
ABSTRACT
Background: Vascular endothelial cell dysfunction, characterized by cell apoptosis and migration, plays a crucial role in ischaemia/reperfusion (I/R) injury, a common aspect of cardiovascular diseases. Recent studies have suggested that non-coding RNAs, such as circular RNAs (circRNA), play a role in cell dysfunction in I/R injury, although the detailed mechanism is unclear.
Methods: Human umbilical vein endothelial cells (HUVECs) were used for in vitro I/R model. Protein expression was detected by western blotting (WB) and immunocytochemistry. The CRISPR/Cas9 system, WB, cell viability assays, Hoechst staining and a 3D migration model were used to explore functional changes. RNA expression was evaluated using quantitative real-time PCR and a FISH assay combined with lentivirus transfection regulating circRNAs and miRNAs. A mouse myocardial I/R model using C57 mice was established to confirm the in vitro findings.
Results: In HUVECs, I/R induced a significant time-dependent decrease in HECTD1 associated with an approximately 45% decrease in cell viability and increases in cell apoptosis and migration, which were attenuated by HECTD1 overexpression. I/R-induced upregulation of endoplasmic reticulum stress was also attenuated HECTD1 overexpression. Moreover, miR-143 mimics inhibited HECTD1 expression, which was restored by circDLGAP4 overexpression, providing insight as to the molecular mechanism of I/R-induced HECTD1 in endothelial cell dysfunction.
Conclusion: Our results suggest a critical role for circDLGAP4 and HECTD1 in endothelial cell dysfunction induced by I/R, providing novel insight into potential therapeutic targets for the treatment of myocardial ischaemia.
Introduction
Cardiovascular disease is the leading cause of mortality in advanced and industrialized countries, far exceeding cancer and other diseases [Citation1,Citation2]. Ischaemia/reperfusion (I/R) injury is an important aspect of cardiovascular disease. Myocardial I/R injury is a major potential threat in the surgical treatment of coronary heart disease and is thus a clinical problem that should be solved imminently [Citation3,Citation4]. Vascular endothelial cell dysfunction plays a crucial role in I/R injury, which is characterized by the overproduction of inflammatory factors such as cytokines and chemokines [Citation5,Citation6] or a change in cell migration and apoptosis [Citation5,Citation7]. HECTD1, which encodes a novel protein homologous to the E6-AP C-terminal (HECT) domain-containing E3 Ub ligase, plays an important role in the cell apoptosis and migration in endothelial cells [Citation8] and macrophages [Citation9]. Whether HECTD1 is involved in I/R-induced endothelial cell dysfunction remains unclear.
Tissue-specific circular RNAs (circRNAs) are involved in a variety of disease processes [Citation10–Citation12]. Regarding their resistance to RNA exonucleases or RNase R, circRNAs are more stable than linear RNAs, making them suitable targets for clinical intervention [Citation13]. A recent study found that the circRNA hsa_circ_0016347 promotes proliferation, invasion and metastasis in osteosarcoma cells [Citation14]. A recent study from our laboratory suggested that the circRNA DLGAP4 regulated the endothelial-mesenchymal transition via miR-143 [Citation15], which contains the binding site for Hectd1. However, there have been few studies on the dysfunction of endothelial cells caused by circRNAs during I/R. Therefore, exploring the roles of circRNAs and their underlying mechanisms in I/R-induced endothelial cell dysfunction will reveal new perspectives on myocardial inflammation and will provide new therapeutic strategies for relieving I/R-induced myocardial inflammation. Such exploration will also enable the reduction of treatment-related complications and provide new targets for clinical treatment, supporting the development of ideal therapeutic drugs.
Materials and methods
Reagents
Foetal bovine serum (FBS), normal goat serum, Dulbecco’s modified Eagle’s medium (DMEM; #1200–046), and 10X MEM (11430–030) were all obtained from Life Technologies. Amphotericin B (BP2645) and GlutaMAX Supplement (35050–061) were omycin (15140–122) was obtained from Fisher Scientific. PureCol type I bovine collagen (3 mg/mL) was obtained from Advanced Biomatrix. The antibodies for HECTD1 (SC-134976, rabbit), Bax (CST-2772, rabbit), Bcl2l1 (CST-2764, rabbit), ERN1 (CST-3294, rabbit), ATF6, and GAPDH (SC-32233, mouse) were obtained from Santa Cruz Biotechnology. UltraCruz Transfection Reagent (SC-395739) for CRISPR/Cas9 Activation Plasmids was purchased from Santa Cruz Biotechnology, Inc. HUVECs were purchased from Genomeditech.
In vitro cell-simulated I/R model
The simulated I/R model was developed with a modified version of a method described previously [Citation6,Citation7,Citation16]. First, normal culture medium was replaced with serum-free DMEM before the start of the experiment. Following pregassing with 95% N2 and 5% CO2 for a minimum of 5 min, ischaemic buffer (1 mM NaH2PO4, 24 mM NaHCO3, 2.5 mM CaCl2, 118 mM NaCl, 16 mM KCl, 0.5 mM sodium EDTA, and 20 mM sodium lactate, pH 6.8, 37°C) was added to the cells, which were then placed in a sealed chamber containing deoxygenation reagent, leading to the consumption of O2 and the production of CO2. This AnaeroPack system created near-anaerobic conditions, with an O2 concentration of < 1% and a CO2 concentration of approximately 5% following 1 h of incubation at 37°C. To directly determine the efficacy of the equilibration system, the PO2 of the medium was measured directly in a few of the experiments using a phosphorescence decay method. The actual supernatant PO2 value was 4.5 ± 0.3 mm Hg (mean ± SEM). Cells in a 24-well plate were treated with the ischaemic buffer solution (1 mL/well) for 2 h before being incubated in glucose-containing DMEM at 37°C in 95% O2 and 5% CO2 (reperfusion) for different times.
Surgical preparation of myocardial I/R
Acute myocardial ischaemia and reperfusion were performed in C57 mice as briefly described. First, mice were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg). Then, mice were ventilated via tracheal intubation on a Harvard rodent respirator (tidal volume 7.2 µl/g, body mass; respiratory rate 200 breaths per min). The body temperature was maintained constant between 36.8 and 37°C, owing to the thermo-regulated surgical table connected to a rectal probe. The chest was opened by a left lateral thoracotomy and a reversible coronary artery snare occluder was placed around the left coronary artery. All animals underwent an ischaemic insult of 30 min. Reperfusion was achieved by loosening the knot for different times. The sham-operated group (SHAM) underwent a sham operation without coronary artery ligation.
Western blotting
Western blotting assays were performed as previously described [Citation9] to determine cellular protein levels.
Real-time quantitative PCR
The expression of circDLGAP4, miR-143, and hectd1 mRNA (Table S1) was determined by real-time quantitative PCR (qRT-PCR) as previously described [Citation17].
Targeted gene editing with CRISPR/Cas9 technology
A HECTD1 CRISPR/Cas9 overexpression plasmid was purchased from Santa Cruz Biotechnology, and transfection was performed according to the manufacturer’s instructions.
Lentiviral transduction of endothelial cells
Endothelial cells were transduced with Lv-circDLGAP4 and Lv-GFP lentiviruses (Hanbio, Inc., Shanghai, CN) as previously described [Citation5]. Briefly, endothelial cells (P3–5) were cultured in a 24-well plate (1 × 104 cells/well) in 10% FBS in DMEM for 24 h. The medium was then replaced with 1 mL of fresh medium with 8 µg/mL polybrene. Then, 50 μl of lentivirus solution (107 IU/mL) was added to each well, and the plates were incubated at 37°C and 5% CO2 for 24 h. Following incubation, the treatment medium was replaced with fresh 10% FBS in DMEM, and the cells were cultured at 37°C and 5% CO2 until >50% confluence was reached. The transduced cells were selected using puromycin by replacing the medium with 10 µg/mL puromycin in 10% FBS in DMEM and culturing the cells at 37°C and 5% CO2 for 24 h. The cells were subsequently washed twice with fresh 10% FBS in DMEM. Pure and transduced endothelial cell cultures were expanded and stored in liquid nitrogen as previously described.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [Citation6,Citation9].
FISH
Endothelial cells were seeded on coverslips that were pretreated with 1 × poly-L-lysine at a density of 1 × 104 cells/well in a 24-well plate. After experimental treatment, the cells were washed twice with cold 1× DEPC PBS, fixed in 4% paraformaldehyde in 1× DEPC PBS for 20 min at room temperature, permeabilized with 0.25% Triton X-100 for 15 min and prehybridized in hybridization solution for 1 h at 37°C. The cells were then incubated with the labelled probe (Table S1) in hybridization solution at 37°C overnight. The following day, the coverslips were washed and incubated in blocking buffer for 1 h at room temperature. The samples were then incubated with primary antibodies (anti-digoxigenin conjugated to horseradish peroxidase or FITC) at 4°C overnight. The next day, the cells were incubated with the appropriate fluorescent secondary antibodies (cyanine 5) and mounted with DAPI [Citation18]. A fluorescence microscope was used to capture the cell images.
CCK-8 assay
Cell viability was measured using a CCK-8 assay (Dojindo, Tokyo, Japan) following the manufacturer’s protocol.
Hoechst staining
Cells were fixed in 4% paraformaldehyde and then stained with 5 μM Hoechst 33,324 (Invitrogen) for 15 min at room temperature. A fluorescence microscope was used to capture cell images after the cells were washed twice with PBS. The number of apoptotic cells was counted using ImageJ software (NIH).
Bromodeoxyuridine labelling
Cells were prepared on 1× poly-L-lysine-coated glass cover slips. Bromodeoxyuridine (BrdU; 10 µM) (Yeasen, 40204ES60) in Dulbecco’s PBS was added 4 h before fixation. After fixing in 4% PFA at 4°C overnight, the cells were denatured in 2 N HCl at room temperature for 30 min and then rinsed in 0.1 M borate buffer (pH 8.0, 300 μL/well) for 10 min. After incubation in blocking solution (PBS containing 10% normal goat serum and 0.3% Triton X-100) for 1 h, the cells were treated with a mouse anti-BrdU antibody (1:200; SC-32,323, Santa Cruz Biotechnology) overnight at 4°C. After being washed with PBS, the cells were incubated with donkey anti-mouse secondary antibodies conjugated to Alexa Fluor 576 for 2 h at room temperature. The cells were then washed three times in PBS and mounted using mounting solution (ProLong Gold antifade reagent with DAPI; P36931, Life Technologies). The slides were imaged using a fluorescence microscope. Five fields per well were randomly selected for cell counting.
In vitro scratch assay
An in vitro scratch assay was used to evaluate cell migration in a 2D culture system as previously described [Citation5–Citation7]. Digital images of the cell gaps were captured at different time points, and the gap widths were quantitatively evaluated using ImageJ software.
Nested-matrix model and cell migration assay
A 3D migration model that can simulate the in vivo environment better than other methods was used, as described previously, with some modifications [Citation9,Citation17]. The number of cells in each field that had migrated from the nested matrix and the maximum migration distance per field were averaged.
Ethics statement
All animal procedures were performed in strict accordance with the ARRIVE guidelines, and the animal protocols were approved by the Institutional Animal Care and Use Committee of Southeast University.
Statistics
The data are presented as the means ± SEM. Unpaired numerical data were compared using an unpaired t-test (two groups) or analysis of variance (ANOVA; more than two groups) with SigmaPlot 11.0. Tukey’s test was used for post-hoc comparisons. A P-value of P < 0.05 was regarded as statistically significant.
Results
I/R mediates a decline in HECTD1 in endothelial cells
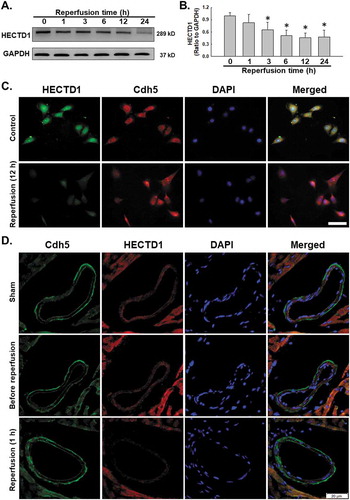
It has been reported that HECTD1 is involved in the regulation of the endothelial-mesenchymal transition and cell metastasis in the processes of fibrosis and tumorigenesis [Citation19], indicating the role of HECTD1 in endothelial cell dysfunction. Therefore, we first examined the role of HECTD1 in endothelial cells subjected to I/R, in which the exposure of endothelial cells to reperfusion resulted in a significant time-dependent decrease in cellular HECTD1 expression (–) associated with an increase in apoptosis (Supplementary Fig. S1), as confirmed using immunocytochemistry (). To further validate our in vitro findings for HECTD1, a mouse acute I/R model was employed. Immunohistochemistry revealed that HECTD1 expression decreased in mouse cardiac vascular tissue after acute reperfusion. The colocalization of HECTD1 with the vascular endothelial cell marker VE-cad also decreased (), confirming the previous in vitro findings for HECTD1 in endothelial cells.
Figure 1. I/R decreased HECTD1 expression. (A) Representative western blots showing that I/R induced HECTD1 expression in a time-dependent manner in HUVECs. (B) Densitometric analyses of HECTD1 levels from five independent experiments; *P < 0.05 compared with the 0 h group. (C) Representative images of immunocytochemical staining showing that I/R induced HECTD1 expression in HUVECs. Scale bar, 100 μm. (D) Representative images of immunohistochemical staining showing that Cdh5 colocalization with HECTD1 decreased in mouse heart vessels after I/R. Scale bar, 20 μm.
HECTD1 is involved in endothelial cell dysfunction stimulated by I/R
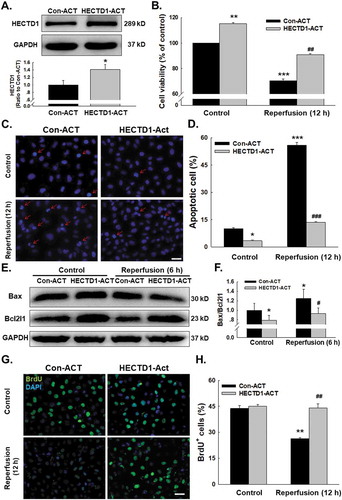
Abnormal angiogenesis, which is characterized by migratory and proliferative phenotypes and the differentiation of endothelial cells into an angiogenic phenotype, is an important aspect of endothelial cell dysfunction and is a feature of ischaemic heart disease. Apoptosis of endothelial cells is the initial step in the angiogenesis and regression of neovessels [Citation5,Citation7]. To determine whether HECTD1 was involved in endothelial cell viability, the CRISPR/Cas9 system was applied. As shown in , transfection with the HECTD1 CRISPR activation plasmid (ACT) upregulated the expression of HECTD1 in endothelial cells. The reperfusion-induced decline in endothelial cell viability was abolished by HECTD1 ACT (). Because the Bax/Bcl2l1 pathway plays a crucial role in I/R-mediated apoptosis, we next examined the involvement of this pathway in HECTD1-mediated endothelial cell apoptosis using Hoechst 33342, a nuclear dye that specifically stains nuclei. As shown in –, endothelial cells in the control group were characterized by regular and round nuclei. In contrast, condensation and fragmentation of nuclei characteristic of apoptotic cells were evident in endothelial cells subjected to reperfusion for 12 h. Overexpression of HECTD1 significantly ameliorated I/R-induced cell death. This finding was confirmed via western blotting, which showed that I/R stimulation caused a transient increase in the Bax/Bcl2l1 expression ratio in endothelial cells, with a peak response at 6 h; this effect was reversed by HECTD1 ACT (–). The same effect of HECTD1 ACT was observed in cleaved caspase-9 (Supplementary Fig. S2A-B) To further elucidate the role of HECTD1 in I/R-induced endothelial cell apoptosis, BrdU, a synthetic nucleoside used to detect cellular proliferation, was employed. The number of BrdU-positive cells was significantly reduced following exposure to I/R, whereas this effect was attenuated by transfection with HECTD1 ACT (–). Interestingly, in the control group, HECTD1-ACT induced increased cell viability, decreased cell apoptosis, and no change in cell proliferation, indicating that the increase in cell viability may be due to the change in cell apoptosis but not cell proliferation.
Figure 2. Involvement of HECTD1 in cell viability changes induced by I/R in HUVECs. (A) Representative western blot and densitometric analyses showing the efficacy of HECTD1 CRISPR ACT in increasing the level of the HECTD1 protein in HUVECs. *P < 0.05 vs. the control group, n = 5. (B) CCK-8 assay showing that HECTD1 ACT attenuated the I/R-induced decrease in HUVEC viability. **P < 0.01 vs. the control group, **P < 0.001 vs. the control group; ##P < 0.01 vs. the I/R group, n = 5. (C) Representative images of Hoechst 33342 staining of HUVECs after I/R. (D) Hoechst 33342 staining demonstrating that apoptosis induced by I/R was attenuated by upregulation of HECTD1 expression with ACT in HUVECs; n = 5; *P < 0.05 vs. the control group, ***P < 0.001 vs. the control group; ###P < 0.001 vs. the I/R group. (E) Representative western blot showing the effect of specific upregulation of HECTD1 expression with ACT on I/R-induced apoptosis marker expression. (F) Densitometric analyses of five separate experiments suggesting that the I/R-induced changes in the Bax/Bcl2l1 ratio are attenuated by HECTD1 ACT. *P < 0.05 vs. the control group; #P < 0.05 vs. the I/R group. (G) Representative images of the BrdU labelling assay in HUVECs after I/R. (H) BrdU labelling assay results demonstrating that the decrease in proliferation induced by I/R was attenuated by HECTD1 ACT; n = 5; **P < 0.01 vs. the control group; ##P < 0.01 vs. the I/R group.
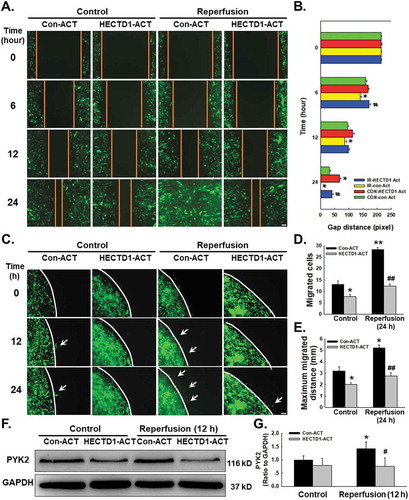
Endothelial cell migration is a critical component of angiogenesis. Functional assays of cell migration were conducted to determine how downregulation of HECTD1 expression is associated with I/R-induced endothelial cell migration (Supplementary Fig. S3A). As shown in –, the results of the scratch assay revealed an increase in endothelial cell migration following I/R that was abolished by HECTD1 ACT. Consistent results were obtained with the nested-matrix model of the cell migration assay, as shown in –. To further understand the mechanism of endothelial cell migration, we subsequently measured the expression of PYK2, a migration-related protein (Supplementary Fig. S3B-C). As shown in –, I/R-induced upregulation of PYK2 expression was eliminated by HECTD1 ACT.
Figure 3. Involvement of HECTD1 in cell migration changes induced by I/R in HUVECs. (A) Representative images from a scratch assay showing that the I/R-induced increase in cell migration was attenuated by HECTD1 ACT. (B) Quantification of the scratch gap distances from six independent experiments; *P < 0.05 vs. the corresponding time point in the control group; #P < 0.05 vs. the corresponding time point in the I/R group. (C) Representative images from a 3D assay showing that the I/R-induced increase in cell migration was attenuated by HECTD1 ACT. Quantification of the migrated cell number (D) and maximum migrated distance (E) from six independent experiments; *P < 0.05 vs. the corresponding time point in the control group; **P < 0.01 vs. the corresponding time point in the control group; ##P < 0.01 vs. the corresponding time point in the I/R group. (F) Representative western blot showing that the I/R-induced increase in PYK2 expression was attenuated by HECTD1 ACT. (G) Densitometric analysis of PYK2 expression from 5 experiments; *P < 0.05 vs. the control group; #P < 0.05 vs. the I/R group.
Involvement of HECTD1 in I/R-mediated endoplasmic reticulum stress in endothelial cells
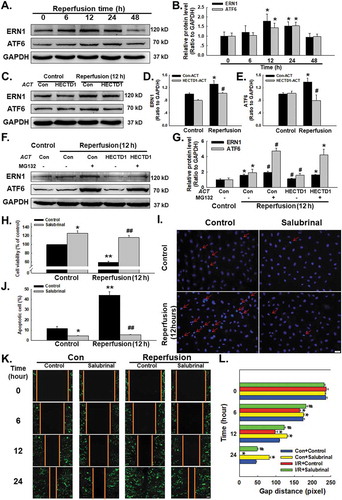
ERstress that disrupts ER function can occur in response to a wide variety of cellular stressors that lead to the accumulation of unfolded and misfolded proteins in the ER and trigger the unfolded protein response, which has been associated with cardiovascular diseases such as I/R injury [Citation20]. Whether HECTD1 mediated I/R induces endothelial dysfunction via ER stress deserves further study. As shown in –, the exposure of endothelial cells to I/R significantly upregulated the expression of the ER stress markers ERN1 and ATF6 in a time-dependent manner, as well as upregulated BIP, eIF2α and CHOP (Supplementary Fig. S4A-B). Moreover, transfection of cells with HECTD1 ACT markedly inhibited the elevation in ERN1 and ATF6 expression induced by 12 h of reperfusion (-). Because HECTD1 encodes a novel protein homologous to the E6-AP C-terminal (HECT) domain-containing E3 ubiquitin (Ub) ligase, we were interested in whether HECTD1 regulates ER stress through the ubiquitination pathway. We used a pharmacological strategy to further explore the effect of HECTD1 and found that the ubiquitination blocker MG132 could relieve the inhibition of ER stress induced by HECTD1 overexpression (-). These results suggest that HECTD1 is involved in I/R-stimulated endothelial cell dysfunction associated with ER stress.
Figure 4. Involvement of HECTD1 in I/R-induced ER stress in HUVECs. (A) Representative western blot showing that I/R increased the expression of ERN1 and ATF6. (B) Densitometric analysis of ERN1 and ATF6 protein expression levels from five independent experiments; *P < 0.05 vs. the control group. (C) Representative western blot depicting the effect of HECTD1 ACT on ERN1 and ATF6 protein expression. Densitometric analysis of ERN1 (D) and ATF6 (E) protein expression levels from five independent experiments; *P < 0.05 vs. the control group; #P < 0.05 vs. the I/R group. (F) Representative western blot showing that MG132 abolished the effects of HECTD1 ACT on I/R-induced ERN and ATF6 changes. (G) Densitometric analysis of ERN1 and ATF6 protein expression levels from five independent experiments; *P < 0.05 vs. the control group, #P < 0.05 vs. the I/R group. (H) CCK-8 assay results showing that salubrinal attenuated the I/R-induced decrease in HUVEC cell viability. *P < 0.05 vs. the control group, **P < 0.01 vs. the control group; ##P < 0.01 vs. the I/R group, n = 5. (I) Representative images of Hoechst 33342 staining of HUVECs after I/R. (J) Hoechst 33342 staining demonstrating that apoptosis induced by I/R was attenuated by salubrinal in HUVECs; *P < 0.05 vs. the control group, **P < 0.01 vs. the control group; ##P < 0.01 vs. the I/R group, n = 5. (K) Representative images from a scratch assay showing that the I/R-induced increase in cell migration was attenuated by salubrinal. (L) Quantification of the scratch gap distances from six independent experiments; *P < 0.05 vs. the corresponding time point in the control group; #P < 0.05 vs. the corresponding time point in the I/R group.
Endoplasmic reticulum stress plays a critical role in I/R-mediated endothelial cell dysfunction
When ER stress is prolonged or severe, it induces apoptosis to eliminate unhealthy cells, contributing to the processes of cardiovascular diseases [Citation21]. To further understand the role of ER stress in I/R, a specific ER stress blocker, salubrinal (20 ng/m), was applied to pretreat endothelial cells for 2 h, which reversed the decrease in cell viability induced by 12 h of exposure to I/R (). Hoechst 33,342 staining illustrated that the percentage of apoptotic cells declined sharply with salubrinal treatment, confirming that ER stress plays a major role in the apoptosis induced by I/R (-). In addition, the increased migration induced by I/R in endothelial cells was abolished by pretreatment of cells with salubrinal, indicating the involvement of ER stress in this process (-).
Expression profile of circdlgap4 in endothelial cells after exposure to I/R
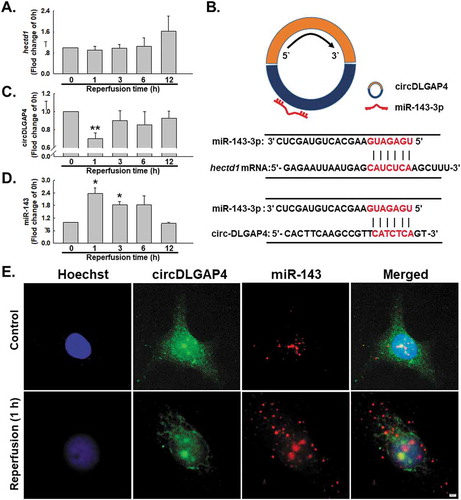
After confirming the downstream mechanism of HECTD1 in I/R-induced endothelial dysfunction, it is reasonable to determine the regulation mechanism of HECTD1. To our surprise, hectd1 mRNA did not change significantly after I/R exposure (), indicating nontranscriptional regulation may be involved in the induction of HECTD1 by I/R. Previous data from our laboratory revealed the upstream regulation mechanism of HECTD1 via noncoding RNA–circRNA, guiding us to perform a bioinformatics analysis, in which the 3ʹ UTR region of hectd1 was shown to have a binding site for miR-143, while miR-143 and circDLGAP4 also have binding sites (). To further elucidate the possible regulatory mechanism, the expression of both circDLGAP4 and miR-143 after I/R exposure were detected, revealing that there was a significant decrease in circDLGAP4 in endothelial cells subjected to reperfusion for 1 h, while miR-143 showed a 2.5-fold increase (-). The expression of circDLGAP4 and miR-143 in I/R-stimulated endothelial cells was confirmed with the FISH assay ().
Figure 5. I/R induces changes in circDLGAP4 in HUVECs. (A) As shown in the qRT-PCR analysis, I/R had no effect on hectd1 expression (n = 5). **P < 0.01 vs. the 0 h group. (B) Bioinformatics analysis showing that circDLGAP4 contains one site complementary to miR-143-3p and that the hectd1 3ʹ UTR contains one miR-143-3p binding site. (C) As shown in the qRT-PCR analysis, I/R decreased circDLGAP4 expression (n = 5). **P < 0.01 vs. the 0 h group. (D) As shown in the qRT-PCR analysis, I/R increased miR-143 expression (n = 5). **P < 0.01 vs. the 0 h group. (E) FISH assay results showing circDLGAP4 and miR-143-3p expression in HUVECs after I/R exposure.
CircDLGAP4/miR-143 regulates the protein expression of HECTD1
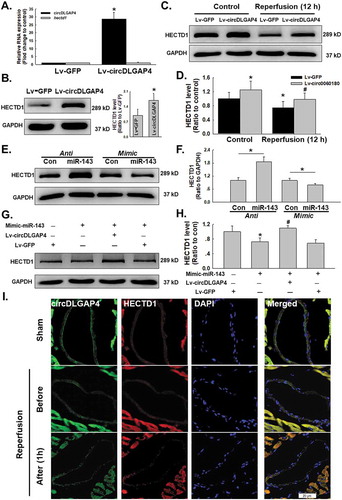
Having confirmed the expression pattern of circDLGAP4 and the HECTD1 protein, we next determined whether circDLGAP4 regulates endothelial dysfunction induced by I/R via HECTD1. The circDLGAP4 lentivirus was transfected into endothelial cells, which increased the expression of circDLGAP4 in endothelial cells, while there was no clear change in the hectd1 mRNA level (). Interestingly, the protein expression of HECTD1 was markedly upregulated after the overexpression of circDLGAP4 (). Moreover, circDLGAP4 overexpression in endothelial cells attenuated the decrease in HECTD1 protein expression induced by 12 h of reperfusion (–). Next, an inhibitor (anti-miR-143) and mimic (mimic-miR-143) of miR-143 were employed to explore the detailed mechanism. The results showed that the protein expression of HECTD1 was significantly increased after treatment with anti-miR-143, whereas mimic-miR-143 suppressed HECTD1 protein expression (–). To further demonstrate that the effect of miR-143 on circDLGAP4 regulated HECTD1 protein expression, Lv-circDLGAP4 and mimic-miR-143 were co-transfected into endothelial cells. As shown in –, the negative regulation of mimic-miR-143 on HECTD1 protein was abolished by the overexpression of circDLGAP4, confirming that circDLGAP4 regulates HECTD1 protein expression via miR-143. On the other hand, regulation of miR-143 did not change the circDLGAP4 expression (Fig. S7A). Moreover, salubrinal has no effect on circDLGAP4 and miR-143 expression (Fig. S7B), suggesting that the circDLGAP4/miR-143 pathway was upstream of ER stress. To validate our in vitro findings, FISH combined immunohistochemical staining were applied to the hearts of mice after I/R, and the results showed that HECTD1 co-localized with circDLGAP4 in the heart vessels of sham mice. Two hours of ischaemia did not affect the expression of HECTD1 and circDLGAP4, while 1 hour of reperfusion decreased HECTD1 expression and increased circDLGAP4 expression ().
Figure 6. Involvement of circDLGAP4 in I/R-induced changes in HECTD1 in HUVECs. (A) As shown in the qRT-PCR analysis, overexpression of circDLGAP4 had no effect on hectd1 expression (n = 5). **P < 0.01 vs. the 0 h group. (B) As shown in the western blot assay, overexpression of circDLGAP4 increased HECTD1 protein levels (n = 5). **P < 0.01 vs. the 0 h group. (C) Representative western blot showing that Lv-circDLGAP4 attenuated the I/R-induced decrease in HECTD1. (D) Densitometric analysis of HECTD1 protein expression levels from five independent experiments; *P < 0.05 vs. the control group; #P < 0.05 vs. the I/R group. (E) Representative western blot showing the effects of anti-miR-143 and mimic-miR-143 transfection on HECTD1 levels in HUVECs. (F) Densitometric analysis of HECTD1 protein expression levels from five independent experiments; *P < 0.05 vs. the control group. (G) Representative western blot showing the effects of cotransfection of mimic-miR-143 and Lv-circDLGAP4 on HECTD1 protein expression in HUVECs. (H) Densitometric analysis of HECTD1 protein expression levels from five independent experiments; *P < 0.05 vs. the control group; #P < 0.05 vs. the mimic-miR-143 group. (I) Representative images of FISH combined with immunohistochemical staining showing that HECTD1 colocalized with circDLGAP4 in the heart vessels of mice after I/R.
CircDLGAP4 is involved in endothelial cell dysfunction caused by I/R exposure
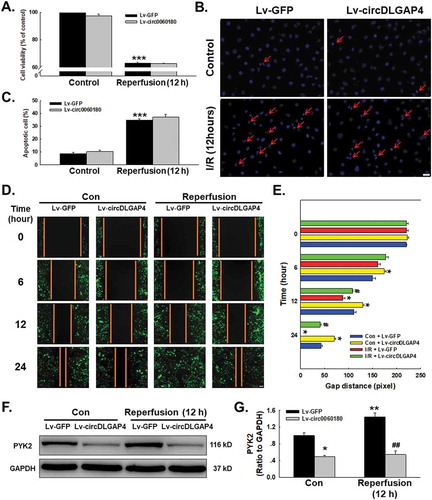
Due to the multiple downstream molecular targets of circRNAs, and because we confirmed the regulatory mechanism of circDLGAP4 on HECTD1, we next determined whether circDLGAP4 also regulates I/R-induced endothelial cell dysfunction. What intrigued us was that transfection of Lv-circDLGAP4 into endothelial cells did not attenuate the decrease in cell viability induced by I/R (). This consequence was consistent with the results of Hoechst 33342 staining, which revealed Lv-circDLGAP4 had no effect on apoptosis in endothelial cells subjected to reperfusion for 12 h (-). On the other hand, circDLGAP4 lentivirus transfection attenuated the I/R-induced increase in endothelial cell migration (-); this finding was confirmed by the fact that the I/R-induced upregulation of the expression of the migration-related protein PYK2 was abolished by the overexpression of circDLGAP4 (-). This finding suggested that there may be different mechanisms involved in cell apoptosis and migration in terms of endothelial dysfunction, indicating that I/R-induced endothelial dysfunction may be complex.
Figure 7. Involvement of circDLGAP4 in I/R-induced cell functional changes in HUVECs. (A) CCK-8 assay showing that Lv-circDLGAP4 had no effect on the I/R-induced decrease in HUVEC viability. ***P < 0.001 vs. the control group, n = 5. (B) Representative images of Hoechst 33342 staining of HUVECs after I/R. (C) Hoechst 33342 staining demonstrating that apoptosis induced by I/R was not affected by Lv-circDLGAP4 in HUVECs; ***P < 0.001 vs. the control group, n = 5. (D) Representative images of a scratch assay showing that the I/R-induced increase in cell migration was attenuated by Lv-circDLGAP4. (E) Quantification of the scratch gap distances from six independent experiments; *P < 0.05 vs. the corresponding time point in the control group; #p < 0.05 vs. the corresponding time point in the I/R group. (F) Representative western blot showing that the I/R-induced increase in PYK2 expression was attenuated by Lv-circDLGAP4. (G) Densitometric analysis of PYK2 expression from 5 experiments; *p < 0.05 vs. the control group, **p < 0.01 vs. the control group; ##p < 0.01 vs. the I/R group.
Discussion
Myocardial I/R injury is a major cause of morbidity and mortality in several important clinical scenarios, including cardiac arrest, percutaneous coronary artery intervention, acute myocardial infarction, and cardiac surgery [Citation22], and can lead to a battery of damage that includes arrhythmia, myocardial enzyme release and intramyocardial haemorrhage. Several mechanisms have been proposed to mediate reperfusion injury, such as the failure of free radical scavenging, calcium overload, leukocyte action, and microvasculature injury, which contribute to the pathogenesis of I/R [Citation22]. Moreover, the endothelial cell barrier damage involved in abnormalities of the microvasculature plays a critical role in inflammatory responses. It has been reported that patients with ischaemic myocardial disease may benefit from an enhancement of angiogenic processes and the establishment of collateral circulation. However, the direct effect of I/R on endothelial cells has been less extensively researched than that on cardiomyocytes. The aim of the current study was to investigate the role of HECTD1 in endothelial cell dysfunction after I/R, such as cell apoptosis and migration.
Endothelial cell dysfunction is involved in angiogenesis via different mechanisms, such as oxidative stress, inflammation, ER stress, ubiquitination and autophagy [Citation5,Citation23,Citation24]; these mechanisms affect endothelial cell activation, apoptosis, and migration [Citation7]. Mounting evidence has suggested the role of oxidant and oxygen radical formation, which is a critical central mechanism of reperfusion injury, in these processes [Citation24,Citation25]. However, the mechanisms underlying I/R-induced endothelial cell dysfunction remain poorly understood. HECTD1 plays an important role in the Ub-proteasome system [Citation9]. Furthermore, HECTD1 is also important for the regulation of cell migration via PIPKIγ90 [Citation19] and is involved in neural tube defects and abnormal cranial mesenchyme via Hsp90 [Citation26]. In addition, HECTD1 expression is upregulated in fibroblasts obtained from primary sites, lymph nodes and the bone marrow of patients with breast cancer [Citation27]. Previous research by our group revealed that HECTD1 participates in the regulation of pulmonary fibrosis through ubiquitination [Citation9] and plays an important role in the endothelial-mesenchymal transition (EndoMT) [Citation8], indicating that HECTD1 might be involved in angiogenic processes in response to I/R. Interestingly, I/R induced endothelial cell apoptosis and migration, which seem to be opposing processed. Actually, endothelial cell activation, proliferation, migration, and apoptosis are involved in angiogenesis induced by I/R, which may be due to the role of these functions at different time intervals after I/R. A previous study from our laboratory suggested that changes in cell viability occurred earlier than those in cell migration [Citation7]. In the current study, HECTD1 was found to inhibit the endothelial cell apoptosis and migration induced by I/R, suggesting a key role for HECTD1 in inflammatory responses.
Various biological events, such as autophagy, ER stress and ubiquitination, are involved in endothelial cell dysfunction [Citation5,Citation28]. ER stress is an evolutionarily conserved cell stress response that has been associated with numerous diseases, including cardiovascular diseases, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, diabetes, renal failure, fatty liver disease, irritable bowel syndrome, and many others [Citation21,Citation29–Citation35]. ER stress plays a critical role in the development of pathological myocardial inflammation and heart failure. ER accumulation of unfolded proteins is detected by at least three ER transmembrane sensors, including PERK (protein kinase-like kinase), ATF6, and IRE1, which activate the PERK-eIF2α-ATF4-CHOP, pro-ATF6 to cleaved ATF6 (cATF6) and CHOP, and IRE1-spliced XBP1 (sXBP1) signalling pathways, respectively [Citation20,Citation36]. This activation results in the upregulation of genes encoding ER chaperones and ER-associated degradation components [Citation37]. When ER stress is prolonged or severe, it induces apoptosis to eliminate unhealthy cells, contributing to the process of myocardial inflammation [Citation38]. The current findings suggested that ER stress mediated apoptosis induced by I/R, indicating that prolonged ER stress plays a pathological role but does not have a protective effect.
Ubiquitination is involved in the regulation of apoptosis, gene expression, transcription, and inflammatory immunity and is closely linked to the pathogenesis of tumours and cardiovascular diseases. Studies have shown that HECTD1 acts as an E3 ubiquitinated ligase that affects pulmonary fibrosis through ubiquitination [Citation9]; it also regulates the migration of brain interstitial cells by regulating the secretion of Hsp90 [Citation26]. Another study has revealed a major role for HECTD1 in the regulation of the endothelial-mesenchymal transition and cell metastasis [Citation19]. Therefore, HECTD1 is suspected to be involved in the regulation of endothelial cell dysfunction caused by I/R. The current study suggested that MG132, a proteasome inhibitor of ubiquitination, significantly reduced the HECTD1-mediated inhibition of ER stress. Moreover, although Ub and Ub-k48 expression was not significantly altered in endothelial cells stimulated by I/R, Ub-k63 expression increased upon exposure to I/R (Supplementary Fig. S5). Therefore, HECTD1 may affect ER stress through its own characteristic E3 Ub ligase activity, or it may indirectly affect ER stress through ubiquitination of a downstream protein; this conjecture needs further confirmation. Furthermore, previous research by our group confirmed that I/R stimulation can induce autophagy in endothelial cells [Citation5]. Therefore, in this study, CRISPR/Cas9 technology was applied to explore whether HECTD1 is also involved in the regulation of autophagy. As shown in Supplementary Figure S6, overexpression of HECTD1 had no distinct effect on the expression of autophagy-related proteins. These results reveal that HECTD1 regulates endothelial cell dysfunction through ER stress, but not through autophagy.
Many of the functions of circRNAs have been elucidated over the last few years. CircRNAs can act as gene expression regulators via different regulatory mechanisms [Citation12]. CircRNAs are involved in the regulation of transcription and alternative splicing [Citation39]. They can also interact with RNA-binding proteins (RBPs) [Citation40], act as miRNA sponges [Citation41,Citation42], or even be translated into proteins [Citation43]. The present study found that miR-143 is the main sponge target of circDLGAP4. Previous studies have suggested that miR-143 was associated with acute ischaemic stroke (AIS) [Citation44] and endothelial-mesenchymal transition (EndoMT) [Citation15], indicating a role of miR-143 in endothelial dysfunction. The current study provides another example of miR-143 in disease, in which upregulation of miR-143 mediated endothelial cell dysfunction via inhibition of HECTD1. Interestingly, in contrast to that of circDLGAP4, the expression of another circRNA, hsa_circ_0031485, which is a byproduct of the splicing of its host gene hectd1, was not significantly altered by I/R (Supplementary Fig. S8). However, a different expression pattern caused by I/R stimulation does occur for certain circRNAs, which in turn affects downstream protein expression and cellular function [Citation12]. Another interesting phenomenon was that circDLGAP4 was involved only in the regulation of endothelial cell migration, without affecting cell viability and apoptosis. One explanation was that circRNAs not only affect the levels of many mRNAs in the nucleus but also adsorb miRNAs in the cytoplasm and directly interact with specific proteins to affect their expression at the transcriptional or posttranscriptional levels. There may be other targets regulated by circDLGAP4 that have a compensatory effect on the endothelial cell dysfunction influenced by the HECTD1 protein. Another possibility is that the multitargeting miRNAs of circDLGAP4 may cause different functional changes in endothelial cell. Moreover, circDLGAP4 significantly attenuates neurological deficits and decreases infarct areas and blood-brain barrier damage in a transient middle cerebral artery occlusion mouse stroke model [Citation15]. The observed differential expression of circRNAs in human tissues and diseases suggests that circRNAs might play important physiological and pathological roles.
In summary, our findings have determined that HECTD1 plays a vital role in apoptosis and cell migration in endothelial cells through ER stress in response to I/R. Furthermore, circDLGAP4 can regulate the expression of HECTD1 by sponging miR-143.
Conclusion
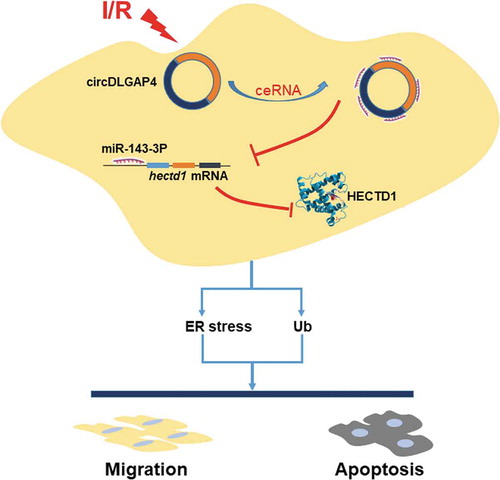
Our results suggest a critical role for circDLGAP4 and HECTD1 in endothelial cell dysfunction induced by I/R, providing novel insight into potential therapeutic targets for the treatment of myocardial ischaemia ().
Figure 8. Schematic diagram showing the mechanisms by which circDLGAP4/HECTD1 mediates I/R-induced endothelial dysfunction.
Author summary
Noncoding RNA-circRNA and its downstream molecules play a role in endothelial cell dysfunction induced by ischaemia/perfusion injury, which is commonly observed in several types of cardiovascular disease, providing novel insight into potential therapeutic targets for the treatment of myocardial ischaemia.
Supplemental Material
Download MS Word (3.2 MB)Acknowledgments
This study was partially supported by the resources and facilities of the core laboratory of the Medical School of Southeast University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322.
- Altamirano F, Wang ZV, Hill JA. Cardioprotection in ischaemia-reperfusion injury: novel mechanisms and clinical translation. J Physiol. 2015;593(17):3773–3788.
- Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100.
- Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59(11):969–978.
- Zhu T, Yao Q, Wang W, et al. iNOS induces vascular endothelial cell migration and apoptosis via autophagy in ischemia/reperfusion injury. Cell Physiol Biochem. 2016;38(4):1575–1588.
- Xie X, Zhu T, Chen L, et al. MCPIP1-induced autophagy mediates ischemia/reperfusion injury in endothelial cells via HMGB1 and CaSR. Sci Rep. 2018;8:1.
- Zhu T, Yao Q, Hu X, et al. The role of MCPIP1 in Ischemia/Reperfusion injury-induced HUVEC migration and apoptosis. Cell Physiol Biochem. 2015;37(2):577–591.
- Fang S, Guo H, Cheng Y, et al. circHECTD1 promotes the silica-induced pulmonary endothelial–mesenchymal transition via HECTD1. Cell Death Dis. 2018;9:3.
- Zhou Z, Jiang R, Yang X, et al. circRNA mediates silica-induced macrophage activation via HECTD1/ZC3H12A-dependent ubiquitination. Theranostics. 2018;8(2):575–592.
- Santer L, Bar C, Thum T. Circular RNAs: a novel class of functional RNA molecules with a therapeutic perspective. Mol Ther. 2019;27(8):1350–1363.
- Patop IL, Wust S, Kadener S. Past, present, and future of circRNAs. Embo J. 2019;38(16):e100836.
- Han B, Chao J, Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther. 2018;187:31–44.
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19(2):141–157.
- Jin H, Jin X, Zhang H, et al. Circular RNA hsa-circ-0016347 promotes proliferation, invasion and metastasis of osteosarcoma cells. Oncotarget. 2017;8(15):25571–25581.
- Bai Y, Zhang Y, Han B, et al. Circular RNA DLGAP4 ameliorates ischemic stroke outcomes by targeting miR-143 to regulate endothelial-mesenchymal transition associated with blood-brain barrier integrity. J Neurosci. 2018;38(1):32–50.
- Yan L, Zhu TB, Wang LS, et al. Inhibitory effect of hepatocyte growth factor on cardiomyocytes apoptosis is partly related to reduced calcium sensing receptor expression during a model of simulated ischemia/reperfusion. Mol Biol Rep. 2011;38(4):2695–2701.
- Yang X, Wang J, Zhou Z, et al. Silica-induced initiation of circular ZC3H4 RNA/ZC3H4 pathway promotes the pulmonary macrophage activation. Faseb J. 2018;32(6):3264–3277.
- Chaudhuri AD, Yelamanchili SV, Fox HS. Combined fluorescent in situ hybridization for detection of microRNAs and immunofluorescent labeling for cell-type markers. Front Cell Neurosci. 2013;7:160.
- Li X, Zhou Q, Sunkara M, et al. Ubiquitylation of phosphatidylinositol 4-phosphate 5-kinase type I by HECTD1 regulates focal adhesion dynamics and cell migration. J Cell Sci. 2013;126(12):2617–2628.
- Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107(9):1071–1082.
- Yao Y, Lu Q, Hu Z, et al. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat Commun. 2017;8(1):133.
- He X, Li S, Liu B, et al. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc Natl Acad Sci U S A. 2017;114(23):E4582–E4591.
- Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70(2):181–190.
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53(1):31–47.
- Wildhirt SM, Weismueller S, Schulze C, et al. Inducible nitric oxide synthase activation after ischemia/reperfusion contributes to myocardial dysfunction and extent of infarct size in rabbits: evidence for a late phase of nitric oxide-mediated reperfusion injury. Cardiovasc Res. 1999;43(3):698–711.
- Sarkar AA, Zohn IE. Hectd1 regulates intracellular localization and secretion of Hsp90 to control cellular behavior of the cranial mesenchyme. J Cell Biol. 2012;196(6):789–800.
- Del Valle PR, Milani C, Brentani MM, et al. Transcriptional profile of fibroblasts obtained from the primary site, lymph node and bone marrow of breast cancer patients. Genet Mol Biol. 2014;37(3):480–489.
- Kim PK, Hailey DW, Mullen RT, et al. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105(52):20567–20574.
- Chiang CK, Hsu SP, Wu CT, et al. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med. 2011;17(11–12:1295–1305.
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917.
- Aridor M. Visiting the ER: the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv Drug Deliv Rev. 2007;59(8):759–781.
- Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol. 2011;23(2):239–252.
- Lee AH, Scapa EF, Cohen DE, et al. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496.
- Fu HY, Sanada S, Matsuzaki T, et al. Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ Res. 2016;118(5):798–809.
- Hetz C, Martinon F, Rodriguez D, et al. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91(4):1219–1243.
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529.
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664.
- Fu HY, Okada K, Liao Y, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122(4):361–369.
- Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–264.
- Yang ZG, Awan FM, Du WW, et al. The circular RNA interacts with STAT3, increasing its nuclear translocation and wound repair by modulating Dnmt3a and miR-17 function. Mol Ther. 2017;25(9):2062–2074.
- Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453–461.
- Guo JU, Agarwal V, Guo H, et al. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15(7):409.
- Legnini I, Di Timoteo G, Rossi F, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66(1):22–37.e9.
- Tiedt S, Prestel M, Malik R, et al. RNA-seq identifies circulating miR-125a-5p, miR-125b-5p, and miR-143-3p as potential biomarkers for acute ischemic stroke. Circ Res. 2017;121(8):970–980.