
ABSTRACT
Cas12e proteins (formerly CasX) form a distinct subtype of Class II type V CRISPR-Cas effectors. Recently, it was shown that DpbCas12e from Deltaproteobacteria and PlmCas12e from Planctomycetes can introduce programmable double-stranded breaks in mammalian genomes. Thus, along with Cas9 and Cas12a Class II effectors, Cas12e could be harnessed for genome editing and engineering. The location of cleavage points in DNA targets is important for application of Cas nucleases in biotechnology. DpbCas12e was reported to produce extensive 5ʹ-overhangs at cleaved targets, which can make it superior for some applications. Here, we used high throughput sequencing to precisely map the DNA cut site positions of DpbCas12e on several DNA targets. In contrast to previous observations, our results demonstrate that DNA cleavage pattern of Cas12e is very similar to that of Cas12a: DpbCas12e predominantly cleaves DNA after nucleotide position 17–19 downstream of PAM in the non-target DNA strand, and after the 22nd position of target strand, producing 3–5 nucleotide-long 5ʹ-overhangs. We also show that reduction of spacer sgRNA sequence from 20nt to 16nt shifts Cas12e cleavage positions on the non-target DNA strand closer to the PAM, producing longer 6–8nt 5ʹ-overhangs. Overall, these findings advance the understanding of Cas12e endonucleases and may be useful for developing of DpbCas12e-based biotechnology instruments.
Introduction
RNA-guided effector nucleases from Class II CRISPR-Cas bacterial defenсe systems are widely used as biotechnology instruments. These enzymes found numerous applications in targeted genome editing, regulation of transcription, and epigenetic modulation [Citation1]. SpCas9 nuclease from Streptococcus pyogenes was the first Cas nuclease used for genome editing in eukaryotes [Citation2,Citation3]. It belongs to type II of Class II CRISPR-Cas nucleases and remains the best-characterized and most widely used Cas protein to date [Citation1,Citation4]. In addition to SpCas9, other type II Cas nucleases were successfully used in genome engineering [Citation5–Citation8].
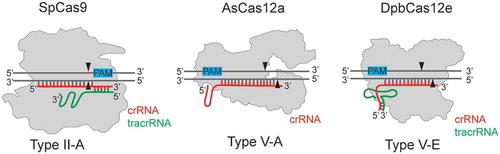
Besides Cas9 enzymes, CRISPR-Cas effectors of other Class II types have found biotechnological applications [Citation9,Citation10]. Thus, Cas12a proteins belonging to type V-A CRISPR-Cas effectors possess specific features distinguishing them from the Cas9 proteins, due to their distinct domain organization. For instance, unlike Cas9, Cas12a is able to catalyse maturation of crRNA in the absence of other factors, which facilitates multiplex genome editing [Citation11,Citation12]. In 2017 Burstein et al. discovered new Cas protein belonging to Class II type V-E CRISPR-Cas systems – Cas12e (formerly CasX) [Citation4,Citation13]. Although Cas12e proteins demonstrate some similarities to Cas12a in domain organization, which will be discussed later, Cas12e and Cas12a effectors are quite distinct. For instance, in opposite to Cas12a which requires only crRNA for DNA recognition, Cas12e are dual-RNA-guided effectors () [Citation4,Citation13].
Figure 1. Cas12a and Cas12e belong to Class II Type V CRISPR-Cas effectors, subtypes V-A and V-E, correspondingly.
In contrast to Cas12a, Cas12e enzymes require tracrRNA in addition to crRNA for DNA target recognition. crRNA indicated in red, tracrRNA indicated in green. PAM sequences are shown with blue rectangles. SpCas9 – Cas9 from Streptococcus pyogenes (1,368 amino acids), AsCas12a – Cas12a from Acidaminococcus sp. (1,307 amino acids), DpbCas12e – Cas12e from Deltaproteobacteria (986 amino acids). The pairing between DNA and RNA molecules, as well as indicated positions of DNA cleavage sites shown are schematic.
In 2019 Jun-Jie Liu et al. using electron microscopy determined the domain composition of DpbCas12e protein from Deltaproteobacteria and also, demonstrated that DpbCas12e, as well as PlmCas12e from Planctomycetes, have genome editing activity in human cells [Citation14]. Analysis of DpbCas12e-sgRNA-DNA complex revealed non-target-strand binding (NTSB) and target-strand loading (TSL) domains [Citation14]. The TSL domain is located in a position analogous to that of the so-called ‘Nuc’ domain in Cas12a [Citation14,Citation15]. These domains perform similar functions in Cas12e and Cas12a enzymes: after non-target DNA strand cleavage by the RuvC domain, they bend sgRNA-DNA duplex. This conformational change allows the target DNA strand to be cleaved by the RuvC domain [Citation14]. Thus, both Cas12e and Cas12a, rely on a single nuclease domain for double stranded DNA cleavage, in contrast to Cas9, which uses distinct domains, HNH and RuvC, to cleave each DNA strand [Citation14,Citation16,Citation17]. In Cas12e and Cas12a, a large structural change alters accessibility of DNA strands for the RuvC nuclease and in this way compensates the lack of the second nuclease domain [Citation14,Citation17].
Possibly due to the similarity of the DNA cleavage mechanism, both Cas12a and Cas12e generate products with staggered ends. In contrast, Cas9 proteins mainly produce blunt ends [Citation14,Citation18,Citation19]. Interestingly, Jun-Jie Liu et al. found that DpbCas12e produces staggered ends about 10-nucleotides long [Citation14], which is longer than 3–5nt overhangs usually produced by Cas12a proteins. The 5ʹ-overhangs produced by Cas12a and Cas12e, potentially can be used for in vivo or in vitro insertion of DNA fragments into genome through direct DNA ligation [Citation20]. Determination of precise positions of Cas12e DNA cleavage is important for applications of these nucleases in biotechnology [Citation20,Citation21].
In most studies published to date, mapping of Cas nucleases cut sites was performed using Sanger sequencing of fragments produced in in vitro DNA cleavage reactions [Citation22,Citation23], which does not reveal the entire distribution of DNA along the target. High throughput sequencing (HTS) allows much more comprehensive, both quantitative and qualitative, characterization of cleavage sites. Although HTS has been used for both Cas9 and Cas12a-induced DSB (double-stranded DNA breaks) determination, the goal of most of these experiments was the evaluation of off-targeting effects in vivo or in vitro but not the precise determination of DNA cleavage sites positions [Citation18,Citation19,Citation24,Citation25,Citation26]. The cell-based assays for in vivo mapping of DSBs miss information about the precise DNA cleavage positions due to modulation of initial DSBs by endogenous cell nucleases and reparation processes.
Here, we performed HTS mapping of DpbCas12e cut sites produced during in vitro DNA cleavage reactions using six different dsDNA targets. We determined DpbCas12e cut sites positions distribution and found the average length of 5ʹ-overhangs generated by this nuclease. Our results show that Cas12e DNA cleavage pattern is very similar to that produced by the Cas12a proteins. We also show that the reducing of the length of spacer segment of sgRNA from 20nt to 16nt can significantly increase the length of 5ʹ-overhangs generated by Cas12e nuclease. These findings can inform development of new DpbCas12e-based genome engineering tools.
Materials and methods
Plasmids
For expression of DpbCas12e, AsCas12a and SpCas9 in E. coli pET21a-based genetic vectors carrying the corresponding genes were cloned. The maltose binding protein (MBP) was added to DpbCas12e N-termini through TEV protease cleavage site to increase solubility of DpbCas12e nuclease. The plasmids maps are presented in the Supplementary Table S1.
Recombinant proteins purification
For recombinant SpCas9 and AsCas12a proteins purification competent E. coli Rosetta cells were transformed with pET21a_SpCas9, and pET21a_AsCas12a plasmid and grown till OD600 = 0.6 in 500 ml LB media supplemented with 100 μg/ml ampicillin. The protein synthesis was induced by adding 1 mM IPTG. After 6 hours of incubation at 22°C the cells were centrifugated at 4000 g and the pellet was lysed by sonication in lysis buffer containing 50 mM Tris-HCl pH = 8 (4°C), 500 mM NaCl, 1 mM beta-mercaptoethanol and 10 mM imidazole supplemented with 1 mg/ml lysozyme (Sigma-Aldrich L6876). The cell lysate was centrifuged at 16,000 g (4°C) and filtered through 0.22 μm filters. The lysate was applied to 1 ml HisTrap HP column (GE Healthcare) and SpCas9 or AsCas12a were eluted by 300 mM imidazole. After affinity chromatography the sample was applied on a Superdex200 Increase 10/300 GL (GE Healthcare) column equilibrated with a buffer containing 50 mM Tris-HCl pH = 8 (4°C), 500 mM NaCl, 1 mM DTT. Fractions containing SpCas9 or AsCas12a monomers were pooled and concentrated using 30 kDa Amicon Ultra-4 centrifugal unit (Merc Millipore, UFC803008). Glycerol was added to a concentration of 10% and the proteins were flash-frozen in liquid nitrogen and stored at −80°C. The purity of the protein was assessed by denaturing 10% PAGE.
For recombinant DpbCas12e purification competent E. coli Rosetta cells were transformed with pET21a_DpbCas12e plasmid and grown till OD 600 = 0.6 in 500 ml L of TB (Terrific broth) media supplemented with 100 g/ml ampicillin according to DpbCas12e purification protocol described earlier (Jun-Jie Liu et al.) The protein synthesis was induced by addition of 1 mM IPTG. After 18 hours of incubation at 16°C the cells were centrifugated at 4000 g and the pellet was lysed by sonication in the lysis buffer containing 50 mM Hepes-HCl pH = 7.5, 500 mM NaCl, 10% glycerol, 1 mM beta-mercaptoethanol supplemented with 1 mg/ml lysozyme (Sigma-Aldrich L6876). The cell lysate was centrifuged at 16,000 g and processed through 0.22 μm filters. The lysate was applied to the 1 ml HisTrap HP Column (GE Healthcare) and DpbCas12e was eluted by 300 mM imidazole. After affinity chromatography the sample was either digested with TEV protease for 16 hours at 4°C to cleave the MBP-tag or applied straight on the Superdex200 Increase 10/300 GL (GE Healthcare) column equilibrated with a buffer containing 50 mM Hepes-HCl pH = 7.5, 500 mM NaCl, 1 mM DTT and 10% glycerol. Fractions containing DpbCas12e monomers were pooled and concentrated using 30 kDa Amicon Ultra-4 centrifugal unit (Merc Millipore, UFC803008) to a concertation of about 2 mg/mL, aliquoted and flash-frozen in liquid nitrogen with subsequent storage at −80°C. The purity of the protein was assessed by denaturing 10% PAGE.
In vitro DNA cleavage assays and samples preparation for HTS
DNA cleavage reactions were performed using the recombinant proteins MBP_DpbCas12e (or DpbCas12e where indicated), AsCas12a or SpCas9, in vitro synthesized guide RNAs and linear dsDNA targets. dsDNA targets were prepared by PCR amplifications of pUC19 plasmid (Targets 1, 2, 6), and human grin2b gene (Targets 3, 4, 5) using primers listed in the Supplementary Table S2. The full sequences of DNA targets are presented in the Supplementary Table S1.
Guide RNAs were synthesized in vitro using HiScribe T7 High Yield RNA Synthesis Kit (NEB, E20140). The sequences of guide RNAs used in this study are presented in the Supplementary Table S3. To pre-form an active ribonucleoprotein complexes the recombinant proteins were mixed with guide RNAs in 1x reaction buffer (20 mM HEPES, pH 7.5, 10 mM MgCl2, 150 mM KCl, 1% glycerol, 1 mM DTT for DpbCas12e or 1x CutSmart buffer (NEB B7204 S, 1 mM DTT) and incubated at room temperature for 10 min. All RNAs used in this study are listed in Supplementary Table S3. Further the DNA targets were added to ribonucleoprotein complexes to the final concentration of the components in the cleavage reactions: 50 nM DNA, 400 nM recombinant protein (1600 nM in case of DpbCas12e), 2 µM guide RNA and 1x reaction buffer in 20 µl final volume. The DNA cleavage reaction mix was incubated at 37°C for 30 min. The reaction was stopped by the adding of 0.5 µl proteinase K (Thermo Fisher Scientific, EO0491) and the subsequent incubation for 30 min at 37°C. The DNA cleavage reaction products were separated by 1.5% agarose gel electrophoresis. The cleaved DNA fragments were isolated from the gel and purified by GeneJET Gel Extraction Kit (Thermo Fisher Scientific, K0692) (two DNA fragments produced by the nuclease cleavage were isolated from agarose gel as one mix in case of each sample). DNA fragments were eluted with 40 µl water.
End repair was performed using modified protocol for DNA blunting used in ChIP assays described by Blecher-Gonen et al. 25 µl of end repair buffer (1х T4 DNA ligase buffer (NEB, B0202), 0.1 mg ml-1 BSA, 0.1 mM dNTPs), 1 µl T4 PNK (Thermo Fisher Scientific, EK0031) and 1 µl T4 polymerase (NEB, M0203) were added to each 40 µl sample (total reaction volume was 67 µl). The reactions were incubated in a thermal cycler using the following settings: 15 min at 15°C, 15 min at 25°C, chilling to 4°C. Agencourt RNAClean XP (Beckman Coulter) beads in 2.5x ratio were used to cleanup the DNA after the end-repair reaction. The DNA fragments were eluted in 40 µl of 10 mM Tris-HCl (pH 8.0).
For A-base addition reaction 6 µl 10xNEB buffer 2 (NEB, B7002), 0.1 µl dATP (100 mM) and 10.9 µl of nuclease-free water were added to each 40 µl DNA sample volume. This yielded to the following final concentration: 1xNEB buffer 2 (NEB, B7002) and 167 µM dATP. 3 µl of Klenow Fragment (3ʹ→5ʹ exo-) (NEB, M0212) were added to the samples and the reactions were incubated at 37°C for 30 min in the thermal cycler. Agencourt RNAClean XP (Beckman Coulter) beads in 2.5x ratio were used to cleanup the DNA after the reaction. The DNA fragments were eluted in 60 µl of 10 mM Tris-HCl (pH 8.0). The further sample preparation for high throughput sequencing (HTS) was performed using NEBNext Ultra II DNA Prep Kit for Illumina (NEB, E7645) starting with the adaptor ligation step. The samples were sequenced using Illumina platform with pair-end 150 cycles (75 + 75) or 300 cycles (150 + 150).
Computational Sequence analysis
HTS of each DNA cleavage reaction products produced in average 200 000 sequencing paired-end reads. To determine the cut sites positions of the cleaved DNA molecules the following pipeline was used. Forward and reverse reads (R1 and R2) were mapped against whole DNA target molecule using BWA (Li, Durbin, 2009). All unmapped reads were discarded from the analysis. The PAM sequence, a spacer and 10 nucleotides after the spacer were chosen as the region of interest. Reads matching the reference upstream the region of interest contained the information about target DNA strand (TS) cleavage position. Reads matching the reference downstream the region of interest contained information about non-target DNA strand (NTS) cleavage position. The number of analysed reads for each target is listed in Supplementary File S2 and Supplementary File S3. These reads were compared to the reference to determine the cut site positions. The number of nucleotides after the PAM sequence (for TS cleavage position determination) or before the end of the region of interest (for NTS cleavage position determination) were counted (Supplementary File S2, Supplementary File S3). To estimate the range of possible lengths of overhangs and calculate the maximal probabilities of their generation, distances between DSBs were computed as Distance = [cut site position on TS] – [cut site position on NTS]. Distances were calculated between all possible TS and NTS DNA cleavage positions. Relative frequencies of generated overhangs were calculated as a sum of [relative frequency of cut site positions on TS] x [relative frequency of cut site positions on NTS] for all combinations of TS and NTS producing the overhangs of certain length.
Results
Mapping of DpbCas12e and AsCas12a cut sites
For precise determination of Cas12e DNA cut site positions and comparison with Cas12a, we performed DNA cleavage reactions of several different targets in vitro. Recombinant versions of wild-type DpbCas12e from Deltaproteobacteria and AsCas12a from Acidaminococcus sp. were purified from E. coli cells (Supplementary Fig. S1). Due to a poor solubility of DpbCas12e, it was purified as an N-terminal fusion to MBP (maltose binding protein). The widely used SpCas9 nuclease from Streptococcus pyogenes, which predominantly produces double-strand DNA breaks with blunt endings, was used as a control. DpbCas12e, AsCas12a, and SpCas9 effector nucleases require different protospacer adjacent motifs (PAMs) for efficient target recognition and cleavage. DpbCas12e and AsCas12a require upstream PAMs with, respectively, 5ʹ-TTCN-3ʹ and 5ʹ-TTTV-3ʹ consensus; the SpCas9 PAM is 5ʹ-NGG-3ʹ and is located downstream of the target. To compare the DpbCas12e and AsCas12a DNA cut site positions, we used DNA targets with a ‘TTTCCN’ sequence at the 5ʹ flank, which allows recognition of almost the same target by both enzymes (shifted by one nucleotide). Downstream, the targets were flanked by the SpCas9 PAM. Thus, all three Cas proteins were able to recognize almost identical DNA target sequences (shifted by several nucleotides for efficient recognition of cognate PAMs).
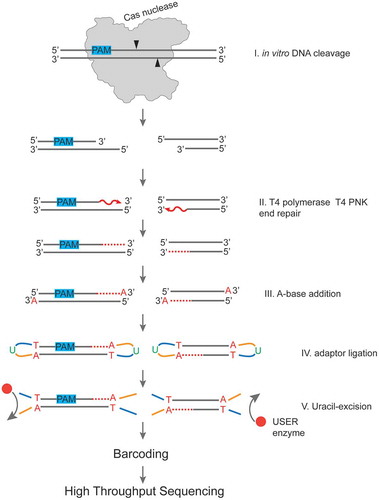
Six different 20-nt targets with an appropriate PAM were chosen randomly (Supplementary Table S1) and used to study cleavage by DpbCas12e or AsCas12a loaded with appropriate sgRNAs. Incubation of target-bearing linear DNA fragments with DpbCas12e-sgRNA or AsCas12a-crRNA led to efficient DNA cleavage and generation of products of expected lengths. The SpCas9-sgRNA ribonucleoprotein complex cleaved only four out of six DNA targets. All successful DNA cleavage reactions were processed according to procedure outlined in to determine cut site positions using high throughput sequencing on Illumina platform. In brief, the cleavage products were separated from uncleaved DNA, blunt-ended with T4 polynucleotide kinase and T4 polymerase, which produces 5ʹ-P and 3ʹ-OH ends and fills-in 5ʹ-overhangs, respectively. Next, DNA was 3ʹ A-tailed using the exonuclease deficient Klenow DNA polymerase fragment. This allowed to avoid unwanted processing/modification of DNA ends formed after cleavage prior to sequencing. For further steps we used components of the NEBNext Ultra II kit starting from adaptors ligation step and bypassing the DNA ends preparation step. In brief, Illumina NEBNext adaptors were ligated to DNA through TA cloning. Next, uracil residues incorporated in the adaptors were cut out by the USER enzyme, a component of NEBNext Ultra II which combines uracil DNA glycosylase and endonuclease VIII activity. Next, the samples were barcoded, according to the NEBNext Ultra II protocol, and sequenced on Illumina platform. The distribution of cleavage sites was revealed by analysing on average 100 000 reads for each cleavage reaction.
Figure 2. A workflow of sample preparation for determination of positions of DNA cleavage sites produced by Cas nucleases in vitro.
The cleaved DNA fragments generated by Cas nuclease during in vitro DNA cleavage reaction (Step I) are blunted using T4 PNK and T4 DNA polymerase (Step II). A-base is added to 3ʹ ends (Step III) for further ligation of Illumina NEBNext sequencing adaptors containing uridine (Step IV). Uridines are cleaved out using the NEB USER enzyme, which combines uracil DNA glycosylase and endonuclease VIII activity. Next, the samples are barcoded to produce DNA libraries ready for high throughput sequencing.
The analysis of the results showed that, as expected, unlike SpCas9, which mainly generated blunt-end cut sites, DpbCas12e and AsCas12a produced DNA cleavage products with staggered ends (). The use of HTS allowed us to observe not only the most frequent cleavage sites but distributions of cut site positions. In agreement with previous studies [Citation27,Citation28] multiple cleavage sites by AsCas12a were observed – the nuclease predominantly cleaved DNA after nucleotide positions 17–19 downstream of PAM in the non-target DNA strand (further NTS) and after positions 21–23 in the target strand (further TS) (, Supplementary Fig. S2).
Due to flexibility in cleavage sites, Cas12a cuts exhibit a wide distribution of 2–6nt overhangs within each target (). Similarly, DpbCas12e predominantly cleaved DNA after nucleotides 17–19 downstream of its PAM in the NTS (, Supplementary Fig. S2). In neither of the six DNA targets tested did we detect NTS cleavage after positions 12–14 position, previously reported by Jun-Jie Liu et al. In the target strand, DpbCas12e predominantly cleaved after the 22nd position.
We also performed AsCas12a and DpbCas12e DNA cleavage reaction of the same 20nt protospacer sequence used by Jun-Jie Liu et al, incorporated into a long 500nt linear DNA target (Supplementary Table S1). The cleavage of this target also resulted in producing 3–5nt overhangs for both nucleases (Supplementary Fig. S3). It is important to mention, that Jun-Jie Liu et al. used a much shorter DNA fragment, which could possibly affect the cleavage position [Citation14]. Overall, we conclude that the DpbCas12e nuclease produces 3–5nt 5ʹ-overhangs ().
Since experiments described above were conducted with MBP fusion of DpbCas12e, we determined the DNA cleavage pattern of DpbCas12e without MBP. As a result, no significant differences between DpbCas12e without MBP and the MBP-fused protein were observed (Supplementary Fig. S4).
Figure 3. Determination of DpbCas12e, AsCas12a, and SpCas9 cut sites positions by high throughput sequencing of in vitro DNA cleavage reaction products. A) Histograms showing mapping the cut sites on the target and non-target DNA strands in case of DpbCas12e, AsCas12a, and SpCas9. Results for three different DNA targets are shown, PAMs are indicated with red rectangles. The numbering of nucleotide positions starts from the end of the PAM and is shown along the target DNA sequence. For each DNA target sequence, the histograms of cut site positions frequency in percentage for the corresponding DNA strand are shown. Each column represents the fraction of DNA cleavage events after the corresponded nucleotide. ‘NTS’ stands for non-target DNA strand, ‘TS’ – for target DNA strand. The most frequent cut site positions are shown with black triangles. Mean values obtained from three independent experiments with standard deviations are shown. B) The overhang lengths produced by DpbCas12e, AsCas12a, and SpCas9. Histograms of differences between cut site positions on DNA TS and NTS were calculated based on DNA cleavage data obtained for all six targets (panel A and Supplementary Fig. S2). Distances between DSB ends were calculated as Distance = [cut site position on TS] – [cut site position on NTS]. Distances were calculated between all possible TS and NTS DNA cleavage positions. Relative frequencies of generated overhangs were calculated as a sum of [relative frequency of cut site positions on TS] x [relative frequency of cut site positions on NTS] for all combinations of TS and NTS producing overhangs of a certain length.
![Figure 3. Determination of DpbCas12e, AsCas12a, and SpCas9 cut sites positions by high throughput sequencing of in vitro DNA cleavage reaction products. A) Histograms showing mapping the cut sites on the target and non-target DNA strands in case of DpbCas12e, AsCas12a, and SpCas9. Results for three different DNA targets are shown, PAMs are indicated with red rectangles. The numbering of nucleotide positions starts from the end of the PAM and is shown along the target DNA sequence. For each DNA target sequence, the histograms of cut site positions frequency in percentage for the corresponding DNA strand are shown. Each column represents the fraction of DNA cleavage events after the corresponded nucleotide. ‘NTS’ stands for non-target DNA strand, ‘TS’ – for target DNA strand. The most frequent cut site positions are shown with black triangles. Mean values obtained from three independent experiments with standard deviations are shown. B) The overhang lengths produced by DpbCas12e, AsCas12a, and SpCas9. Histograms of differences between cut site positions on DNA TS and NTS were calculated based on DNA cleavage data obtained for all six targets (panel A and Supplementary Fig. S2). Distances between DSB ends were calculated as Distance = [cut site position on TS] – [cut site position on NTS]. Distances were calculated between all possible TS and NTS DNA cleavage positions. Relative frequencies of generated overhangs were calculated as a sum of [relative frequency of cut site positions on TS] x [relative frequency of cut site positions on NTS] for all combinations of TS and NTS producing overhangs of a certain length.](/cms/asset/de7246a4-fd4d-43f5-b122-415d5a8db7dd/krnb_a_1777378_f0003_c.jpg)
The influence of sgRNA spacer segment length on DpbCas12e cleavage sites
Previous studies of Cas12a showed that the spacer length of crRNA can influence the position of Cas12a DNA cleavage sites [Citation20]. Using crRNA with a spacer length of 18nt caused a shift of cut site position in NTS to positions 13–15 instead of position 18 observed when Cas12a effector was charged with crRNAs whose spacer segments were 20 nucleotides or longer [Citation20]. Due to DNA cleavage pattern similarity between Cas12a and Cas12e, we were interested to determine if sgRNA spacer length can also affect the position of DNA cleavage site by DpbCas12e. DpbCas12e charged with sgRNAs of different spacer length (16, 18, 20, 22 and 24nt) was used to cleave three different DNA targets. Analysis of cleavage products by HTS showed that DpbCas12e begins to cleave NTS closer to PAM when shorter than 20nt spacer sgRNAs are used: 16nt spacer sgRNAs produced 6–8nt 5ʹ-overhangs, while sgRNAs with 20, 22, and 24nt spacer segments led to cleavage after target positions 17–19, producing 3–5nt overhangs (, Supplementary File S3).
Figure 4. The influence of sgRNA spacer length on cleavage by DpbCas12e. A) Results of mapping of in vitro cleavage sites produced in three different DNA targets by DpbCas12e complexed with sgRNAs of different spacer lengths, PAMs are indicated with red rectangles; the numbering of nucleotide positions starts from the end of the PAM and is shown along the target DNA sequence. Heatmaps for each DNA target sequence show the positions of cut sites for the corresponding DNA strand for sgRNAs with indicated spacer lengths. Each heatmap cell intensity represents the fraction of DNA cleavage events after the corresponding nucleotide. The heatmaps are drawn based on mean values obtained from three independent experiments. B) The range of lengths of 5ʹ-overhangs, produced by DpbCas12e in complex with sgRNAs with 16, 18, 20, 22, or 24nt spacers. The differences between cut site positions on DNA TS and NTS were calculated based on data shown in panel A. Distance between DSB ends was calculated as Distance = [cut site position on TS] – [cut site position on NTS]. Distances were calculated between all possible TS and NTS DNA cleavage positions. Relative frequencies of generated overhangs were calculated as a sum of [relative frequency of cut site positions on TS] x [relative frequency of cut site positions on NTS] for all combinations of TS and NTS producing overhangs of a certain length. The most abundant distances of 0–8nt were used to plot a stacked barchart. The length of each sector of the columns represents the fraction of overhangs of certain length.
![Figure 4. The influence of sgRNA spacer length on cleavage by DpbCas12e. A) Results of mapping of in vitro cleavage sites produced in three different DNA targets by DpbCas12e complexed with sgRNAs of different spacer lengths, PAMs are indicated with red rectangles; the numbering of nucleotide positions starts from the end of the PAM and is shown along the target DNA sequence. Heatmaps for each DNA target sequence show the positions of cut sites for the corresponding DNA strand for sgRNAs with indicated spacer lengths. Each heatmap cell intensity represents the fraction of DNA cleavage events after the corresponding nucleotide. The heatmaps are drawn based on mean values obtained from three independent experiments. B) The range of lengths of 5ʹ-overhangs, produced by DpbCas12e in complex with sgRNAs with 16, 18, 20, 22, or 24nt spacers. The differences between cut site positions on DNA TS and NTS were calculated based on data shown in panel A. Distance between DSB ends was calculated as Distance = [cut site position on TS] – [cut site position on NTS]. Distances were calculated between all possible TS and NTS DNA cleavage positions. Relative frequencies of generated overhangs were calculated as a sum of [relative frequency of cut site positions on TS] x [relative frequency of cut site positions on NTS] for all combinations of TS and NTS producing overhangs of a certain length. The most abundant distances of 0–8nt were used to plot a stacked barchart. The length of each sector of the columns represents the fraction of overhangs of certain length.](/cms/asset/51965df1-37cb-4c5a-95d3-8a5f5c72cb0b/krnb_a_1777378_f0004_c.jpg)
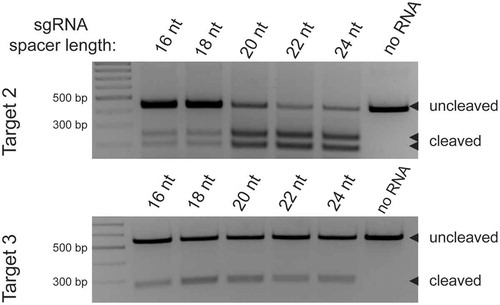
The reduction of the sgRNA spacer length may compromise the DNA cleavage efficiency of the effector. Indeed, using of sgRNAs with spacer lengths shorter than 20 nt led to lower DpbCas12e DNA cleavage efficiency, although the effect was not dramatic and the nuclease activity was sufficient for effective introduction of double-stranded breaks ().
Figure 5. DpbCas12e DNA in vitro cleavage using sgRNAs of different spacer length. Above – a gel showing the results of in vitro cleavage of Target 2 using sgRNAs of 16nt, 18nt, 20nt, 22nt or 24nt spacer length. Below – similar gel for Target 3.
Discussion
In this work we determined DNA cleavage sites produced by DpbCas12e in in vitro reactions. Analysis of cleaved DNA fragments by HTS allowed us to show not only the most frequent cut site positions but revealed the distribution of DNA cleavage sites along the targeted DNA molecules. In agreement with the previous results, our data demonstrates that in contrast to SpCas9, DpbCas12e and AsCas12a produce staggered ends at cut sites. The DNA cleavage positions in case of each nuclease slightly vary depending on the target sequence, though the overall cleavage pattern remains the same. We show that DpbCas12e and AsCas12a, CRISPR-Cas effectors of distinct subtypes V-E and V-A, have similar distribution of DNA cut site positions. Both enzymes introduce cuts after nucleotides 17–19 downstream of PAM in the non-target DNA strand. The target DNA strand cleavage position is after nucleotides 21–23 for AsCas12a and predominantly after nucleotide 22 for DpbCas12e. This DNA cleavage pattern leads to generation of 3–5nt overhangs by both enzymes. We did not observe 10nt 5ʹ-overhangs in DNA cleaved by DpbCas12e, that were reported earlier, and that could have been advantageous for certain biotechnological applications.
Nevertheless, we show that the length of 5ʹ-overhangs generated by DpbCas12e can be modulated: using of sgRNAs with shorter, 16 nucleotides spacer segment increased the 5ʹ-overhangs of cleavage products by 3nt, producing long 6–8nt staggered overhangs instead of 3–5nt overhangs, produced when 20nt spacer segment was used. Longer overhangs generated at cut sites may be potentially advantageous for in vitro and in vivo ligation of DNA fragments into Cas12e-generated breaks in double-stranded DNA. Indeed, this strategy was successfully used by Chao Lei et al. for incorporation of DNA fragments into plasmids in vitro using Cas12a nuclease [Citation20].
Overall, our data on cut site position determination can be useful for potential development of DpbCas12e-based biotechnology instruments as well as for understanding of the mechanisms of Cas12e nucleases action in molecular details. The approach applied here for DNA cleavage pattern determination is general and can be used for characterization of cleavage sites by different Cas and non-Cas nucleases.
Data availability
Raw sequencing data have been deposited with the National Center for Biotechnology Information Sequence Read Archive under BioProject ID PRJNA605170
Supplemental Material
Download Zip (1.1 MB)Acknowledgments
We thank Aleksandr Koshkin for insightful comments and suggestions. We are grateful to the Skoltech Genomics Core facility for sequencing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016;85:227–264.
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823.
- Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9:467–477.
- Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191.
- Kim E, Koo T, Park SW, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500.
- Lee CM, Cradick TJ, Bao G. The neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Mol Ther. 2016;24:645–654.
- Harrington LB, Paez-Espino D, Staahl BT, et al. A thermostable Cas9 with increased lifetime in human plasma. Nat Commun. 2017;8:1424.
- Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53.
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR–Cas13. Nature. 2017;550:280–284.
- Fonfara I, Richter H, Bratovič M, et al. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–521.
- Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–34.
- Burstein D, Harrington LB, Strutt SC, et al. New CRISPR–Cas systems from uncultivated microbes. Nature. 2017;542:237–241.
- Liu -J-J, Orlova N, Oakes BL, et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019;566:218–223.
- Yamano T, Nishimasu H, Zetsche B, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962.
- Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949.
- Swarts DC, van der Oost J, Jinek M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Molecular Cell. 2017;66:221-233.e4. doi:10.1016/j.molcel.2017.03.016
- Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017;8:15058.
- Wienert B, Wyman SK, Richardson CD, et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. 2019;364:286–289.
- Lei C, Li S-Y, Liu J-K, Zheng X, Zhao G-P, Wang J. The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res. 2017;6:e74.
- Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157.
- Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771.
- Teng F, Cui T, Feng G, et al. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 2018;4:63.
- Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12:237–243.
- Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat Methods. 2017;14:607–614.
- Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR–Cas9 cleavage. Nat Methods. 2017;14:600–606.
- Li S-Y, Zhao G-P, Wang J. C-Brick: a new standard for assembly of biological parts using Cpf1. ACS Synth Biol. 2016;5:1383–1388.
- Kim D, Kim J, Hur JK, et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34:863–868.