP (70S ribosome) being acetylated, denatured, then deutero-acetylated. (b) Cartoon diagram showing a charged lysine interacting with the negative backbone of RNA and a solvent exposed uncharged lysine. The charged lysine is resistant to the first acetylation step and will be modified during the second acetylation with a deuter-acetyl (red bold acetyl group). (c) Left: Sample MS spectra containing two doubly charged peptides of m/z 498.796 and m/z 500.277 corresponding to the peptide VGFGYKAR from E. coli protein RS5. In the m/z 498.796 version, the lysine is modified with an acetyl group, whereas in the m/z 500.277 version, the lysine is modified with a deuteron-acetyl group, leading to a 3 Dalton mass difference. Right: Theoretical modeling of the predicted isotope pattern for the lighter version using MS-Isotope [Citation25] indicates a fourth isotope peak would contribute 5% peak intensity to the first isotope of the deuterated monoisotopic peak signal, and would have an insignificant effect on overall quantification](/cms/asset/3a1cc0c6-c997-482c-a0c5-a676c83df92e/krnb_a_1632777_f0001_c.jpg)
We tested the method by probing two different RNPs: the E. coli 30S ribosome subunit and human spliceosomes. We chose the E. coli 30S subunit as an initial benchmark because its structure is well characterized by X-ray crystallography. As a control, we also probed total protein isolated from E. coli 30S ribosome proteins in the absence of 16S rRNA, which we expected to have a very different conformation. Human spliceosomes are more representative of a more challenging and dynamic sample. We are able to assemble spliceosomes in vitro trapped in distinct conformations that are expected to exhibit structural differences. To better model the situation for challenging samples, we analyzed low picomole amounts of complexes for both E. coli 30S ribosome subunits and human spliceosomes. After two-step chemical probing, MS/MS analysis of individual samples reported lists of peptide sequences and associated modifications, including lysine acetylation, N-terminal methionine loss/acetylation, methionine oxidation, and pyroglutamate formation. Additional key data included peak intensity for each peptide, which is indicative of relative abundance, and the associated parent protein. We selected the peptides from well-characterized components of 30S ribosomal subunit and human spliceosomes [Citation26] and examined the modification state of all lysine residues. Across ten samples analyzed (triplicates of intact 30S ribosome subunit and of isolated 30S proteins, and duplicate preparations of two spliceosome conformations), >96% of lysine residues present in the MS/MS peptide sequences were reported as modified (). Correspondingly, >96% of peptides obtained from trypsin digest ended in arginine or the final amino acid of the protein. Assuming equal modification rates at each step and considering that any accessible residue in the RNP that escaped modification in the first step would likely be modified in the second step, we estimate probing efficiency for each reaction to be above 94%. Importantly, the high rate of modification produced a large number of comparable peptides between replicates and between different conformations of the same complex.
Table 1. MS/MS peptide characteristics
For each sample, we next extracted peptides with modified lysines from the MS/MS results to compile a list of individual lysine residues and their position in the parent protein. We scored each of these as acetylated or deutero-acetylated based on the associated modification reported for the peptide. Within an individual sample, a subset of lysines were were reported multiple times when their parent peptide carried additional modifications, such as oxidized methionine, and we recorded these peptides as independent measurements of the modification state. Nearly 70% of these lysines carried the same acetyl modification, as expected for a stable conformation and efficient modification in both probing steps. In the cases for which a given lysine was identified in multiple peptides with both acetyl and deutero-acetyl modification, we compared the mass spectrometry peak intensity for the parent peptides. For a subset of these lysines, the peak intensity for the peptide with one modification greatly exceeded that of the other. We scored the lysine as having the predominant modification if the peak intensity ratio indicated enrichment of more than five-fold (2–6% of all scored lysines across different samples). Otherwise, we flagged lysines observed with both modifications as mixed. We hypothesized that mixed modifications may be due to incomplete acetylation of the native RNP, but a comparison of modifications between replicates, as discussed below, suggests they more often reflect heterogeneity in the composition or conformation of the RNP being probed.
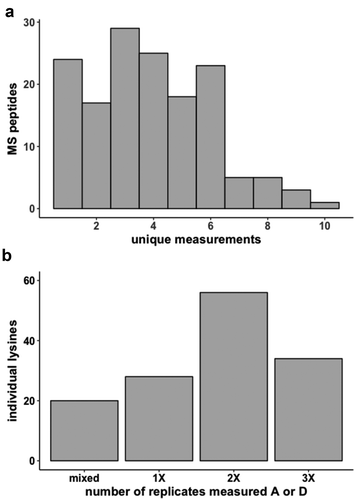
We assessed probing reproducibility by comparing three replicate experiments for the 30S ribosome subunit. As a result of the two-step strategy, over half of the MS peptides measured overall were present in all three replicates, and 118 individual lysine residues were observed more than once ()). Of these, we found that 78% exhibited the same modification state across replicates ()). The remaining were identified as mixed in one or more samples, or exhibited opposing modifications (acetyl vs. deutero-acetyl). Notably, mixed and opposing modifications were not randomly distributed among all residues, as would be expected if inefficient probing alone were the cause.
Figure 2. 30S ribosome subunit two-step acetylation reproducibility (a) Frequency at which MS peptides are uniquely measured (b) MS lysine modification reproducibility across replicates. A is acetyl-lysine, D is deutero-acetyl-lysine
To better understand the source of mixed modifications, we calculated conditional probabilities for three possible lysine orientations in the 30S ribosome subunit: solvent exposed, solvent inaccessible, and equally present in both conformations. As previously stated, based on the number of unmodified lysines detected, we estimate the modification efficiency is approximately 94% at both probing steps. We compared a statistical model in which probing inefficiency accounted for nearly all our observations of mixed modification to a model in which multiple conformations of the lysine was the primary cause. The analysis indicated that 80% of differentially modified lysines observations were more likely due to mixed conformations present in the sample, rather than to probing inefficiency. We used the conditional probabilities to ultimately classify individual lysines in each complex as primarily solvent accessible (A), primarily buried (D) or present in both conformations (M) (Supplemental Table 2).
Comparing lysine probing patterns of the 30S ribosome subunit to crystallographic structures
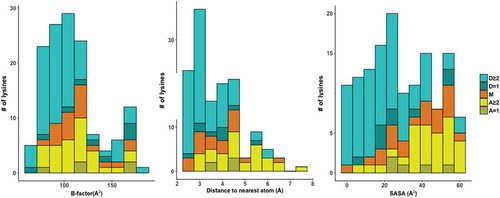
As a quantitative verification of principle of the two-step probing strategy, we compared our probing results for the 30S E. coli ribosome to a high-resolution model of the 70S E. coli ribosome subunit derived from X-ray crystallography [Citation27]. Our results demonstrate consistency of lysine probing patterns with the structural model. In the structure, a majority of deutero-acetyl-lysines are modeled in conformations consistent with ionic interactions with the phosphate backbone of rRNA, while lysines with acetyl or mixed modification state are generally directed toward solvent (). To evaluate the relationship between lysine reactivity and structure, we also compared the observed modification state to three parameters: solvent accessible surface area (SASA), distance to the nearest atom and B-factor (the positional uncertainty of an atom), all relative to the primary lysine amine derived from the same ribosome crystal structure (, Supplemental Table 3). Consistent with the primary amine being constrained by interactions that reduce lysine reactivity in the native ribosome, values for deutero-acetyl-lysine trended lower for these three parameters relative to acetyl-lysine and mixed.
Figure 3. MS/MS lysines colored to match their acetylation state displayed on the 30S subunit of the 70S ribosome. 16S RNA is blue, 30S proteins are grey, deutero-acetyl lysine is cyan, acetyl lysine is yellow and mixed is yellow
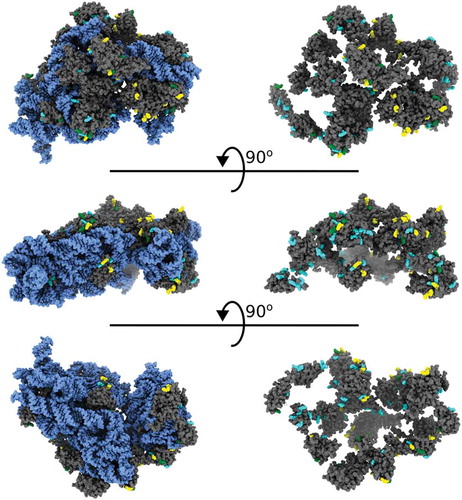
Figure 4. Stacked histograms of primary amine B-factor (left), distance to nearest atom (middle) and SASA scores (right) of individual MS/MS lysines separated by modification state and the number of times a given lysine was measured across replicates. A is acetyl-lysine, D is deutero-acetyl-lysine and M is mixed. Shading indicates whether the lysine was observed in one (= 1, darker) or more than one (≥2, lighter) replicates
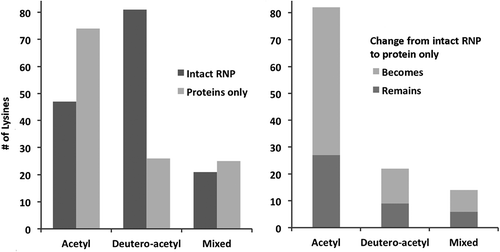
Next, we compared probing results from total protein isolated from the 30S subunit. As expected, in the absence of the 16S rRNA, lysines from these proteins were identified with a higher proportion of acetyl modifications relative to the intact RNP (). We also saw an increased proportion of lysines with mixed modification, which may be due to conformational heterogeneity of the predicted intrinsically disordered regions in most ribosome proteins in the absence of rRNA. Still, 80% of lysines in free 30S proteins exhibit the same modification state across replicates (Supplemental Table 4). Together, these data indicate our probing strategy can discern the loss of 30S protein interactions in the absence of 16S rRNA, and indicates that the proteins largely maintain a consistent structure in terms of lysine accessibility. This detailed comparison of the ribosome structure and probing data served as an important benchmark for the two-step acetylation strategy, and was made possible by the availability of a high-resolution structure. The strong correlation between probing results and structure indicates that lysine probing can provide architectural and functional detail when applied to more challenging molecules for which limited resolution precludes modeling side chains positions.
Figure 5. Modification changes between intact 30S ribosome subunit versus isolated 30S ribosome proteins only. Left panel: Number of lysines with indicated modification states. Right panel: Number of lysines that remain or become the indicated modification state in isolated 30S ribosome proteins relative to the intact RNP
Comparing multiple conformations of the spliceosome
The spliceosome is an example of a dynamic RNP for which several structural models exist at mid-range resolution (4–9 Å), and epitomizes a ‘real-world’ challenge for structure probing. The relatively low-resolution maps precluded detailed interpretation of side chain interactions. Furthermore, large portions of most spliceosome components are not included in the model, a situation that underscores the need for orthogonal biochemical data. The limitations of the spliceosome structures are a result of known heterogeneity in spliceosome preparations, which is compounded by the restricted amount of material that is available. We expected that in addition to indicating the local structural environment of a lysine residue, chemical probing would reflect similarities and differences in lysine environment between different functional conformations of a macromolecular complex. To identify residues that change reactivity due to a structural change, we probed purified spliceosomes stalled before and after the second chemical step of splicing (termed catalytic and product spliceosomes, respectively [Citation28,Citation29]). As expected, we generated a large number of identical peptides from different forms of human spliceosomes isolated from in vitro splicing reactions as a result of the two-step acetylation strategy (Supplemental Table 1). Comparing catalytic and product human spliceosomes, 69% of the 404 lysines observed in both samples had the same modification (Supplemental Table 4). Some proteins shared between the complexes showed no differences at all, while others had more modification changes ()). This result is consistent with cryo-EM models of similar states of yeast spliceosomes, which exhibited a high degree of similarity in overall architecture [Citation3].
Figure 6. Modification changes between catalytic and post-catalytic spliceosomes. (a) Histogram depicting the distribution of the percentage of overall modification differences for the 58 proteins present in both complexes. (b) Structural alignment of human Cwc15 from catalytic (cyan with blue lysine residues) and product (orange with pink lysine residues) spliceosomes and a table listing the MS/MS modification state of corresponding lysines. D is deutero-acetyl, A is acetyl and M is mixed while C and P concerns the corresponding catalytic and product spliceosome MS/MS data sets. Note that lysine 22 (K22) from the catalytic spliceosome lies within an unmodeled region. (c) Close-up view of the relative positions of U5 snRNA stem-loop I (lavender), 5′ exon (magenta), and differentially modified Prp8 lysine residues K670 and K672 (green) in catalytic and post-catalytic spliceosome conformations. The last nucleotide of the 5ʹ exon is highlighted in yellow, and U96 of U5 snRNA is in cyan
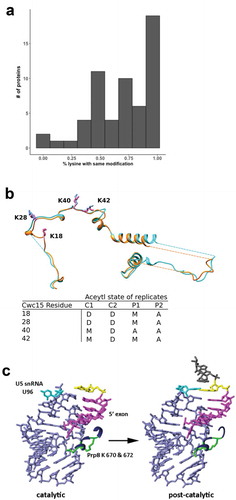
Lysines that change in accessibility as the spliceosome carries out exon ligation may indicate interaction sites for incoming/outgoing components or reflect conformational changes that regulate spliceosome progression. For example, we identified such a difference in the protein Cwc15 between catalytic and post-catalytic spliceosomes. Cwc15 is associated with the Prp19 complex, a spliceosome subcomplex that regulates catalytic conformations. Two adjacent loops in Cwc15 each contain two lysines (K:18/28/40/42) that our data predict to be inaccessible in catalytic spliceosomes, and then accessible in the product spliceosomes following exon ligation () bottom). Cwc15 is modeled in a nearly identical conformation in cryo-EM structures of human catalytic [Citation1] and product [Citation30] spliceosomes. Nevertheless, our probing data suggest that in solution, the lysines in this region change molecular interactions at some point during exon ligation () top). Our data therefore have identified specific residues for future targeted manipulations, such as mutagenesis, to support or reject a functional role for the changes in lysine accessibility. The data also indicates that nearby surfaces or binding partners of these lysines in Cwc15 may have functional relevance to splicing.
Discussion
The two-step chemical probing method described here specifically measures changes in lysine interactions. The method is useful when assessing structural variations of any protein complex in solution, even in the context of dynamic entities such as the spliceosome. Given the continuous negative charge of a RNA backbone, lysine probing is particularly helpful in analyzing RNPs. Our approach was specifically developed to address the challenges of characterizing complex assemblies that are difficult to manipulate and only available in very small quantities, such as spliceosomes. However the strategy can also be employed to characterize interactions between individual proteins and RNA. Importantly, the results will complement other structural methodologies typically only applicable to molecules that are more straightforward to manipulate in vitro and often readily available significant quantities.
When employing our methodology, a few points should be considered. First, low-picomole amounts of material are sufficient to capture a large portion of the sequence of a given protein, but more material will allow for greater coverage, especially if used in conjunction with other proteases for generating peptides for the MS/MS analysis. Second, for all of the data presented here, the first probing step of native complexes occurred over the course of an hour. Because of this, interactions that quickly fluctuate between two states are likely to be identified as solvent accessible. A potential extension of two-step probing would be to carry out a time course during the first probing period to differentiate more dynamic lysine residues. Third, more sophisticated mass spectrometry strategies, like multiple reaction monitoring, could be used to better quantify the relative ratios of differentially modified lysines. Finally, modifying reagents against other amino acids can be additionally incorporated into the protocol if their respective chemistries occur at a pH at which the target complex is purified.
Technological advances in cryo-EM have brought structure determination of many multi-megadalton biological assemblies within reach. Now, more than ever, interpretations of the models need further testing to support or reject putative molecular mechanisms suggested by structural architecture. This need is further intensified by the knowledge that structures derived by both cryo-EM or X-ray crystallography typically represent only the most stable and highly populated conformations that are sampled in aqueous solution. With X-ray crystallography, macromolecules are constrained by the crystalline lattice. While a cryo-EM preparation theoretically includes all conformations of the molecule that exist at the time of rapid freezing, non-abundant conformations are often lost as a result of image processing. In contrast, chemical probing reports on all aqueous conformations, and can add an additional layer of information for characterizing flexible regions of proteins and nucleic acids, which are relevant to their function. For example, we previously identified a change in lysine reactivity in a handful of residues within the essential spliceosome scaffolding protein Prp8 in complexes arrested before and after spliceosome activation [Citation31]. Cryo-EM models of pre-activated and catalytic spliceosomes both modeled the lysines in close proximity to the U5 small nuclear RNA (snRNA) near its interaction with the 5´ exon of the pre-mRNA substrate [Citation2,Citation4,Citation5,Citation16,Citation21]. Notably, the register of the interaction between U5 snRNA and 5´ exon differed between the two structures ()) [Citation15,Citation16]. Based on the structural difference and the change in reactivity of the Prp8 residues, we hypothesized that Prp8 may have an intermediate conformation (not yet displayed in a cryo-EM structure). We used yeast genetics to assess the importance of the side chains, and the results support a model in which the Prp8 lysines play a role in positioning the 5′ exon of the pre-mRNA for catalysis. The study demonstrated how intersection of three experimental approaches (lysine probing, cryo-EM models and genetics) can help pinpoint novel functional roles of individual residues in complicated molecular machines.
Materials and methods
30S ribosome subunit reconstitution
Purified 30S E. coli ribosomal RNA and ribosomal proteins (provided by the Noller lab at UCSC) in 10 mM Tris-HCl pH 7.5, 100 mM NH4Cl, 10 mM MgCl2, 6 mM BME were assembled into 30S subunits via incubation at 42°C for 15 mins. After reconstitution, 30S ribosome subunits were exchanged into 20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2 and 1 mM DTT via Slide-A-Lyzer 10 KDa mini-dialysis device (Thermo Fisher Scientific) for 5 hours at 4°C.
In vitro spliceosome assembly and purification
Catalytic and poduct pre-mRNA substrates are derivatives of the AdML transcript AND tagged with three MS2 sites in either the intron or at the 3′ end, respectively. T7 runoff transcription was used to generate G(5′)ppp(5′)G-capped radiolabeled pre-mRNA, which was gel-purified and preincubated with a 50-fold excess of MS2-MBP fusion protein. Catalytic spliceosomes arrested after first-step chemistry were assembled with a pre-mRNA containing a mutant GG 3′ splice site as previously described [Citation29]. Post-catalytic spliceosomes arrested after second-step chemistry were assembled with a pre-mRNA containing a truncated 3′ exon as previously described [Citation28].
Both complexes were assembled in in vitro splicing reactions containing 10 nM pre-mRNA, 80 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg/mL tRNA, and 40% HeLa cell nuclear extract at 30°C for 60 minutes. The reactions were fractionated by size exclusion chromatography using Sephacryl S-400 resin (GE Healthcare) in 20 mM HEPES pH 7.9, 150 mM potassium chloride, 5 mM EDTA, 0.05% v/v NP-40, and then bound to amylose resin. After washing in the same buffer, spliceosomes were eluted from the amylose resin in 20 mM HEPES pH 7.9, 150 mM potassium chloride, 5 mM EDTA, and 1 µM maltose.
Chemical probing by two-step acetylation
For the first acetylation step, one to five picomoles of reconstituted 30S E. coli ribosome subunits (20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2 and 1 mM DTT), isolated 30S ribosome proteins (20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2 and 1 mM DTT) or human spliceosomes (20 mM HEPES pH 7.9, 150 mM KCl, 5 mM EDTA, 1 µM maltose) were incubated in 5 mM N-hydroxysulfosuccinimide (Pierce Technology) at room temperature for one hour. Reactions were quenched by addition of 1/10th volume 1 M Tris pH 7.9, and then subjected to SDS-PAGE. After staining with Coomassie-G (5% w/v aluminum sulfate 14–18 hydrate, 10% v/v ethanol, 0.02% w/v CBB G-250, and 2% v/v phosphoric acid), the entire protein lane was excised into 6–9 gel slices. For the second acetylation step, gel slices were incubated with shaking agitation under the following sequence of conditions: 1) water for 10 minutes at 37°C, 2) 10 mM ammonium bicarbonate at room temperature for 10 minutes, 3) 50 mM ammonium bicarbonate:acetonitrile (1:1) for 45 minutes at 37°C two times sequentially, 4) water two times sequentially for 10 minutes each, and 5) 100% acetonitrile at 37°C for 5 minutes twice, after which the gel slice is visibly dehydrated and no longer translucent. After complete removal of the acetonitrile, 20 μl of D6-acetic anhydride (Acros Organics) mixed with 40 μl of 100 mM ammonium bicarbonate was added to the dehydrated gel slices, which were continuously agitated to evenly distribute the anhydride solution. Upon absorption of the D6-acetic anhydride mixture, the rehydrated gel slices were submerged in 100 mM ammonium bicarbonate, and the solution pH adjusted to 7–8 with 1 M ammonium bicarbonate. After 60 minutes at 37°C, the gel slices were washed with water three times and submitted for LC-MS/MS analysis.
Protein digestion and mass spectrometric analysis
For protein digestion, gel pieces were washed in 25 mM ammonium bicarbonate/60% v/v acetonitrile. Disulfide bonds were reduced using 10 mM dithiothreitol in 25 mM ammonium bicarbonate for 30 minutes at 60°C, and then free sulfhydryls were alkylated using 20 mM iodoacetamide in 25 mM ammonium bicarbonate for an hour. Gel pieces were washed again using 25 mM ammonium bicarbonate/60% v/v acetonitrile, and then digested overnight using 200 ng TPCK-treated porcine trypsin (Promega) in 25 mM ammonium bicarbonate. Peptides were extracted using 50% v/v acetonitrile/1% v/v formic acid. Extracted peptides were dried down by vacuum centrifugation, and resuspended in 0.1% v/v formic acid prior to mass spectrometric analysis.
Peptides were analyzed by LC-MS/MS using a NanoAcquity (Waters) UPLC system interfaced with either an LTQ Orbitrap Velos or QExactive Plus (both Thermo) mass spectrometer. Peptides were separated using an integrated 75 μm x 15 cm PEPMAP reverse phase column and spray tip (EasySpray source). For E. coli ribosome samples, a gradient from 2 to 30% v/v solvent B (0.1% v/v formic acid in acetonitrile) over solvent A (0.1% acetonitrile in water) at a flow rate of 400 nL/min for 72 min was used, followed by ramping to 50% B over 2 min, before returning to starting conditions. For spliceosome samples, a gradient from 2 to 27% v/v solvent B (0.1% v/v formic acid in acetonitrile) over solvent A (0.1% acetonitrile in water) at a flow rate of 400 nL/min for 27 min was used.
With the Velos, survey scans were measured at a resolution of 30,000 full width at half maximum (FWHM). The six most intense precursor ions were automatically selected for HCD fragmentation analysis by data-dependent acquisition and measured at a resolution of 7,500 FWHM. For QExactive data, survey scans were measured at a resolution of 70,000 FWHM, and were followed by data-dependent acquisition of the top ten most intense precursors at a resolution of 17,500 FWHM.
Raw data were converted to mgf format peak list files using in-house software based on the Raw_Extract script in Xcalibur version 2.4. For ribosome samples, the data were searched with Protein Prospector version 5.21.23 against a database of all E. coli proteins in SwissProt downloaded on 18 May 2017, plus decoy versions of all of these sequences (a total of 23,044 entries searched). For spliceosome samples, the data were searched using Protein Prospector version 5.18.22 against a database of all human proteins in SwissProt downloaded on 9 May 2016 supplemented with entries for E. coli maltose-binding periplasmic protein, enterobacteria phage MS2 coat protein and pig trypsin, plus decoy versions of all of these sequences (a total of 20,204 entries searched). Searches were performed with tolerances of ±15 ppm on precursor ions and ±30 ppm on fragment ions. Carbamidomethylation of cysteine residues was searched as a constant modification, and variable modifications considered were acetylation or deutero-acetylation of uncleaved lysines, methionine oxidation, pyroglutamate formation from N-terminal glutamines, and protein N-terminal methionine removal and/or acetylation. Results were threshholded to an estimated 1% protein FDR according to target:decoy database searching [Citation32]. Intensities of peaks were extracted from the raw data using Protein Prospector by summing together signal over a period −10 to +20 seconds from the time the peak was selected for MS/MS.
Assignment of lysine modifications
Mass spectrometry data for each sample were returned as a list of sequenced peptides with associated protein identifiers. The lists were first filtered for peptides of proteins characterized as stable components of the associated RNP complex (Supplemental Table 1), and then for peptides containing modified lysine residues. From these peptides a catalog of lysine residues was compiled, which were flagged as acetylated (A) or deutero-actylated (D), along with the number peptides supporting the modification state (Supplemental Table 2). In cases where both modifications were identified in an individual sample, we compared the mass spectrometry peak intensity for the parent peptides. In cases where less than 5-fold enrichment for a particular modification was observed, the lysine was flagged as mixed (M).
We next compared results for replicate experiments and initially flagged lysines as A or D in cases where all measurements were in agreement, and M if opposing modifications were present. To estimate the likelihood of mixed modifications arising from inefficiency in probing vs. multiple lysine conformations, we calculated conditional probabilities for three lysine conformations (solvent accessible, buried, and equally present in both states) using Bayes theorem formulated as:
Prior probability P(A) was designated as the frequency of peptides identified with A or D modification, and assuming a low probability (1%) of mixed conformations. Posterior probability P(B|A) for each conformation was calculated as binomial probability mass function for number of specific modification observations per total observations across replicas with modification likelihood based on an estimated probing efficiency of 94% for both steps. This efficiency was derived from the relative frequency of peptides ending in unmodified lysine vs. arginine in the MS data. P(Ā) is 1-P(A), and P(B|Ā) is the sum of posterior probabilities of the other two conformations. These values are reported in Supplemental Table 2.
Analysis of lysine modification relative to ribosome structure and Cwc15 alignment
Structural analyses and generation of molecular graphics were performed with UCSF Chimera [Citation33] and ChimeraX [Citation34], which are developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH R01-GM129325 and P41-GM103311. Lysine modification data for E. coli ribosomes was compared to the structure of the 30S subunit extracted from RSCB entry 4YBB. Distances from lysine primary amines to their nearest neighbor were determined with the Chimera ‘findclash’ command. Solvent accessible surface area for the primary amine was calculated with the FreeSASA program [Citation35]. B-factors were extracted from the PDB file. The values reported are average measurements from the two 30S subunits in the crystal’s asymmetric unit. Data were plotted using ggplot2 in RStudio. Cwc15 structures were extracted from human catalytic (RSCB entry 5XJC) and human product (RSCB entry 6QDV) spliceosomes structures. The two proteins were then aligned using Chimera ‘matchmaker’ command.
Supplemental Material
Download Zip (1.1 MB)Acknowledgments
We thank Laura Lancaster and Harry Noller for providing reconstituted 30S E. coli ribosome subunits and isolated 30S proteins.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Zhang X, Yan C, Hang J, et al. An atomic structure of the human spliceosome. Cell. 2017;169:918–929 e914.
- Plaschka C, Lin PC, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–621.
- Bai R, Yan C, Wan R, et al. Structure of the post-catalytic spliceosome from saccharomyces cerevisiae. Cell. 2017;171:1589–1598 e1588.
- Bertram K, Agafonov DE, Dybkov O, et al. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell. 2017;170:701–713 e711.
- Yan C, Wan R, Bai R, et al. Structure of a yeast activated spliceosome at 3.5 A resolution. Science. 2016;353:904–911.
- Wan R, Yan C, Bai R, et al. The 3.8 A structure of the U4/U6.U5 tri-snRNP: insights into spliceosome assembly and catalysis. Science. 2016;351:466–475.
- Wan R, Yan C, Bai R, et al. Structure of a yeast catalytic step I spliceosome at 3.4 A resolution. Science. 2016;353:895–904.
- Rauhut R, Fabrizio P, Dybkov O, et al. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science. 2016;353:1399–1405.
- Galej WP, Wilkinson ME, Fica SM, et al. Cryo-EM structure of the spliceosome immediately after branching. Nature. 2016;537:197–201.
- Yan C, Hang J, Wan R, et al. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182–1191.
- Zhang X, Yan C, Zhan X, et al. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018;28:307–322.
- Zhan X, Yan C, Zhang X, et al. Structure of a human catalytic step I spliceosome. Science. 2018;359:537–545.
- Plaschka C, Lin PC, Charenton C, et al. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559:419–422.
- Liu S, Li X, Zhang L, et al. Structure of the yeast spliceosomal postcatalytic P complex. Science. 2017;358:1278–1283.
- Wilkinson ME, Fica SM, Galej WP, et al. Postcatalytic spliceosome structure reveals mechanism of 3ʹ-splice site selection. Science. 2017;358:1283–1288.
- Fica SM, Oubridge C, Galej WP, et al. Structure of a spliceosome remodelled for exon ligation. Nature. 2017;542:377–380.
- Nguyen TH, Galej WP, Bai XC, et al. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature. 2015;523:47–52.
- Jiang J, Wang Y, Susac L, et al. Structure of telomerase with telomeric DNA. Cell. 2018;173:1179–1190 e1113.
- Chu F, Thornton DT, Nguyen HT. Chemical cross-linking in the structural analysis of protein assemblies. Methods. 2018;144:53–63.
- Robinson PJ, Trnka MJ, Bushnell DA, et al. Structure of a complete mediator-RNA polymerase II pre-initiation complex. Cell. 2016;166:1411–1422 e1416.
- Oganesyan I, Lento C, Wilson DJ. Contemporary hydrogen deuterium exchange mass spectrometry. Methods. 2018;144:27–42.
- Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009;28:785–815.
- Limpikirati P, Liu T, Vachet RW. Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods. 2018;144:79–93.
- Kim OT, Yura K, Go N. Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 2006;34:6450–6460.
- Chalkley RJ, Hansen KC, Baldwin MA. Bioinformatic methods to exploit mass spectrometric data for proteomic applications. Methods Enzymol. 2005;402:289–312.
- Cvitkovic I, Jurica MS. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41:D132–D141.
- Noeske J, Wasserman MR, Terry DS, et al. High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol. 2015;22:336–341.
- Ilagan JO, Chalkley RJ, Burlingame AL, et al. Rearrangements within human spliceosomes captured after exon ligation. RNA. 2013;19:400–412.
- Jurica MS, Licklider LJ, Gygi SR, et al. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA. 2002;8:426–439.
- Fica SM, Oubridge C, Wilkinson ME, et al. A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science. 2019;363:710–714.
- MacRae AJ, Mayerle M, Hrabeta-Robinson E, et al. Prp8 positioning of U5 snRNA is linked to 5ʹ splice site recognition. RNA. 2018;24:769–777.
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612.
- Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25.
- Mitternacht S. FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Res. 2016;5:189.