
ABSTRACT
Enhancers are distal genomic elements critical for gene regulation and cell identify control during development and diseases. Many human cancers were found to associate with enhancer malfunction, due to genetic and epigenetic alterations, which in some cases directly drive tumour growth. Conventionally, enhancers are known to provide DNA binding motifs to recruit transcription factors (TFs) and to control target genes. However, recent progress found that most, if not all, active enhancers pervasively transcribe noncoding RNAs that are referred to as enhancer RNAs (eRNAs). Increasing evidence points to functional roles of at least a subset of eRNAs in gene regulation in both normal and cancer cells, adding new insights into the action mechanisms of enhancers. eRNA expression was observed to be widespread but also specific to tumour types and individual patients, serving as opportunities to exploit them as potential diagnosis markers or therapeutic targets. In this review, we discuss the brief history of eRNA research, their functional mechanisms and importance in cancer gene regulation, as well as their therapeutic and diagnostic values in cancer. We propose that further studies of eRNAs in cancer will offer a promising ‘eRNA targeted therapy’ for human cancer intervention.
1. Introduction
The development of high-throughput sequencing technologies in the last decade yielded exciting insights into genome and transcriptome regulation. One of the most unexpected findings is the widespread transcription of the human genome, with more than 85% capable of transcribing in different cell types [Citation1,Citation2]. Such pervasive transcription of genomic regions other than protein-coding genes generated tremendous non-coding RNA (ncRNA) species far more than previously recognized [Citation1,Citation2]. Among these, RNAs generated from enhancers (i.e. eRNAs) have attracted a particular interest due to their potential roles in mediating enhancer functions and gene transcription, and their frequent overlap with disease-associated noncoding risk loci [Citation3–Citation6].
As a major category of regulatory DNA elements, enhancers are commonly known to control target gene expression by forming spatial chromatin loops with target promoters [Citation7]. Besides the long-established functions in cell development, enhancers were increasingly realized to directly drive human diseases, including cancer [Citation8,Citation9]. A few recent studies have systematically studied the expression landscapes of eRNAs in large cohorts of human cancer samples, suggesting potentially broad roles of eRNAs in tumorigenesis [Citation6,Citation10]. In this review, we summarize the studies on eRNAs associated with tumorigenesis, their regulated expression, roles and mechanisms, and we aim to discuss the potential clinical utility of eRNAs in cancer diagnosis, prognosis and therapeutic intervention.
2. eRNAs mark an additional layer of enhancer function
Although individual studies of enhancer-derived RNAs using locus-specific approaches can date back to the early 1990s [Citation11], the global revelation of pervasive RNA polymerase II (Pol II) occupation and RNA transcription at active enhancers took place in 2010 [Citation12,Citation13]. Since then, mounting evidence demonstrated the wide existence and potential functions of eRNAs in different cell lineages and in response to various stimuli [Citation14–Citation21]. To date, an estimated ~65,000 enhancers were found to be transcriptionally active in the human genome across multiple tissues and cell types, generating an extremely large number of eRNAs with largely uncharted functions [Citation4,Citation16].
eRNAs can be either bidirectionally or unidirectionally transcribed from active enhancers, which are generally marked by histone modifications including acetylation of histone H3 lysine 27 (H3K27ac) and mono-methylation of H3K4 (H3K4me1), with length varying from several hundreds to thousands of nucleotides (nt), depending on both primary sequences and chromatin status, which have been discussed in several previous studies and reviews [Citation3–Citation5,Citation22,Citation23] []. The majority of eRNAs is found to be non-polyadenylated (~90% by estimation), having lower abundance and shorter half-lives as compared to mRNAs [Citation4]. These features make them less detectable by conventional oligo-dT-based RNA-seq or even ribo-depleted total RNA-seq [Citation3,Citation4,Citation12,Citation13,Citation22]. To identify a complete eRNA catalogue, the FANTOM5 Consortium measured eRNA transcription by CAGE (Cap Analysis of Gene Expression) in a wide range of cell and tissue types [Citation4,Citation16]. Many other studies used nascent RNA sequencing methods such as Global Run-On sequencing (GRO-seq), Precision nuclear Run-On sequencing (PRO-seq) or Transient Transcriptome sequencing (TT-seq) to enrich eRNAs (and other nascent RNAs) [Citation19,Citation21,Citation24–Citation28]. Results from these methods depicted the landscapes of eRNAs and provided important insights into their functions and regulation.
Figure 1. Mechanistic similarity between enhancer RNAs (eRNAs) and some enhancer-derived lncRNAs.
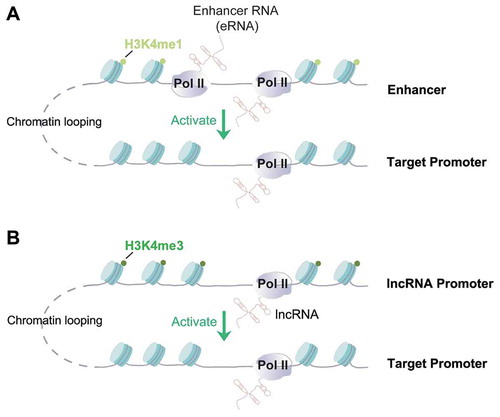
Functionally, eRNAs can be considered an integral component of active enhancers, which facilitate gene activation and/or enhancer-promoter loops through interacting with transcriptional activators and co-activators [Citation18,Citation19,Citation29–Citation33]. Although accumulated evidence agreed upon a strong correlation between eRNA transcription and enhancer activity, it remains challenging to discern whether any function of eRNAs comes from the transcripts per se or the act of enhancer transcription [Citation5]. Most strategies so far in the literature, such as genomic deletion [Citation34,Citation35], transcription terminator insertion [Citation36,Citation37], transcription suppression by CRISPRi [Citation38], will affect both transcription activity and eRNA transcripts. A common strategy to study the roles of the eRNA transcripts is to knock down eRNA by short hairpin RNAs (shRNAs), small interfering RNAs (siRNAs) or antisense oligonucleotides (ASOs or locked nucleic acids, LNAs). The past few years have witnessed strong evidence supporting the function of eRNAs per se using these knockdown methods [Citation15,Citation18,Citation19,Citation39–Citation42]. However, caution needs to be taken for interpreting these results as the cleavage of nascent transcripts using these tools may trigger transcription termination and interfere with the transcription activity of RNA polymerase [Citation43]. Despite these technical difficulties, the functions of eRNAs in regulating enhancer activity and target gene transcription are increasingly recognized and the underlying mechanisms began to emerge (see below).
It is noteworthy that although most eRNAs fit the above descriptions (e.g. non-polyadenylated, short-lived and functions in cis) [Citation4], exceptions exist. Several studies reported that specific eRNAs can be polyadenylated and have relatively higher stability [Citation3,Citation30,Citation39]. More importantly, depletion of these poly-A eRNAs, such as one transcribed from an enhancer close to KLK3 gene in prostate cancer cells [Citation30], affected the expression of hundreds of genes, suggesting their functions in trans. For another example, Tsai et al. observed that an eRNA derived from a Myogenic Differentiation 1 (MyoD) enhancer colocalized with, and mediated the activation of a target gene in trans, which was located on another chromosome [Citation44]. Because eRNAs were defined mainly based on their site of production (i.e. enhancers) rather than based on RNA features [Citation3,Citation4,Citation13,Citation14,Citation21,Citation22], it is inevitable that the categorization of eRNAs and of lncRNAs has overlaps. lncRNAs denote non-coding RNAs that are >200nt long, therefore, eRNAs can perhaps be considered a class of lncRNAs based on length. However, eRNAs possess more variable RNA features such as lack of splicing and polyadenylation [Citation4], and their transcription start sites contain less clear features of gene or lncRNA promoters (i.e. H3K4me3, see ref [Citation5] for more discussion of such features). Indeed, it was reported that a significant portion of annotated lncRNAs (~20%) can be mapped to enhancer regions [Citation45]. Some enhancer-generated lncRNAs have been shown to function in a very similar manner in chromatin/gene control as eRNAs, but can control genes beyond immediate neighbourhood of the enhancer, sometimes even on another chromosome. For example, lncRNA that enhances eNOS expression (LEENE) is a spliced lncRNA generated from an enhancer element in human endothelial cells, which can control eNOS gene expression on a different chromosome [Citation46]. One of the first reported activating lncRNAs called HOXA Distal Transcript Antisense RNA (HOTTIP) is actually transcribed from an enhancer-like region and is brought to its target gene promoter via chromatin looping [Citation47,Citation48]. Similarly, LncRNA downstream of Cdkn1b (Lockd) is an lncRNA located close to Cdkn1b gene and played a role to activate the latter [Citation37]. The transcription start region of Lockd showed typical features of a promoter, i.e. high trimethylation of H3K4 (H3K4me3), and it physically interacts with Cdkn1b gene via chromatin looping, which mimics what enhancers typically do [Citation37,Citation49] (). The similar molecular mechanisms of eRNA and enhancer-derived lncRNA blurred their boundaries (). Interestingly, their functional similarity may be interpreted as a support to a hypothesis that lncRNAs may be evolutionarily originated from eRNAs, but became stabilized and gained functions in trans during evolution [Citation50,Citation51]. In the following section, we will highlight recent studies focusing on the roles of eRNAs in cancer. In most cases, we do not distinguish eRNAs from enhancer-derived lncRNAs.
3. eRNA regulation and dysregulation in cancer
Enhancer malfunction is now recognized as a key process in tumorigenesis [Citation9,Citation52]. Besides commonly identified genetic mutations, epigenetic alterations of enhancers are also recognized as driving causes of cancer initiation and progression [Citation53,Citation54]. Moreover, special high-density clustering of enhancers (so-called super- or stretch-enhancers) are found to be de novo established and drove the proliferation of tumour cells [Citation55–Citation57]. Aberrant eRNA expression is highly associated with enhancer malfunction, and was involved in dysregulation of oncogenes [Citation58,Citation59], tumour suppressor genes [Citation40], as well as in abnormal cellular responses to external signals, such as hormone [Citation19,Citation28,Citation60], inflammation [Citation61–Citation63], hypoxia [Citation64] and other stimuli [Citation65–Citation69]. We will discuss these areas below.
3.1. eRNA and oncogene activation
eRNAs are involved in the oncogene activation process, which can be highlighted by a role of enhancer-derived lncRNAs in MYC gene activation (more cases discussed in later sections). MYC is a key proto-oncogene, and its dysregulation contributes to the development of many cancer types [Citation70]. In CpG island methylator phenotype (CIMP) colorectal tumour, cMYC oncogene expression was driven by an active super-enhancer, and it also highly correlated with the presence of a noncoding transcript from this enhancer that was named CCAT1 (colon cancer-associated transcript 1) [Citation58]. Data generated from The Cancer Genome Atlas project (TCGA) showed that the expression correlation between cMYC and CCAT1 exists in multiple tumour types [Citation58], suggesting a potential role of CCAT1 in regulating cMYC gene expression. Relevant to this, Chen group reported that a longer form of CCAT1, named CCAT1L, modulates the long-range interaction between the cMYC promoter and this CCAT1-producing super-enhancer [Citation59]. Moreover, in cis overexpression of CCAT1L promotes cMYC expression and facilitates tumorigenesis [Citation59], while inhibition of CCAT1 by BET inhibitor led to the reduction of both cMYC expression and cell growth [Citation58]. These results support a direct oncogenic function of this enhancer-derived lncRNA, which also supports the possibility of targeting this MYC eRNAs for cancer intervention (see section 5).
3.2. eRNA and tumour suppressor gene function
eRNAs are also involved in the proper function of tumour suppressors. P53 is a key tumour suppressor gene that acts as a transcription factor. Activation of p53 by treating breast cancer cells with Nutlin-3a stimulated the expression of thousands of eRNAs [Citation40]. Knockdown of one of these eRNAs attenuated the enhancer activity and target gene expression, suggesting the functional role of eRNA in p53-induced transcription enhancement [Citation40]. Interestingly, some of the eRNA-generating enhancers were not bound by activated p53. Further analysis showed that these enhancers were activated by a regulatory RNA named lncRNA activator of enhancer domains (LED) [Citation71]. Inhibition of LED by siRNA significantly affected the cell cycle arrest phenotype of these cancer cells, which was accompanied by a reduced expression level p53 target genes, such as CDKN1A [Citation71]. Thus, LED is required for p53 to fully exert its tumour suppressor functions [Citation71]. Consistent with this tumour suppressive role, LED is often inactivated by DNA hypermethylation in cancer cells, which could be reversed by DNA demethylating agent such as 5-Azacytidine [Citation71]. Together, these results demonstrated the importance of eRNAs in mediating functions of key tumour suppressor genes.
3.3. eRNA and cancer signalling responses
Many cell signalling events converge on chromatin, and act via enhancers to control target gene expression. Sex hormones such as oestrogen and androgen are important drivers of hormone receptor-positive breast and prostate cancers [Citation72,Citation73]. Female sex hormone oestrogen stimulus activates hundreds of genes, which were preceded by massive enhancer activation [Citation19,Citation21,Citation74,Citation75]. The expression of eRNAs in response to oestrogen stimulus was found to correlate with epigenetic features of active enhancers [Citation19,Citation21,Citation74], and some specific eRNAs are functionally required for oestrogen-induced enhancers to achieve their activation of target genes [Citation19]. Mechanistically, inhibition of two eRNAs can lead to a decrease of spatial interactions between the enhancer and its target gene, and may even disrupt a super-long distance chromatin loop spanning 27Mb on chromosome 21 [Citation19]. These results demonstrated that eRNAs are functionally important for cellular signalling responses to oestrogen, at least in part, by facilitating gene expression and chromatin looping. Androgen signalling in human prostate cancer cells is also reported to be modulated by eRNAs [Citation28,Citation30]. Indeed, liganded androgen receptor (AR) activates thousands of eRNAs [Citation28], and two studies found that an eRNA transcribed in the vicinity of a key prostate gene PSA (a.k.a. Kallikrein Related Peptidase 3 (KLK3)) regulates AR target genes in trans [Citation30], and facilitated the progression of castration resistance of prostate cancer [Citation76]. Mechanistically, one study found that PSAe functions together with Cyclin T1 to stimulate the transcription of its target gene [Citation76]. Sequence analysis showed that PSAe contains an RNA motif with strong similarity to the TAR RNA of HIV virus, which the authors elegantly showed to bind Cyclin T1 and activate the Positive transcription elongation factor b (P-TEFb) to promote target gene elongation [Citation76]. Interestingly, the other paper found that PSAe was also involved in mediating AR-dependent looping between this enhancer and its target promoter by forming a ribonucleoprotein complex consists of PSAe, AR and MED1 (Mediator complex subunit1) [Citation30]. Importantly, PSAe selectively enhances AR-regulated gene expression and promote prostate cancer castration resistance, which can be suppressed by siRNA, suggesting the feasibility of improving prostate cancer treatment by PSAe inhibition [Citation30,Citation76].
Inflammation plays an important roles in the initiation and progression of tumorigenesis [Citation77]. Inflammatory signals are known to induce large programs of enhancer activation and eRNA production [Citation63,Citation78]. In cancer cells, Rhanamoun et al. discovered that tumour-promoting p53 mutants abnormally activated a cohort of enhancers in response to pro-inflammatory TNF-α signalling [Citation61,Citation62]. At these enhancers, co-binding of mutant p53 and Nuclear Factor Kappa B (NF-κB) induced eRNA synthesis, one of which was required for the activation of important inflammation genes such as C-C Motif Chemokine Ligand 2 (CCL2) [Citation61,Citation62]. Therefore, eRNAs are directly involved in the immune response of cancer cells. Similar to inflammation, hypoxia activates a complex signalling network in cancer cells and promotes cell survival and propagation [Citation79]. Hypoxia-Induced Factor-1 (HIF-1), the key sensor of the hypoxic environment of solid tumours and a driver of tumour progression [Citation80,Citation81], acts on chromatin, at least in part, via controlling enhancer activation [Citation82]. Many other cancer related signalling pathways such as Wnt, Notch, and Hippo pathways also orchestrate nuclear events including chromatin remodelling and recruitment of transcription factors/cofactors to function through enhancer control [Citation5,Citation66–Citation69]. For example, Notch regulatory elements are more often located in enhancers rather than promoters [Citation68,Citation69], where they can mediate breast cancer gene activation and drug resistance [Citation83]. Interestingly, a recent study found that Hippo pathway effectors, YAP/TEAD, facilitated hormone-induced eRNA transcription in breast cancer cells [Citation84], suggesting that many of these aforementioned signalling events may cross-talk to each other at enhancers in cancer gene regulation. Albeit a direct role of eRNAs in regulating these signalling events is currently lacking, it is promising to pursue the function of eRNAs in these enhancer-driven signalling events critical for cancer progression.
4. Mechanisms underlying eRNA functions in gene regulation and in cancer
4.1. The role of eRNA–protein interaction in transcriptional regulation
A common theme underlying the functions of eRNAs, and perhaps of many other regulatory RNAs, is that eRNAs interact with specific protein partners and modulate their activity. A number of studies have reported eRNA interaction with transcription factors, cofactors and RNA-binding proteins (RBPs) (). Rosenfeld and colleagues provided the first evidence that oestrogen-regulated eRNAs can bind cohesion complex proteins, including RAD21 Cohesin Complex Component (RAD21) and Structural Maintenance Of Chromosomes 3 (SMC3), and proposed that eRNA:Cohesin interaction stabilizes chromatin looping in breast cancer cells () [Citation19]. Following this work, there has been growing appreciation that eRNA modulates chromatin looping via its interaction with other protein factors such as Mediator Complex Subunit 1 (MED1) in prostate cancer cells [Citation30], Heterogeneous Nuclear Ribonucleoprotein U (hnRNPU) in gastric cancer cells [Citation85], CCCTC-Binding Factor (CTCF) in colon cancer cells [Citation59], and Mediator Complex Subunit 12 (MED12) in T-cell acute lymphoblastic leukaemia cells [Citation86]. Besides cancer, cohesin-eRNA interaction was also found important for a trans-acting eRNA in muscle to regulate target gene activation [Citation44]. However, contradicting evidence also exists, which showed that the reduction of some eRNAs may not affect chromatin looping [Citation31,Citation75]. The discrepancy between these findings needs to be further investigated, but can be partially attributed to different eRNA knockdown or inhibition strategies, or may also reflect locus-specific regulation of looping dynamics.
Table 1. A list of reported eRNA binding proteins and underlying mechanisms.
Another mechanism discovered to underlie eRNA functions is that eRNAs interact with transcriptional coactivator such as CBP/p300 and Bromodomain Containing 4 (BRD4) (). Berger and co-workers found that multiple eRNAs in mouse embryonic fibroblasts bind an RNA binding region (RBR) within the catalytic histone acetyl-transferase (HAT) domain of CBP, which facilitates CBP binding with its substrate histones and stimulates the HAT activity [Citation87]. Rahnamoun et al. showed that the bromodomain (BD) of BRD4, a key transcription coactivator and oncogene, directly interacts with eRNAs in human colon cancer cell lines, particularly under inflammatory stimulus, and eRNA-BRD4 interaction promotes BRD4 tethering to acetylated chromatin regions to induce transcription activation of inflammation genes [Citation62]. These studies showed that eRNAs can interact with transcription cofactors directly to regulate histone acetylation and gene activation.
Figure 2. Mechanisms underlying eRNA-RBP interaction and gene transcriptional regulation.
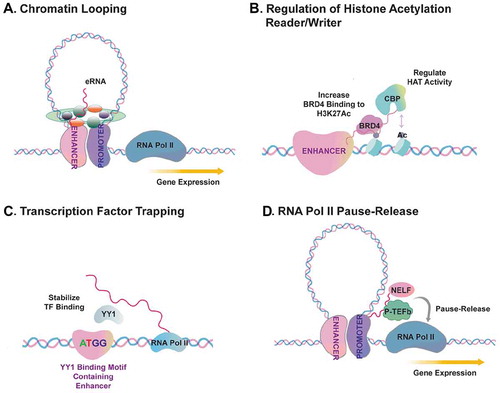
As a third mechanism, Young and colleagues proposed a role of eRNAs in transcriptional regulation via a process dubbed ‘transcription factor trapping’ () [Citation32]. In this study by Sigova et al., they revealed that eRNA is important to trap transcription factor Yin-Yang1 (YY1) in enhancer region in murine embryonic stem cells [Citation32]. Reduced transcription of eRNA blocked the YY1 occupancy on the enhancer region [Citation32]. By contrast, the artificial tethering of a specific eRNA to enhancers promoted YY1 occupancy, although the quantity of YY1 binding increase was very mild [Citation32]. This study suggested that eRNA transcripts may directly modulate TF binding to regulatory DNA elements and this may facilitate positive feed-forward loop to enhance transcriptional circuits of gene activation.
Another important mechanism defined for eRNA functions is that they can modulate RNA Pol II pause-release at the target gene promoters through binding elongation repressor proteins to keep them away from promoters () [Citation31,Citation76,Citation88,Citation89]. This hypothesis has been supported by several studies. Kim and colleagues showed that an eRNA promotes the transition of paused RNA Pol II into the elongation stage in murine neurons, which was achieved by the eRNA’s ‘binding and decoying’ the negative elongation factor (NELF) complex away from immediate early genes [Citation31]. This study found that eRNAs bind the NELF-E subunit of NELF complex via the RNA Recognition Motif (RRM) of the later [Citation31]. In another study, Zhao et al. showed that PSA eRNA, also known as KLK3e, facilitates cis and trans gene transcription via forming a complex with positive transcriptional elongation factor (P-TEFb) [Citation76]. Importantly, this study showed that PSAe contains an RNA secondary structure that is reminiscent of TAR RNA of HIV virus and of the 7SK small nuclear RNA (snRNA) [Citation76]. The structure of eRNAs contributing to protein interaction and P-TEFb activation provided an interesting perspective to understand the molecular basis of eRNA-protein functional interaction. In accord with these studies, several other reports have demonstrated an interaction between eRNA and NELF or P-TEFb in monocytes (SERPINB2e and ADAMEC1e) [Citation88,Citation89]. These evidences together suggest that specific eRNAs regulate gene transcription through interacting with critical proteins during the process of RNA Pol II pause-release.
4.2. eRNA-DNA interaction, R-loop formation, and DNA damage response
eRNAs may not only bind proteins, but also DNAs directly to modulate enhancer function and activity. It is considered that nascent RNA transcripts occasionally hybridize with template DNAs, leaving the non-template DNA single-stranded [Citation90,Citation91]. This special three-stranded nucleic acid structure, referred to as R-loop, can be associated with DNA damage response and genomic instability [Citation92–Citation94]. Basu and colleagues examined the transcriptome of mESCs and B cells after genetic ablation of two major components of the RNA exosome complex, Exosome Component 3 (Exosc3) and Exosome Component 10 (Exosc10), which up-regulated a subset of eRNA expression in these cells [Citation41]. This is consistent with other studies finding that eRNAs are substrates of RNA exosome complex [Citation95,Citation96]. But in addition to that, intriguingly, the authors found that increased eRNA expression resulted in R-loop formation and consequent genomic instability at enhancers [Citation41]. Interestingly, a recent study noticed that the global enhancer activation often positively correlated with tumour aneuploidy [Citation6], suggesting a potential malicious profit of deregulated eRNA transcription/expression and genomic instability for human cancer progression. Further studies will be important to elucidate the functional interplay between eRNA transcription, R loop formation and genomic instability at enhancers and other noncoding genomic regions in cancer cells.
5. eRNAs as potential diagnostic and prognostic markers, and therapeutic targets for human cancers
5.1. eRNA expression in cancer diagnosis and prognosis
Although eRNA expression may be of lower abundance as an entire category, a subset of them can still be detected by conventional oligo-dT-based RNA-Seq. Using large cohorts of RNA-Seq datasets generated from ~10,000 human tumour samples by the Cancer Genome Atlas Consortium (TCGA), which predominantly are polyA RNA-Seq, eRNA landscapes have been systematically characterized by two recent studies [Citation6,Citation10]. These studies demonstrated the existence of cancer-enriched eRNAs and their potential clinical importance as, i) diagnostic markers, ii) prognostic markers, and iii) therapeutic targets or predictive markers for therapeutic response. These studies first tested whether eRNA expression has a unique pattern in different cancer types. For this, in Zhao et al., we identified in total 9,108 eRNAs, out of which, 652 are ubiquitously expressed, which we defined as those expressed in >10 cancer types (~7% of all eRNAs). By contrast, a larger number of eRNAs are cancer type-specific, which account for ~59% of all identified eRNAs (5,332 out of 9,108). These cancer type-specific eRNAs can distinguish cancer types very well in a t-Distributed Stochastic Neighbour Embedding (t-SNE) analysis [Citation10]. This result demonstrated that each cancer type has unique eRNA expression patterns; therefore, eRNAs can be potentially utilized for molecular diagnosis of cancer types. Second, the two studies examined the correlation of eRNA expression and patient survival to reveal the prognostic utility of eRNA expression. Interestingly, in most cancer types, the proportion of enhancers that showed prognostic significance was comparable to, or even higher than, that of protein-coding genes [Citation6]. In addition, some eRNAs showed significant correlation with not only patient survival, but also other important cancer-related clinical features including subtypes, stages, and grades [Citation10]. For example, an enhancer referred to as ‘Enhancer 22’ showed significant correlation with patient survival in multiple cancer types including kidney renal cell clear cell carcinoma, low-grade glioma, uveal melanoma and others [Citation6]. In Zhao et al., they found strong correlation of eRNA expression with many clinical cancer features, which can be exemplified by correlation between eRNA expression and survival (NET1e and TAOK1e), subtype (EN1e), stage (CELF2e), grade (APH1Ae) and smoking history (SCRIBe), respectively, [Citation10]. Consistent with these large-scale analyses, a separate study found that one eRNA, AP001056.1, can serve as a prognostic marker of head and neck squamous cell carcinoma (HNSCC) [Citation97]. In particular, the correlation of this eRNA with HNSCC showed remarkable specificity in that its expression was highly detected only in some anatomic subsites that harbour distinctive somatic mutations, HPV status and survival outcomes [Citation97–Citation99]. Overall, these studies highlighted clinical utility of eRNA expression for cancer diagnosis and prognosis.
5.2. eRNAs serve as potential targets for cancer therapy
Interestingly, many eRNAs showed remarkable over-expression in tumour samples as compared to their adjacent normal tissues [Citation6,Citation10]. This is consistent with multiple studies reporting enhancer over-activation in cancer [Citation100–Citation103]. This phenomenon raises a potential to target eRNAs to overcome enhancer over-activation for cancer therapy.
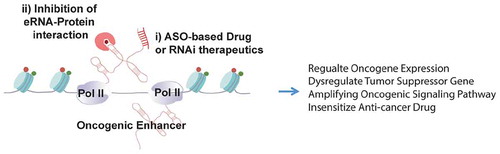
We propose that eRNAs per se may serve as useful and highly precise therapeutic targets for future cancer intervention. This is particularly based on the high specificity of eRNA expression across tissues [Citation4], and across cancer types, as revealed by our and others’ recent work [Citation6,Citation10]. It is also based on effective inhibition of target gene and tumour growth using antisense oligos to target specific eRNAs [Citation10,Citation30,Citation85,Citation86,Citation104,Citation105]. Importantly, the high specificity of eRNAs has a superior advantage to be drug target as its inhibition will in theory not affect other irrelevant tissues. Indeed, in support of this argument, we and colleagues have demonstrated that an eRNA transcribed close to Neuroepithelial cell-transforming gene 1 (NET1) gene, referred to as NET1e, is a breast cancer-specific eRNA [Citation10]. Anti-NET1e LNA treatment strongly inhibited cell growth of NET1e-high breast cancer cells, such as MCF7, while no inhibitory effect was found when anti-NET1e was given to mammary epithelial cells MCF10A or cervical cancer cell line HeLa, both of which contain little or no NET1e RNA [Citation10]. That eRNAs can serve as therapeutic targets are also supported by other studies. For example, suppression of oncogenic eRNAs caused growth inhibitory effects on cells of prostate cancer, bladder cancer, EBV-transformed lymphoblastoid, cervical cancer and gastric cancer [Citation30,Citation85,Citation86,Citation104,Citation105]. Especially, Jiao et al. found a critical oncogenic role of HPSE eRNA using both in vitro assays and an in vivo xenograft model [Citation85]. Although further work is needed to overcome the insufficient knowledge on the mechanisms of eRNAs in conferring oncogenic growth of the cells, these evidences clearly demonstrated that targeting eRNA can be a promising anti-cancer strategy with high precision and tumour type specificity.
In addition, eRNAs may play roles in mediating cancer therapeutic responses. Chen et al. found a correlation between the expression level of Enhancer 9 (chr9:5580709–558,016) and that of Programmed death-ligand 1 (PD-L1) in multiple cancer types [Citation6]. Because PD-L1 gene expression has been used as an important marker for predicting efficacy of cancer immunotherapy, this study suggests a potential value of using eRNA expression as a prediction marker of immunotherapy efficacy [Citation6]. In addition, cells with Enhancer 9 deletion showed a remarkable reduction of PD-L1 expression in mRNA and protein levels [Citation6], supporting that this enhancer directly acts on PD-L1 gene. However, the direct roles of the eRNAs from this enhancer cannot be deduced from this experiment, which deleted the entire enhancer region. Recently, we and colleagues also demonstrated that the expression level of NET1e, a breast cancer enriched eRNA, is associated with drug response, for example, with IC50 of Obatoclax and BEZ235 [Citation10]. Importantly, when we mimicked the cancer-associated NET1e overexpression by using CRISPR-a in cis induction, we detected an increased IC50 of Obatoclax and BEZ235 in breast cancer cells, supporting a direct role of this eRNA in drug response [Citation10]. These results together demonstrated that eRNA expression in cancer cells can be used as a predictive marker for therapeutic response to clinically used drugs, and manipulating eRNA expression can be a promising strategy to alter drug response ().
Figure 3. A potential application of eRNA-targeting therapeutics in cancer intervention.
6. Conclusions and future perspectives
Despite rapid progress of eRNA studies in the past decade, much remains unknown in terms of eRNA functions in cancer and the underlying mechanisms. For example, the exact role of eRNA transcription versus transcripts is still a matter of debate under certain circumstances. How common an eRNA carries biology function is also unclear, especially in consideration of their extremely large number in the human transcriptome (>65,000 enhancers showed eRNA transcription) [Citation4,Citation16]. The two recent endeavours significantly advanced the studies of eRNAs in cancer by providing a comprehensive landscape of eRNAs in a large cohort of tumour samples [Citation6,Citation10], providing a blueprint for functional investigation of eRNA functions and mechanisms. Indeed, by targeting specific eRNAs, it is hopeful that we can achieve cancer type- or patient-specific therapy for cancer intervention [Citation6,Citation10]. Further work using in vivo models is required to test this concept on clinically relevant eRNAs. A major challenge for future research is to understand the molecular mechanisms of eRNA action in cancer to potentially facilitate a better therapeutic intervention. Indeed, eRNA-centric unbiased proteomic methods need to be applied to clinically relevant eRNAs to characterize their proteomic partners (e.g. ChIRP, RAP or iDRiP [Citation106–Citation108]), and functional studies may follow to characterize eRNA–protein interactions in potentially causing cancer phenotypes (). Besides eRNA-protein interactions, the structural and chemical regulation of eRNAs are poorly explored. For example, RNA epitranscriptomic modifications such as A-to-I editing, methylation by N6-adenosine methylation (m6A), 5-cytosine methylation (m5C), hydroxy-methyl cytosine (5hmC) or methyl-1 adenosine (m1A) modify various mRNAs or regulatory RNAs, but their existence and functions on eRNAs are largely unknown [Citation109–Citation116], except by one study [Citation117]. We also have little understanding of the secondary structures of eRNAs that may impact transcription apparatus, which has been implied by studies of nascent RNAs [Citation118]. Beyond the cis functions, how eRNAs potentially participate in higher-order chromatin/nuclear organization via RNA-protein, RNA-DNA or RNA–RNA interactions [Citation119,Citation120], or how they may modulate the dynamics or function of nuclear phase-separated condensates will also be significant questions for future studies [Citation121–Citation123]. To successfully delineate these outstanding questions of the basic eRNA mechanisms will not only advance our understanding of gene regulation, but also pave the way for eRNA-based novel cancer diagnostic or therapeutic strategies.
Acknowledgments
We regret for not citing all relevant papers due to space and citation limits. We appreciate the work from Hwajin Shin and Sinhye Lee for helping our generation of the figure illustration. W.L. is a Cancer Prevention and Research Institute of Texas (CPRIT) Scholar in Cancer Research. We thank the funding support from the University of Texas (UT Rising STARs Award), from NIH (K22CA204468, R21GM132778), CPRIT (RR160083, RP180734), and Welch foundation to W.L (AU-2000-20190330).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108.
- Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569.
- Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1–19.
- Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461.
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223.
- Chen H, Li C, Peng X, et al.; Cancer Genome Atlas Research N. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173:386–99 e12.
- Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol. 2016;17:743–755.
- Murakawa Y, Yoshihara M, Kawaji H, et al. Enhanced identification of transcriptional enhancers provides mechanistic insights into diseases. Trends Genet. 2016;32:76–88.
- Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483–493.
- Zhang Z, Lee J, Ruan H, et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat Commun. 2019;10:4562.
- Tuan D, Kong S, Hu K. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci U S A. 1992;89:11219–11223.
- De Santa F, Barozzi I, Mietton F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384.
- Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187.
- Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200.
- Alvarez-Dominguez JR, Hu W, Yuan B, et al. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–581.
- Arner E, Daub CO, Vitting-Seerup K, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347:1010–1014.
- Lai F, Gardini A, Zhang A, et al. Integrator mediates the biogenesis of enhancer RNAs. Nature. 2015;525:399–403.
- Lam MT, Cho H, Lesch HP, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515.
- Li W, Notani D, Ma Q, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520.
- Sigova AA, Mullen AC, Molinie B, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci U S A. 2013;110:2876–2881.
- Hah N, Danko CG, Core L, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634.
- Lam MT, Li W, Rosenfeld MG, et al. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–182.
- Koch F, Fenouil R, Gut M, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18:956–963.
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848.
- Kwak H, Fuda NJ, Core LJ, et al. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339:950–953.
- Schwalb B, Michel M, Zacher B, et al. TT-seq maps the human transient transcriptome. Science. 2016;352:1225–1228.
- Core LJ, Martins AL, Danko CG, et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46:1311–1320.
- Wang D, Garcia-Bassets I, Benner C, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394.
- Lai F, Orom UA, Cesaroni M, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501.
- Hsieh CL, Fei T, Chen Y, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A. 2014;111:7319–7324.
- Schaukowitch K, Joo JY, Liu X, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42.
- Sigova AA, Abraham BJ, Ji X, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350:978–981.
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731.
- Gonen N, Futtner CR, Wood S, et al. Sex reversal following deletion of a single distal enhancer of Sox9. Science. 2018;360:1469–1473.
- Osterwalder M, Barozzi I, Tissieres V, et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018;554:239–243.
- Ho Y, Elefant F, Liebhaber SA, et al. Locus control region transcription plays an active role in long-range gene activation. Mol Cell. 2006;23:365–375.
- Paralkar VR, Taborda CC, Huang P, et al. Unlinking an lncRNA from its associated cis element. Mol Cell. 2016;62:104–110.
- Mumbach MR, Satpathy AT, Boyle EA, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017;49:1602–1612.
- Mousavi K, Zare H, Dell’orso S, et al. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. 2013;51:606–617.
- Melo CA, Drost J, Wijchers PJ, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49:524–535.
- Pefanis E, Wang J, Rothschild G, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774–789.
- Ounzain S, Pezzuto I, Micheletti R, et al. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J Mol Cell Cardiol. 2014;76:55–70.
- Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407.
- Tsai PF, Dell’Orso S, Rodriguez J, et al. A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol Cell. 2018;71:129–41 e8.
- Bonasio R, Shiekhattar R. Regulation of transcription by long noncoding RNAs. Annu Rev Genet. 2014;48:433–455.
- Miao Y, Ajami NE, Huang TS, et al. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun. 2018;9:292.
- Orom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58.
- Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124.
- Espinosa JM. Revisiting lncRNAs: how do you know yours is not an eRNA? Mol Cell. 2016;62:1–2.
- Wu X, Sharp PA. Divergent transcription: a driving force for new gene origination? Cell. 2013;155:990–996.
- Espinosa JM. On the origin of lncRNAs: missing link found. Trends Genet. 2017;33:660–662.
- Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53:859–866.
- Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:6348:eaal2380.
- Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299.
- Hnisz D, Schuijers J, Lin CY, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58:362–370.
- Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334.
- Parker SC, Stitzel ML, Taylor DL, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110:17921–17926.
- McCleland ML, Mesh K, Lorenzana E, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest. 2016;126:639–652.
- Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513–531.
- van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci. 2013;14:14833–14859.
- Rahnamoun H, Lu H, Duttke SH, et al. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun. 2017;8:754.
- Rahnamoun H, Lee J, Sun Z, et al. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol. 2018;25:687–697.
- Hah N, Benner C, Chong LW, et al. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc Natl Acad Sci U S A. 2015;112:E297–302.
- Moreau PR, Ord T, Downes NL, et al. Transcriptional profiling of hypoxia-regulated non-coding RNAs in human primary endothelial cells. Front Cardiovasc Med. 2018;5:159.
- Ko JY, Oh S, Yoo KH. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol Cells. 2017;40:169–177.
- Meng Z, Moroishi T, Guan KL. Mechanisms of hippo pathway regulation. Genes Dev. 2016;30:1–17.
- Galli GG, Carrara M, Yuan WC, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell. 2015;60:328–337.
- Wang H, Zang C, Taing L, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with super enhancers. Proc Natl Acad Sci U S A. 2014;111:705–710.
- Skalska L, Stojnic R, Li J, et al. Chromatin signatures at Notch-regulated enhancers reveal large-scale changes in H3K56ac upon activation. Embo J. 2015;34:1889–1904.
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
- Leveille N, Melo CA, Rooijers K, et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun. 2015;6:6520.
- Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225–240.
- Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–711.
- Hah N, Murakami S, Nagari A, et al. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223.
- Murakami S, Nagari A, Kraus WL. Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev. 2017;31:1535–1548.
- Zhao Y, Wang L, Ren S, et al. Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 2016;15:599–610.
- Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51:27–41.
- Brown JD, Lin CY, Duan Q, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231.
- Muz B, de la Puente P, Azab F, et al. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92.
- Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490.
- Grampp S, Platt JL, Lauer V, et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat Commun. 2016;7:13183.
- Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107.
- Magnani L, Stoeck A, Zhang X, et al. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci U S A. 2013;110:E1490–9.
- Zhu C, Li L, Zhang Z, et al. A non-canonical role of YAP/TEAD is required for activation of estrogen-regulated enhancers in breast cancer. Mol Cell. 2019;75:791–806 e8.
- Jiao W, Chen Y, Song H, et al. HPSE enhancer RNA promotes cancer progression through driving chromatin looping and regulating hnRNPU/p300/EGR1/HPSE axis. Oncogene. 2018;37:2728–2745.
- Tan SH, Leong WZ, Ngoc PCT, et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood. 2019;134:239–251.
- Bose DA, Donahue G, Reinberg D, et al. RNA binding to CBP stimulates histone acetylation and transcription. Cell. 2017;168:135–49 e22.
- Shii L, Song L, Maurer K, et al. SERPINB2 is regulated by dynamic interactions with pause-release proteins and enhancer RNAs. Mol Immunol. 2017;88:20–31.
- Shi L, Li S, Maurer K, et al. Enhancer RNA and NFkappaB-dependent P300 regulation of ADAMDEC1. Mol Immunol. 2018;103:312–321.
- Sanz LA, Hartono SR, Lim YW, et al. Prevalent, dynamic, and conserved r-loop structures associate with specific epigenomic signatures in mammals. Mol Cell. 2016;63:167–178.
- Thomas M, White RL, Davis RW. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc Natl Acad Sci U S A. 1976;73:2294–2298.
- Gan W, Guan Z, Liu J, et al. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011;25:2041–2056.
- Bhatia V, Barroso SI, Garcia-Rubio ML, et al. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–365.
- Paulsen RD, Soni DV, Wollman R, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35:228–239.
- Lubas M, Andersen PR, Schein A, et al. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep. 2015;10:178–192.
- Kilchert C, Wittmann S, Vasiljeva L. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol. 2016;17:227–239.
- Gu X, Wang L, Boldrup L, et al. AP001056.1, A prognosis-related enhancer RNA in squamous cell carcinoma of the head and neck. Cancers (Basel). 2019;11:347.
- Stadler ME, Patel MR, Couch ME, et al. Molecular biology of head and neck cancer: risks and pathways. Hematol Oncol Clin North Am. 2008;22: 1099–1124. .
- Thibaudeau E, Fortin B, Coutlee F, et al. HPV prevalence and prognostic value in a prospective cohort of 255 patients with locally advanced HNSCC: a single-centre experience. Int J Otolaryngol. 2013;2013:437815.
- Zhang X, Choi PS, Francis JM, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48:176–182.
- Bahr C, von Paleske L, Uslu VV, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515–520.
- Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377.
- Corces MR, Granja JM, Shams S, et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362(6413): eaav1898.
- Liang J, Zhou H, Gerdt C, et al. Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc Natl Acad Sci U S A. 2016;113:14121–14126.
- Ding M, Zhan H, Liao X, et al. Enhancer RNA - P2RY2e induced by estrogen promotes malignant behaviors of bladder cancer. Int J Biol Sci. 2018;14:1268–1276.
- McHugh CA, Chen CK, Chow A, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236.
- Minajigi A, Froberg J, Wei C, et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015;349(6245), aab2276.
- Chu C, Zhang QC, da Rocha ST, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416.
- Han L, Diao L, Yu S, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–528.
- Batista PJ, Molinie B, Wang J, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719.
- Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–342.
- Liu N, Dai Q, Zheng G, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564.
- Zhang X, Liu Z, Yi J, et al. The tRNA methyltransferase NSun2 stabilizes p16INK(4) mRNA by methylating the 3ʹ-untranslated region of p16. Nat Commun. 2012;3:712.
- Liu RJ, Long T, Li J, et al. Structural basis for substrate binding and catalytic mechanism of a human RNA: m5Cmethyltransferase NSun6. Nucleic Acids Res. 2017;45:6684–6697.
- Delatte B, Wang F, Ngoc LV, et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351:282–285.
- Roovers M, Wouters J, Bujnicki JM, et al. A primordial RNA modification enzyme: the case of tRNA (m1A) methyltransferase. Nucleic Acids Res. 2004;32:465–476.
- Aguilo F, Li S, Balasubramaniyan N, et al. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1alpha. Cell Rep. 2016;14:479–492.
- Zamft B, Bintu L, Ishibashi T, et al. Nascent RNA structure modulates the transcriptional dynamics of RNA polymerases. Proc Natl Acad Sci U S A. 2012;109:8948–8953.
- Li X, Fu XD. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet. 2019;20:503–519.
- Xiao R, Chen JY, Liang Z, et al. Pervasive chromatin-RNA binding protein interactions enable RNA-based regulation of transcription. Cell. 2019;178:107–21 e18.
- Fay MM, Anderson PJ. The role of RNA in biological phase separations. J Mol Biol. 2018;430:4685–4701.
- Lin Y, Protter DS, Rosen MK, et al. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell. 2015;60:208–219.
- Hnisz D, Shrinivas K, Young RA, et al. A phase separation model for transcriptional control. Cell. 2017;169:13–23.