
ABSTRACT
RNA secondary structure elements in the mRNA 3ʹ-untranslated regions (3ʹUTR) play important roles in post-transcriptional regulation. RNA structure elements in the viral RNA provide valuable model for studying diverse regulation mechanisms. Herpesvirus genomes are double-stranded DNA with GC-rich sequences, which can be transcribed into abundant GC-rich RNAs. It is valuable to explore the structures and function of those GC-rich RNAs. We identified a G2-quadruplex-forming sequence named PQS18-1 in the 3ʹUTR of the unique immediate early gene of Pseudorabies virus (PRV), an important member of Alphaherpesvirinae subfamily. The RNA PQS18-1 was folded into parallel G-quadruplex structure, enhancing gene expression. Both non-G-quadruplex mutant and G3-quadruplex mutant in the 3ʹUTR showed lower gene expression level than the wildtype G2-quadruplex. TMPyP4 destroyed PQS18-1 G2-quadruplex and suppressed gene expression, accordingly reducing PRV replication by one titre in the PK15 cells at 24 h post infection. Our findings indicated that the RNA G2-quadruplex in 3ʹUTR was essential for high expression of IE180 gene, and it could be a specific post-transcription regulation element in response to small molecules or other macromolecules. This study discovers a novel RNA G2-quadruplex in the 3ʹUTR of an immediate early gene of alphaherpesvirus and provides a new nucleic acid target for anti-virus drug design.
Graphical abstract
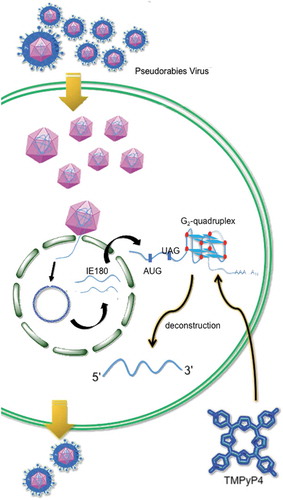
1. Introduction
Post-transcriptional gene regulation in eukaryotic organism can be controlled by structure elements affecting RNA–RNA interaction [Citation1,Citation2,] and RNA-protein interaction [Citation3] in 3ʹ-untranslated regions (3ʹUTR) of messenger RNAs (mRNAs). RNA structure elements in the viral RNA provided valuable models for studying the basic principles of diverse regulation mechanisms [Citation4]. The tRNA-like structure (TLS) in the 3ʹUTR [Citation4] and long-range RNA–RNA interactions in the 3ʹend terminal of viral genomes [Citation5,Citation6,] were necessary for virus proliferation. Moreover, the G-rich sequences acted as cis-elements by forming G-quadruplex structures [Citation7,Citation8,]. G-quadruplexes were increasingly identified in microbial genomes and played diverse roles in influencing virulence, latency, immune evasion of viruses [Citation9]. G-quadruplexes (G4), as special DNA or RNA secondary structures, are composed of π-π-stacked G-quartets via hydrogen-bonded structure in DNA or RNA [Citation10]. Each G-quartet contains four guanines held by eight hydrogen bonds and coordinated with a central cation, such as K+ or Na+. G-quadruplexes in the 3ʹUTR contributed to mRNA stability and localization through being targeted by the cellular proteins [Citation11,Citation12,], prevented binding of microRNA to the target mRNA [Citation13], shortened transcripts by affecting alternative polyadenylation in 3ʹUTRs [Citation14], and suppressed translation processes of several human genes [Citation15–Citation17]. Based on these findings, it could be assumed that formation and disruption of RNA G-quadruplexes in untranslated region of viral genes would play roles in regulating virus life cycles, which remains to be further investigated.
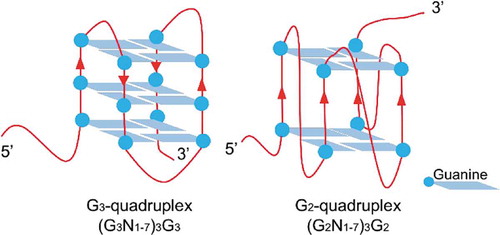
Herpesviridae included many important herpesviruses infecting human and animals. Herpesviruses have double-stranded DNA genomes, most of which are rich of guanine and cytosine. Pseudorabies virus (PRV) belongs to Alphaherpesvirinae subfamily. PRV could cause virulent infection in many kinds of wild and livestock animals, and cause life-long latency in host. PRV has been used as a model species for studying infection cycle, latency and reactivation, which are critical processes for the survival of alphaherpesviruses [Citation18]. It was reported that herpesvirus transcription was regulated in a cascade-like process subdivided into three temporally regulated phases: immediate-early (IE), early (E) and late (L). PRV transcribed one immediate early gene, IE180, the orthologue of the transcription activator ICP4 of Herpes simplex virus type 1 (HSV-1). The transcription of IE180 did not require any viral protein synthesis, but required the regulation by cellular transcription factors, such as NF-microE1 and oct-1 [Citation19]. Further, IE180 protein suppressed its own expression level through inhibiting its own promoter activity [Citation18] and translation initiation. On the other hand, IE180 mRNA could be a valuable target for controlling PRV proliferation and latency establishment. Targeting the 3ʹUTR of IE180 gene by short hairpin RNA (shRNA) was reported to have successfully knocked down the IE180 gene expression, and the transgenic mice that produced the shRNA showed resistance to PRV infection [Citation20]. Our previous study reported that the 3ʹUTR of IE180 gene was rich in extremely high GC, and that high level of GC was predicted to be folded into potential G2-quadruplex cluster [Citation21]. G-quadruplex with three G-tetrads was defined as G3-quadruplex, and G-quadruplex with two G-tetrads was defined as G2-quadruplex () [Citation22]. In the 3ʹUTR region of herpesvirus genes, G2-quadruplexes were in higher density and were more sensitive to cations or molecules, compared to G3-quadruplexes [Citation21]. G2-quadruplexes could be adjusted by small molecules and proteins, and therefore they were suitable to serve as regulating switches. The study of G2-quadruplexes in the 3ʹUTR of viral genes will reveal new functions of G2-quadruplexes, such as acting as translation regulator or specific targets of antiviral therapy.
Scheme 1. Diagram of the G2-quadruplex and G3-quadruplex.
In this work, bioinformatics analysis of 3ʹUTR sequence of IE180 gene from 19 PRV strains was performed, and several putative G2-quadruplex sequences (G2-PQS) were found to exhibit high conservation. One sequence was identified to exhibit stable G2-quadruplex structure. Dual-luciferase assay suggested that the formation of the G-quadruplex in 3ʹUTR promoted the protein translation. Non-G-quadruplex mutation in the 3ʹUTR resulted in the decreased gene expression of reporter gene. G3-PQS mutant had one more G-tetrad than the wildtype G2-PQS, and this mutant also showed lower reporter luciferase level than the wildtype construct. A chemical compound destroying the G-quadruplex exhibited suppressions for the expression of the reporter gene bearing wildtype 3ʹUTR of IE180 gene. Moreover, the chemical compound decreased the expression level of IE180 protein, resulting in the suppression of PRV titre in its host cells at 24 h post infection. This study reports a stable RNA G2-quadruplex that is essential for maintaining high level of gene expression and confirms G2-PQS to be a selectively identifiable target of the chemical compound. This is the first attempt to identify RNA G2-quadruplex acting as an up-regulated cis-element from 3ʹUTR of herpesvirus gene. The abundant G2-quadruplexes in the 3ʹUTR of herpesvirus genes are conducive to the functional study of RNA structures and provide potential antiviral therapeutic targets.
2. Results
2.1. Identification of G2-quadruplex-forming sequences in vitro
2.1.1. Conservation analysis of G2-quadruplex-forming sequences in the 3ʹUTR of IE180 gene
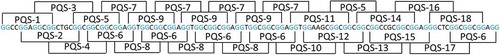
A 470-bp GC-rich untranslated region (GenBank accession number: MN202464) was identified downstream of the open reading frame of IE180 from the PRV Ea strain through 3ʹRACE procedure based on the genome sequence of PRV Ea strain [Citation23]. Putative G-quadruplex-forming sequences (PQSs) with the sequence motif in the form of (G2+N1–7)3G2+ were predicted from a 111-bp G-rich region in the IE180 3ʹUTR. Through systematic analysis, 18 overlapped putative G2-quadruplex sequences with the sequence motif in the form of (G2N1–7)3G2 were identified and 5 of them were repeated more than twice (). No putative G3-quadruplex sequence with the sequence motif (G3+N1–7)3G3+ was found in this G-rich region. The conservation of each PQS was analysed among 19 PRV genomes. PQS2, PQS3, PQS4, PQS5, and PQS18 showed the percentage conservation more than 80% (Fig. S1, ).
Table 1. Summary of structure validation experiments of PQSs from 3ʹUTR of IE180 gene of PRV Ea strain.
Figure 1. Distribution of G2-PQSs in 3ʹUTR of IE180 gene from PRV Ea strain. G-tracts were highlighted in blue. Each putative G-quadruplex-forming sequence contained four G-tracts.
2.1.2. Structure validation of the putative G2-quadruplex sequences in the 3ʹUTR of IE180 gene
PRV genomic DNA replication occurs by a rolling-circle mechanism [Citation18]. G-quadruplexes in herpesvirus genomic DNA can interact with the chemical compound to alter latent DNA replication and episomal persistence [Citation24]. Biophysical methods were applied to test the potential G-quadruplex-forming sequence from the above DNA G2-PQSs. Potential G-quadruplex structures were primarily screened from 18 PQSs by circular dichroism (CD) spectroscopy and thermal difference spectra (TDS). Then, the conserved G-quadruplex-forming sequences were further confirmed through H-NMR spectroscopy ().
At first, circular dichroism (CD) spectroscopy was employed to identify parallel, anti-parallel, or hybrid G-quadruplex structures formed by the putative G2-quadruplex sequences in the 3ʹUTR of IE180 gene. Parallel G-quadruplexes exhibited a negative peak near 240 nm and a positive peak near 260 nm. While anti-parallel G-quadruplexes displayed a negative peak near 260 nm and a positive peak around 290 nm. The hybrid G-quadruplexes always displayed one negative peak at 240 nm and two positive peaks at 260 nm and 290 nm [Citation25], respectively. CD spectra showed that DNA PQS5, PQS6, PQS7, PQS8, and PQS9 exhibited typical peaks of parallel G-quadruplexes, that DNA PQS16 displayed typical peaks of anti-parallel G-quadruplexes, and that DNA PQS18 presented the signals of hybrid G-quadruplex ( and S2).
Figure 2. CD spectra, TDS spectra, and 1H NMR spectra of DNA PQS5 and DNA PQS18. A. CD spectra of DNA PQS5 and DNA PQS18. B. TDS spectra of DNA PQS5 and DNA PQS18. C. 1H NMR spectra of DNA PQS5 and DNA PQS18.
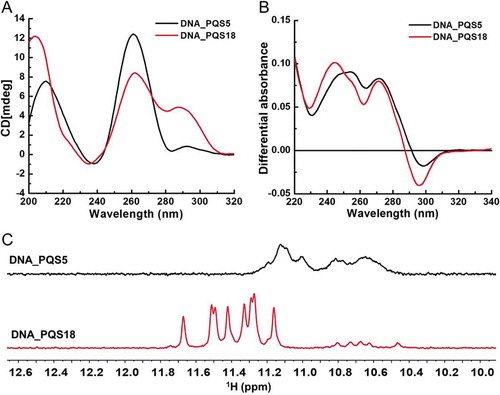
Secondly, the ultraviolet absorbance spectra of each folded and unfolded PQS were recorded at 20°C and 95°C, the temperatures below and above its melting temperature (Tm). The spectra indicating the difference between two spectra under the two temperatures was defined as the thermal difference spectra (TDS), and the TDS of G-quartet was observed to have a specific shape with major positive peaks at 243 ± 2 nm and 273 ± 1 nm, and a negative peak at 295 ± 1 nm [Citation26]. TDS result indicated that the spectra of PQS3, PQS5, PQS6, PQS7, PQS8, PQS9, and PQS18 showed typical peaks of G-quadruplex ( and S3), and six of them were consistent with the CD result (). Among these six G-quadruplex-forming sequences, PQS5 and PQS18 showed conservation rate higher than 80% among 19 PRV strains (Fig. S1).
In previous studies, folded G-quadruplexes were reported to have exhibited sharp imino proton peaks of guanines derived from the G-tetrads at 10.0 ~ 12.0 ppm in 1H NMR spectroscopy [Citation27,Citation28,]. Our 1H NMR spectroscopy results further confirmed that PQS5 and PQS18 were folded into G-quadruplexes, which was consistent with CD and TDS spectra results (). The spectrum of PQS5 showed wide imino peaks at 10.4 ~ 11.2 ppm, and the spectrum of PQS18 exhibited 8 significant imino peaks at 11.0 ~ 11.8 ppm (). The above result indicated that PQS18 was a conserved sequence with a stable G-quadruplex structure.
2.2. Formation of PQS18 G2-quadruplex enhances translation level in vivo
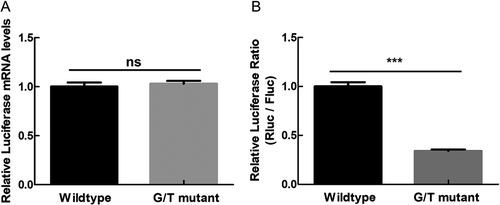
In order to investigate the role of PQS18 in regulating gene expression, we performed dual-luciferase assay with psiCHECK-2 vector (Promega, USA) in the method described in the previous study [Citation21]. The 3ʹUTR of IE180 was inserted into the downstream of the open reading frame of reporter gene Renilla luciferase (hRluc) encoding renilla luciferase. The hluc gene encoded the reference protein, firefly luciferase. The ratio of the renilla luciferase to the firefly luciferase indicated the relative expression level of the reporter gene. The non-G4 mutant (G/T mutant) sequence (Fig. S4) was applied to replace the PQS 18 in the wildtype construct to test the effect of the structure change on gene expression. The G/T mutant was produced by replacing one guanine of each G-tract of PQS18 with thymine in the wildtype construct psiCHECK-2-PQS18 (Table S1). The obtained psiCHECK-2-G/T did not show any difference in the relative mRNA level (), compared with wildtype construct, indicating that the G-quadruplex folded from DNA PQS18 had no effect on plasmid replication and transcription level. The psiCHECK-2-G/T showed about 60% decrease in the relative luciferase activity, compared to the wildtype construct (), suggesting that the RNA PQS18 in the 3ʹUTR was essential element for enhancing translation level of reporter gene. The structure of RNA PQS18 was further confirmed with biophysical methods.
Figure 3. Comparison of transcription level and translation level of Renilla luciferase gene between wildtype construct and G/T mutant construct. A. Relative mRNA level of renilla luciferase gene of wildtype construct and G/T mutant construct. B. Relative luciferase activity of wildtype construct and G/T mutant construct.
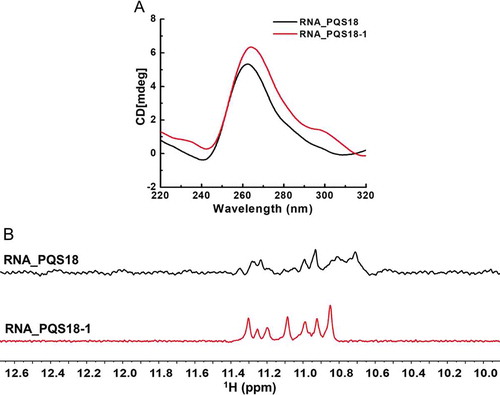
2.2.1. RNA PQS18-1 is folded into a more stable G-quadruplex structure
Circular dichroism spectroscopy results confirmed that RNA PQS18 was folded into parallel G-quadruplex structure (). Interestingly, the RNA PQS18-1 with an adenine at the end of PQS18 showed stronger signal at the positive peak at 263 nm than RNA PQS18 (). 1H NMR spectroscopy results indicated that RNA PQS18-1 was folded into much more stable G-quadruplex structure than RNA PQS18. The RNA PQS18-1 showed significantly sharp and well-resolved imino proton peaks at 10.8–11.4 ppm, while RNA PQS18 presented several wide peaks (). RNA PQS18-1 G-quadruplex was the most stable and conserved structure among the 18 putative G-quadruplexes in 3ʹUTR of IE180 gene, suggesting that it could act as structural regulatory element.
Figure 4. Comparison of CD spectra and 1H NMR spectra between PQS18 and PQS18-1. A. CD spectra of RNA PQS18 and RNA PQS18-1. B. 1H NMR spectra of RNA PQS18 and RNA PQS18-1.
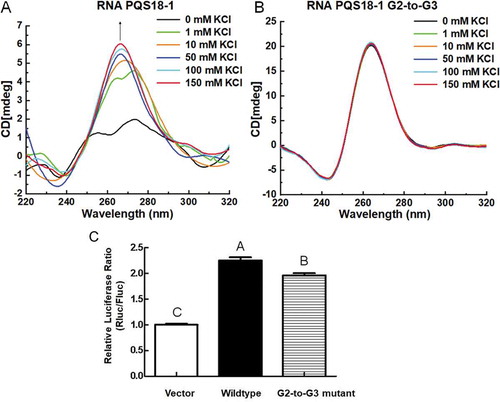
2.2.2. G2-quadruplex of RNA PQS18-1 shows higher gene expression than its G3-quadruplex mutant
G2-quadruplex was comprised of two G-quartets, whereas G3-quadruplex consisted of three G-quartets (). In our previous study, we found that there were much more G2-quadruplex forming sequences than G3-quadruplex forming sequences in the PRV genome, and we also found that G2-quadruplex was more sensitive to cations and small molecules than G3-quadruplex [Citation21]. We further investigated the different roles in altering gene expression played, respectively, by G2-quadruplex structure and G3-quadruplex. RNA PQS18-1-G2-to-G3 mutant was synthesized (5ʹ-GGGCUCGGGCGGGCGGGA-3ʹ) (Table S1). The conformations of RNA PQS18-1 changed with the increasing concentrations of potassium, and exhibited a typical CD spectrum of the parallel G-quadruplex when the added potassium was higher than 50 mM (). Sodium also induced the conformation change (Fig. S5), even though the variance with the increasing concentrations of sodium is different from that induced by potassium. The RNA PQS18-1-G2-to-G3 sequence was folded into very stable parallel G-quadruplex with no response to potassium concentration variation (). The above result showed that the conformation of RNA PQS18-1 was much more sensitive to potassium concentration than that of G2-to-G3 mutant, and that the G2-quadruplex structure was more adjustable than G3-quadruplex structure. The mutant 3ʹUTR containing PQS18-1-G2-to-G3 was cloned into the downstream of the hRluc gene. Compared to that of the wildtype construct, the relative luciferase activity of the reporter gene (hRluc) containing PQS18-1-G2-to-G3 in the 3ʹUTR was decreased significantly ().
Figure 5. CD spectra of RNA PQS18-1 and PQS18-1-G2-to-G3 and their effect on gene expression. A. CD spectra of RNA PQS18-1 in response to a series of concentrations of potassium. B. CD spectra of RNA PQS18-1-G2-to-G3 mutant in response to a series of concentrations of potassium. C. Relative luciferase activity of G2-to-G3 mutant construct. Different capital letters indicated the significant difference at P < 0.05 (Duncan’s multiple comparison test) among the constructs.
G3-quadruplexes in the 3ʹUTR could interact with several cellular proteins [Citation11,Citation29,], while the interaction between G2-quadruplexes and proteins were less documented. Our results indicated that the RNA PQS18-1 G2-quadruplex increased gene expression more significantly than G3-quadruplex, and that the former exhibited a significant conformation change, whereas the latter displayed no conformation change. The conformation change of G2-quadruplexes might be recognized by specific host proteins to enhance the translation process of viral protein, which might be attributed to natural selection during virus evolution.
2.2.3. A chemical compound destroys RNA PQS18-1 G2-quadruplex structure
G-quadruplex ligands were screened, and a chemical compound, TMPyP4 (), destroyed RNA PQS18-1 and displayed antiviral activity. CD spectra of RNA PQS18-1 exhibited the decreased positive peaks at 260 nm with the increasing concentrations of TMPyP4 (). The interaction between the compound and RNA PQS18-1 was examined in 1H NMR experiment. It was reported that the signal of imino proton from the tetrad-guanine would shift in location or change in shape after a ligand bound to G-quadruplex [Citation30,Citation31,]. After addition of TMPyP4, the 1H NMR spectroscopy showed that the typical G-quadruplex imino peaks of RNA PQS18-1 disappeared at 10.0 ~ 12.0 ppm (). The 1H NMR was consistent with the CD spectra, suggesting that the TMPyP4 destroyed the G-quadruplex of RNA PQS18-1.
Figure 6. CD spectra and 1H NMR spectra of RNA PQS18-1 added with TMPyP4 and luciferase activity of wildtype and G/T mutant constructs in response to TMPyP4. A. Structure of TMPyP4. B. CD spectra of RNA PQS18-1 added with TMPyP4 at different molar rates. C. 1H NMR spectra of RNA PQS18-1 added with TMPyP4. D. Relative luciferase activity of wildtype construct and G/T mutant construct in the presence of different concentrations of TMPyP4. Different capital letters indicated the significant difference at P < 0.05 (Duncan’s multiple comparison test) among the treatments with different concentrations of TMPyP4. ns: no significant difference.
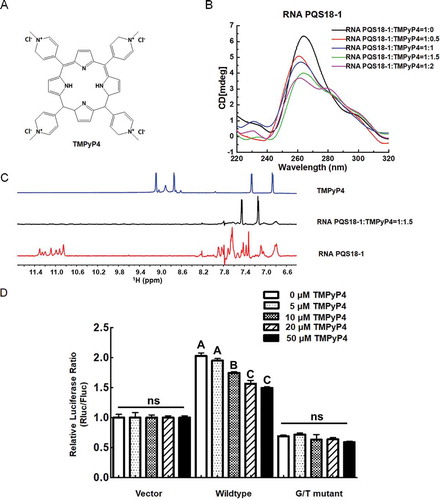
Subsequently, TMPyP4 was applied to the HEK293T cells transfected with wildtype construct psiCHECK-2-PQS18-1. The relative luciferase ratio of wildtype construct was decreased significantly with addition of TMPyP4, whereas the psiCHECK-2-G/T mutant did not respond to TMPyP4 (). These above results indicated that TMPyP4 destroyed PQS18-1 RNA G2-quadruplex and decreased gene expression in a dose-dependent manner.
2.2.4. TMPyP4 shows antiviral activity by destroying RNA PQS18-1 G2-quadruplex
The dual-luciferase assay result indicated that the chemical compound destroyed RNA PQS18-1 G-quadruplex structure, resulting in the decreased gene expression of reporter gene. The qRT-PCR was performed to examine the mRNA level of IE180 after the addition of TMPyP4. The results indicated that with the addition of 20 μM TMPyP4, the mRNA level of IE180 gene in the PRV-infected PK-15 cells was significantly lower than that of the control at 9 h post infection (). Further, the PRV-infected PK-15 cells were treated by TMPyP4 for 12 h, and the protein level of IE180 was analysed by western blot. The analysis result showed that the amount of IE180 in the PRV-infected PK-15 cells was decreased after treatment with TMPyP4 ().
Figure 7. Expression level of IE180 and virus titre of PRV in PRV-infected PK15 cells in the presence of TMPyP4. A. Transcription level of IE180 gene in PRV-infected PK15 cells in the presence of 20 μM TMPyP4. B. Expression level of IE180 protein in PRV-infected PK-15 cells in the presence of TMPyP4. Western blot were performed as previously described [Citation23]. The polyclonal antibody of IE180 was obtained from rabbits immunized with Peptide (seq: AGDGGAPPQRQPRRC). GAPDH monoclonal antibody was purchased from CWBIO, China. The numbers below the image represent the relative level of IE180 of experiment group corresponding to control group via ImageJ software analysis. C. Virus titre of PRV with addition of G-quadruplex ligand TMPyP4 in PK15 cells. The asterisk (**) indicated the significant difference at P < 0.01 (Student t-test) between TMPyP4 treatment group and control group.
![Figure 7. Expression level of IE180 and virus titre of PRV in PRV-infected PK15 cells in the presence of TMPyP4. A. Transcription level of IE180 gene in PRV-infected PK15 cells in the presence of 20 μM TMPyP4. B. Expression level of IE180 protein in PRV-infected PK-15 cells in the presence of TMPyP4. Western blot were performed as previously described [Citation23]. The polyclonal antibody of IE180 was obtained from rabbits immunized with Peptide (seq: AGDGGAPPQRQPRRC). GAPDH monoclonal antibody was purchased from CWBIO, China. The numbers below the image represent the relative level of IE180 of experiment group corresponding to control group via ImageJ software analysis. C. Virus titre of PRV with addition of G-quadruplex ligand TMPyP4 in PK15 cells. The asterisk (**) indicated the significant difference at P < 0.01 (Student t-test) between TMPyP4 treatment group and control group.](/cms/asset/7f3da031-5646-4e81-9830-a8eeb62cee67/krnb_a_1731664_f0007_b.gif)
In order to test the effect of TMPyP4 on virus proliferation, plaque assay was performed with PRV Ea strain, a virulent strain collected from Hubei province in China. The PK-15 cells were infected with PRV Ea strain at an MOI of 0.1, and then were treated with 20 μM TMPyP4 (Fig. S6) for 24 h. Afterwards, the virus was collected from the treated PK-15 cells for plaque assay. The plaque assay indicated that the addition of TMPyP4 at a concentration of 20 μM significantly reduced proliferation by one titre at 24 h post treatment (). The above results indicated that TMPyP4 decreased IE180 expression and reduced PRV proliferation at 24 h post infection.
3. Discussion
This study revealed that RNA G2-quadruplex in the 3ʹUTR of a viral gene was able to act as an important up-regulatory element. One conserved G-quadruplex sequence PQS18-1 was identified from 18 putative G-quadruplex sequences in the 3ʹUTR of IE180 gene of PRV by biophysical assays, and it was folded into stable parallel G2-quadruplex, resulting in the enhanced gene expression. Both G/T mutant sequence and G2-to-G3 mutant sequence of PQS18-1 in the 3ʹUTR of reporter gene resulted in lower relative luciferase activity than the wildtype PQS18-1. Destruction of PQS18-1 RNA G2-quadruplex by the chemical compound resulted in reducing gene expression and suppressing virus proliferation. RNA G-quadruplexes in the 3ʹUTR of human genes [Citation7] either enhanced mRNA stability by improving mRNA alternative polyadenylation [Citation14,Citation22,] and localization [Citation32], preventing miRNA binding [Citation13,Citation33,], or suppressed translation without affecting mRNA level [Citation15,Citation16,]. The PQS18-1 RNA G2-quadruplex was folded in the 3ʹUTR of IE180 gene, and it enhanced protein level, which was different from the reported role of RNA G-quadruplexes found in the 3ʹUTR of human genes as gene expression repressor.
G2-quadruplexes might act as sensors to ions, proteins, or cellular metabolites to regulate gene expression, in turn affecting immune evasion of virus. RNA PQS18-1 was very sensitive to potassium concentration gradient (). Potassium concentration gradient is the critical factor for normal function of nerve [Citation34]. RNA PQS18-1 might play a role in distinguishing the latency site of PRV, trigeminal neurons [Citation18,Citation34,] which exhibited high concentration of potassium and low concentration of sodium for regular neuron activity. G3-quadruplexes from eukaryotic genes have been reported to be bound by DHX36 [Citation29], hnRNPF [Citation35], SLIRP [Citation36], and other RNA-binding proteins with RGG motif [Citation37]. In PRV genome, the number of G2-quadruplex sequences were over seven folds more than that of G3-quadruplex [Citation21]. G2-quadruplex folded from PQS18-1 might be specifically chosen by the PRV for escaping from the cellular immune molecules or proteins. It will be very interesting to find the host proteins interacting with RNA PQS18-1 and reveal their binding mode. Our findings indicated that the RNA G2-quadruplex from 3ʹUTR of viral gene could be post-transcription regulation element with various conformations in response to cellular cation and small molecule. This study would provide an insight into the interaction between herpesvirus and its host cell and reveal the mechanism underlying immune evasion and latency establishment. The binding site between viral RNA G2-quadruplex and cellular factors will be potential target site for antiviral drug development.
In conclusion, we identified a functional RNA G2-quadruplex forming sequence PQS18-1 from the 3ʹUTR region of the important immediate early gene IE180 of PRV. The G2-quadruplex structure could enhance expression of IE180 protein, and destruction of this G2-quadruplex by small molecule could suppress IE180 expression. Study on the interaction between PQS18-1 RNA G2-quadruplex and cell factors may also help to understand IE180 expression regulation and PRV latency establishment in host cells. This study reveals that G2-quadruplex could act as regulatory element to enhance viral gene expression, and it can be potential target for anti-virus therapy.
4. Materials and methods
4.1. 3ʹ-end rapid amplification of cDNA end (RACE)
Nineteen PRV complete genome sequences from different strains were downloaded from the NCBI Genome database (https://www.ncbi.nlm.nih.gov/). The 3ʹUTRs of IE180 gene were not annotated clearly with experimental evidence. The predicted 3ʹUTRs of IE180 gene were of high GC content. The total RNA was extracted from PK15 cells that were infected by PRV Ea strain. The cDNA were reverse-transcribed from total RNA with the primer QT (Table S1). The 3ʹUTR of IE180 gene was amplified from the cDNA through nest PCR with two gene specific primers, GSP1 and GSP2, and two library-specific primers, QO and QI (Table S1), and with Ex Taq DNA polymerase in 2× GC buffer (TAKARA, Japan). Then, the product of inner PCR amplified with GSP2 and QI was cloned into the PMD18-T vector and sequenced.
4.2. Percentage conservation of putative G-quadruplex sequences in the 3ʹUTR of IE180 gene among PRV genomes
The putative G-quadruplex sequences in the 3ʹUTR of IE180 in the PRV Ea strain were searched by modified Quadparser software [Citation38]. The formula of the putative G-quadruplex sequence was defined as G2+N1-7G2+N1-7G2+N1-7G2+, where G is guanine, and N is any nucleotide including guanine. The percentage conservation of the putative G-quadruplex sequences of 19 PRV genomes with G-rich sequences in the 3ʹUTR of IE180 gene was analysed (Table S2). The sequence alignment was performed with DNAMAN software.
4.3. Oligonucleotide folding condition
All DNA oligonucleotides (Table S1) purchased from Takara Biomedical Technology (Beijing, China) were salt-free, purified, and dissolved in ddH2O to achieve the concentration of 100 μM. Oligonucleotides were diluted to 20 μM in 10 mM potassium phosphate buffer (pH 7.0) supplemented with 100 mM KCl, then heated to 95°C for 5 min in a 1.5 mL Eppendorf tube in water bath, subsequently, slowly cooled for ~8 h to room temperature, then used for spectrum analysis or storage at 4°C.
The RNA PQS18 and RNA PQS18-1 were purchased from Takara Biomedical Technology (Beijing, China) with HPLC purification. The RNA was dissolved in nuclease-free water (Thermo Fisher, USA), and diluted to 20 μM in 10 mM potassium phosphate buffer (pH 7.0) supplemented with a series of concentrations of KCl. The samples were heated to 95°C for 5 min in a 1.5 mL Eppendorf tube in water bath, subsequently, slowly cooled for ~8 h to room temperature, then used for spectrum analysis or storage at 4°C.
The RNA PQS18-1 for CD titration was purchased from Takara Biomedical Technology (Beijing, China) with HPLC purification. The meso-Tetra (N-methyl-4-pyridyl) porphine tetrachloride, TMPyP4 (CAS# 92739-63-4), was purchased from Frontier Scientific (Logan, Utah, USA). The RNA was dissolved in nuclease-free water (Thermo Fisher, USA) and diluted to 20 μM in 5 mM KCaCo buffer (pH 6.5) supplemented with 150 mM KCl. The samples were heated to 95°C for 5 min in a 1.5 mL Eppendorf tube in water bath, subsequently, slowly cooled for ~8 h to room temperature. Then, TMPyP4 was added into the RNA sample at different molar ratios and the mixture were incubated at room temperature for 10 min before test.
4.4. Thermal difference spectra
Thermal difference spectra (TDS) were used for distinguishing structures of nucleic acid based on the ultraviolet absorbance spectra [Citation26]. TDS was calculated by subtracting the spectra between 220 and 340 nm of the folded structure at 20°C from that of the unfolded structure at 95°C.
4.5. Circular dichroism
CD spectra were recorded on a Jasco J-810 spectropolarimeter using a 1 mm path length quartz cuvette. The scanning was performed at room temperature (20°C) between 200 and 320 nm. Data were collected at a scanning speed of 100 nm/min with data pitch of 0.5 nm, response time of 1 s, bandwidth of 2 nm, and sensitivity of 100 mdeg. Each spectrum was derived from the average of three scans. Samples containing only buffer were also scanned according to these parameters for background subtraction.
4.6. Nuclear magnetic resonance (NMR) spectroscopy
1D 1H NMR spectroscopy experiments for DNA PQS5, DNA PQS8, and DNA PQS18 were performed on a Bruker 600 MHz (BioSpin, Germany). The 600 μL sample containing 0.2 mM DNA, 10 mM potassium phosphate (pH = 7), 100 mM KCl, and 10% D2O were folded at 298 K. The 1D 1H NMR spectroscopy experiments for RNA PQS18 and RNA PQS18-1 were performed on a Varian INOVA 600 MHz spectrometer (Varian, USA). The 500 μL sample containing 0.15 mM RNA PQS18-1, 20 mM potassium cacodylate (pH = 6.5), 50 mM KCl, and10% D2O were folded at 293 K. The TMPyP4 was solved in D2O as the stock solution. The RNA PQS18-1 and TMPyP4 were mixed at a molar ratio of 1:1.5 in the solution containing 20 mM potassium cacodylate (pH = 6.5), 50 mM KCl, and 10% D2O at 293 K. The NMR experiments for samples in water solution were performed with Watergate or Jump-and-Return water suppression techniques. The mixing time was 300 ms. In 1D experiments, NMR spectra were processed and visualized by MestReNova (mestrelab.com). A total of 256 scans were acquired for each spectrum of DNA with a relaxation delay of 2 s between scans. The 1024 scans were acquired for each spectrum of RNA with a relaxation delay of 2 s between scans.
4.7. Plasmid construction
The basic vector for the dual-luciferase assay experiments was psiCHECK-2 (Promega, USA) reporter vector which contains the renilla luciferase gene as reporter gene and the firefly luciferase gene as control gene. The full-length of the wildtype 3ʹUTR of IE180 gene derived from PRV Ea strain was cloned downstream of the open reading frame of renilla luciferase gene. The G-to-T mutant construct (G/T mutant) was produced from wildtype construct by replacing the PQS18-1 with PQS18-1-G/T-mut (Table S1) by using the ClonExpress II One Step Cloning Kit (Vazyme, China). The G2-to-G3 mutant construct was produced from wildtype construct by replacing the PQS18-1 with PQS18-1-G2mutG3 sequence (Table S1) by using the same kit mentioned above.
4.8. Cell culture
Cell lines, human embryonic kidney (HEK) 293T, porcine kidney cell (PK-15), and bovine kidney cells (MDBK) were provided by the State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University in China. The above cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% foetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin (Invitrogen). Cells were grown at 37°C in a humidified atmosphere with 5% CO2.
4.9. Transfection and dual-luciferase assays
The HEK293T cells were seeded in the 24-well plates at the concentration of 1 × 105 cells/well. The cells were transfected with 0.8 μg of psiCHECK-2 reporter plasmids and Lipofectamine 2000 (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s instructions. The medium was replaced at 6 h post transfection. Subsequently, a series of concentrations of meso-Tetra (N-methyl-4-pyridyl) porphine tetrachloride (TMPyP4) [Citation39] (Frontier Scientific, USA) was added to the cells. The activities of firefly and renilla luciferase were measured at 24 h after addition of TMPyP4 by using the Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI, USA) on a GloMax 20/20 luminometer (Promega, Madison, WI, USA).
4.10. qRT-PCR
The HEK293T cells grown in 24-well plates were transfected with plasmids, as described above with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Total RNA was extracted using Trizol reagent (Invitrogen) at 24 h post transfection. PRV-infected PK15 cells were treated with TMPyP4 at the concentration of 20 μM for 12 h, and the cell samples were collected every 3 h for total RNA extraction. The 1 μg total RNA from each sample was reverse-transcribed into cDNA by using a Transcriptor First Strand cDNA Synthesis Kit (Roche), then 1 μL of the resultant cDNA was used for an SYBR green PCR assay (Applied Biosystems, USA). The individual mRNA transcript in each sample was assayed for three times. The PCR procedure was as follows: pre-denaturation at 95°C for 10 min, then 40 cycles of 15 s at 95°C, 15 s at 58°C and 40 s at 72°C. The primers of firefly luciferase and renilla luciferase were designed by Primer Express software (version 3.0, Applied Biosystems, Carlsbad, CA) (Table S1). The primers of IE180 [Citation40] and β-actin [Citation41] genes were presented (Table S2), and the expression level of IE180 was normalized according to the expression of β-actin gene. The relative gene expression level was calculated with 2−ΔΔCt method.
4.11. Western blot analysis
PK-15 cells were infected with PRV at an MOI of 0.1 for 2 h at 4°C in dishes which were shaken once every 15 min during the infection. After the infection at 4°C, the virus solution was removed and replaced with 2 mL supplemented DMEM containing 20 μM TMPyP4, and the cells were incubated at 37°C for 12 h. Proteins were harvested at 12 h post infection (hpi) and lysed with SDS-PAGE loading buffer. Western blot was performed as previously described [Citation23]. The polyclonal antibody of IE180 was obtained from rabbits immunized with Peptide (seq: AGDGGAPPQRQPRRC). GAPDH monoclonal antibody was purchased from CWBIO, China. The relative expression level of IE180 in the treatment group corresponding to the control group was analysed via ImageJ software.
4.12. Cytotoxicity assay
The cytotoxicity of TMPyP4 to PK15 cells was determined by the MTT assay through the measurement of mitochondrial dehydrogenase enzyme activity from viable cells. The PK15 cells at the density of 1 × 104 cells per well were seeded in the 96-wells microplate. When cell confluence reached 90%, the growth medium was replaced with 100 μL supplemented DMEM containing TMPyP4 at different final concentrations (25 μM, 50 μM, 100 μM, 200 μM), and the cells were incubated at 37°C for 24 h. Then, 50 μL MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution was added into each well and the cells were incubated at 37°C for another 4 h. After the incubation, the supernatant was removed and 150 μL DMSO was added into each well to dissolve the MTT. The relative cell viabilities were analysed by measuring the absorbance at 570 nm on the SynergyTM HTX microplate reader (BioTek, USA).
4.13. Plaque assay
The antiviral activity of TMPyP4 to PRV Ea strain was tested through plaque assay. The PK15 cells at the density of 1.2 × 106 cells per well were seeded in the 6-well plate. When cell confluence reached 80-90%, the cells were placed at 4°C for 1 h, and then the PRV Ea strain virus solution (MOI = 0.1) was added into each well. The 6-well plate was shaken once every 15 min during the incubation. After the incubation at 4°C, the virus solution was replaced with 2 mL supplemented DMEM containing 20 μM TMPyP4, and the cells were incubated at 37°C for 24 h. After incubation, the infectious viral particles were isolated from each well and prepared for viral titre measurement.
MDBK cells at the density of 2 × 105 cells per well were seeded in the 12-well plate. When the confluence reached 90%, the cells were used for the viral titre measurement. The 200 μL virus solution was added into each well and incubate at 37°C for 2 h. Then, the infected cells were covered by 4% sodium carboxymethylcellulose (CMC-Na) coupled with 3% FBS and 1% Penicillin-Streptomycin solution. After incubation at 37°C for another 48 h, the infected cells were fixed and stained with the crystal violet solution (0.35%, W/V in ethanol) at room temperature. After 15-min staining, the crystal violet solution was removed and the plate was washed with tap water. Finally, the viral titre was determined by the plaque assay. Triplicates were set up for each group.
4.14. Statistical analysis
Student’s t-test was used to determine the significant differences in plaque assay. One-way analysis of variance (ANOVA) coupled with Duncan’s multiple comparison was used to determine the significant difference among various treatments of dual-luciferase assay and Q-PCR with the SPSS software (version 16.0).
Author Contributions
Yashu Zhang: methodology, software, validation, formal analysis, investigation, data curation, writing—original draft preparation, visualization. Sisi Liu: Conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft preparation, visualization, project administration, funding acquisition. Hui Jiang: methodology, validation, data curation, writing—original draft preparation, visualization. Hui Deng: methodology, investigation. Chen Dong: visualization. Wei Shen: visualization. Haifeng Chen: data curation. Chao Gao: methodology. Shaobo Xiao: resources, writing-review and editing. Zheng-Fei Liu: Conceptualization, methodology, software, resources, writing-review and editing, supervision, project administration. Dengguo Wei: Conceptualization, methodology, software, resources, writing-review and editing, supervision, project administration, funding acquisition.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (1.2 MB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kulkarni S, Savan R, Qi Y, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498.
- Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3ʹ UTRs via Alu elements. Nature. 2011;470:284–288.
- Matsushita K, Takeuchi O, Standley DM, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–1190.
- Hartwick EW, Costantino DA, MacFadden A, et al. Ribosome-induced RNA conformational changes in a viral 3ʹ-UTR sense and regulate translation levels. Nat Commun. 2018;9:5074.
- Huber RG, Lim XN, Ng WC, et al. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat Commun. 2019;10:1408.
- Nicholson BL, White KA. Functional long-range RNA-RNA interactions in positive-strand RNA viruses. Nat Rev Microbiol. 2014;12:493–504.
- Huppert JL, Bugaut A, Kumari S, et al. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008;36:6260–6268.
- Sauer M, Juranek SA, Marks J, et al. DHX36 prevents the accumulation of translationally inactive mRNAs with G4-structures in untranslated regions. Nat Commun. 2019;10:2421.
- Saranathan N, Vivekanandan P. G-quadruplexes: more than just a kink in microbial genomes. Trends Microbiol. 2019;27:148–163.
- Balasubramanian S, Neidle S. G-quadruplex nucleic acids as therapeutic targets. Curr Opin Chem Biol. 2009;13:345–353.
- Decorsiere A, Cayrel A, Vagner S, et al. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3ʹ-end processing and function during DNA damage. Genes Dev. 2011;25:220–225.
- Subramanian M, Rage F, Tabet R, et al. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep. 2011;12:697–704.
- Rouleau S, Glouzon JS, Brumwell A, et al. 3ʹ UTR G-quadruplexes regulate miRNA binding. RNA. 2017;23:1172–1179.
- Beaudoin JD, Perreault JP. Exploring mRNA 3ʹ-UTR G-quadruplexes: evidence of roles in both alternative polyadenylation and mRNA shortening. Nucleic Acids Res. 2013;41:5898–5911.
- Arora A, Suess B. An RNA G-quadruplex in the 3ʹ UTR of the proto-oncogene PIM1 represses translation. RNA Biol. 2011;8:802–805.
- Crenshaw E, Leung BP, Kwok CK, et al. Amyloid precursor protein translation is regulated by a 3ʹUTR guanine quadruplex. PLoS One. 2015;10:e0143160.
- Bolduc F, Garant JM, Allard F, et al. Irregular G-quadruplexes found in the untranslated regions of human mRNAs influence translation. J Biol Chem. 2016;291:21751–21760.
- Pomeranz LE, Reynolds AE, Hengartner CJ. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev. 2005;69:462–500.
- Kozmík Z, Arnold L, Paces V. Multiple sets of adjacent μE1 and oct- 1binding sites upstream of the pseudorabies virus immediate-early gene promoter. Virology. 1991;182:239–249.
- Daniel-Carlier N, Sawafta A, Passet B, et al. Viral infection resistance conferred on mice by siRNA transgenesis. Transgenic Res. 2013;22:489–500.
- Deng H, Gong B, Yang Z, et al. Intensive distribution of G(2)-quaduplexes in the pseudorabies virus genome and their sensitivity to cations and G-quadruplex ligands. Molecules. 2019;24:774.
- Lightfoot HL, Hagen T, Clery A, et al. Control of the polyamine biosynthesis pathway by G2-quadruplexes. Elife. 2018;7:e36362.
- Wang X, Wu CX, Song XR, et al. Comparison of pseudorabies virus China reference strain with emerging variants reveals independent virus evolution within specific geographic regions. Virology. 2017;506:92–98.
- Madireddy A, Purushothaman P, Loosbroock CP, et al. G-quadruplex-interacting compounds alter latent DNA replication and episomal persistence of KSHV. Nucleic Acids Res. 2016;44:3675–3694.
- Kypr J, Kejnovska I, Renciuk D, et al. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009;37:1713–1725.
- Mergny JL, Li J, Lacroix L, et al. Thermal difference spectra: a specific signature for nucleic acid structures. Nucleic Acids Res. 2005;33:e138.
- Ambrus A, Chen D, Dai J, et al. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry. 2005;44:2048–2058.
- Dai J, Carver M, Hurley LH, et al. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J Am Chem Soc. 2011;133:17673–17680.
- Chen MC, Tippana R, Demeshkina NA, et al. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature. 2018;558:465–469.
- Wang KB, Elsayed MSA, Wu G, et al. Indenoisoquinoline Topoisomerase Inhibitors Strongly Bind and Stabilize the MYC promoter G-quadruplex and downregulate MYC. J Am Chem Soc. 2019;141:11059–11070.
- Morris MJ, Wingate KL, Silwal J, et al. The porphyrin TmPyP4 unfolds the extremely stable G-quadruplex in MT3-MMP mRNA and alleviates its repressive effect to enhance translation in eukaryotic cells. Nucleic Acids Res. 2012;40:4137–4145.
- Rihan K, Antoine E, Maurin T, et al. A new cis-acting motif is required for the axonal SMN-dependent Anxa2 mRNA localization. RNA. 2017;23:899–909.
- Stefanovic S, Bassell GJ, Mihailescu MR. G quadruplex RNA structures in PSD-95 mRNA: potential regulators of miR-125a seed binding site accessibility. RNA. 2015;21:48–60.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ. 2016;40:480–490.
- Huang H, Zhang J, Harvey SE, et al. RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev. 2017;31:2296–2309.
- Williams P, Li L, Dong X, et al. Identification of SLIRP as a G quadruplex-binding protein. J Am Chem Soc. 2017;139:12426–12429.
- Huang ZL, Dai J, Luo WH, et al. Identification of G-quadruplex-binding protein from the exploration of RGG Motif/G-quadruplex interactions. J Am Chem Soc. 2018;140:17945–17955.
- Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–2916.
- Qin Y, Rezler EM, Gokhale V, et al. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007;35:7698–7713.
- Zhao X, Cui Q, Fu Q, et al. Antiviral properties of resveratrol against pseudorabies virus are associated with the inhibition of IkappaB kinase activation. Sci Rep. 2017;7:8782.
- Loving CL, Brockmeier SL, Sacco RE. Differential type I interferon activation and susceptibility of dendritic cell populations to porcine arterivirus. Immunology. 2007;120:217–229.