
ABSTRACT
RNA-binding proteins regulate RNA fate and govern post-transcriptional gene regulation. A new family of RNA-binding proteins is represented by regulatory RNases (Regnase, also known as Zc3h12 or MCPIP), which have emerged as important players in immune homoeostasis. Four members, Regnase1-4, have been identified to date. Here we summarize recent findings on the role of Regnase in the regulation of RNA biology and its consequences for cell functions and inflammatory processes.
Introduction
In recent years, RNAs have emerged as modulators of cellular functions. Their fate is closely associated with that of RNA-binding proteins (RBPs), which bind, process, and degrade respective target RNAs. By these means, RBPs modulate biological processes and can shape both innate and adaptive immune responses [Citation1,Citation2,Citation3]. Growing evidence supports their role in a wide variety of diseases and clinical conditions, including autoimmune disease and also sterile inflammation of the heart and other organs [Citation4, Citation5, Citation6, Citation7].To date, four members of the RegnaseRBP family have been identified. They are also known as Zc3h12a-d or MCPIP1-4. All members share two conserved domains: a CCCH-type zinc finger domain and a Pilt-N-terminus (PIN) like domain that recognizes and binds RNA [Citation4,Citation8,Citation9]. Thereby, they can modulate cellular functions and immune responses. In this review, we summarize recent publications on the four members of the Regnase family and give insight into their contribution, mode of action, and modulatory role in inflammation.
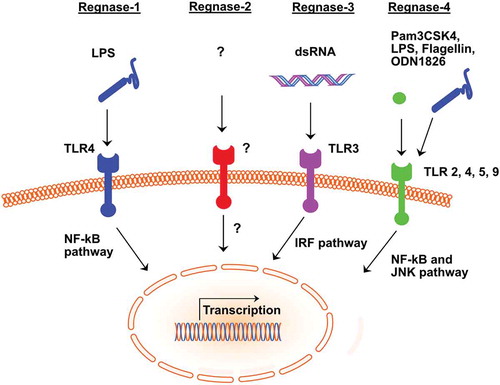
Figure 1. Activation of Regnase 1 to 4 by ligands and their intracellular pathways
Molecular function of RNA-binding proteins
The RBP–RNA interaction is mediated mainly by cis-regulatory elements present in the 3ʹ-untranslated region (UTR) of the respective RNA and the function of trans-acting RNA-binding protein [Citation7, Citation10, Citation11]. AU rich elements (ARE) and stem-loop structures present in the cis-regulatory elements facilitate the anchoring of RNA-binding proteins. The stem-loop structures represent a three-dimensional structure for the interaction of the RNA-binding proteins and the respective RNA. They comprise a pyrimidine-purine-pyrimidine tri-loop sequence in the 3ʹ UTR of inflammatory mRNA that is recognized and bound by RBP, such as Regnase-1 [Citation12]. Regnase-1acts as an endoribonuclease, directly degrading target cytokine mRNA and thereby modulating the local and systemic immune response. Regnase-1 contains a CCCH zinc finger domain and a PIN-like RNase domain (see ). The PIN domain harbours the catalytic center responsible for the endonuclease function. For instance, Regnase-1 destabilizes interleukin-6 (Il-6)mRNA via the conserved cis-regulatory element present in its 3ʹUTR.
Figure 2. Schematic representation of the Regnase family members structure

Regnase-1 in the innate immune system
Regnase-1 is a well-characterized member of this RNA-binding protein family. It is of importance in both adaptive and innate immune responses as well as in haematopoiesis. Mice with global deficiency in Regnase-1 die around 12 weeks of age, presenting with anaemia, elevated serum levels of immunoglobulins, and autoantibody production with increased numbers of plasma cells [Citation13]. In the following, we highlight specific functions of Regnase-1 in different immune cell populations and tissues.
In the innate immune system, biological functions of Regnase-1 have mainly been identified in macrophages, splenocytes, epithelial cells, and epidermal keratinocytes. Macrophages from Regnase-1 global knockout mice show highly increased production of interleukin-6 (Il-6) and Il-12b in response to toll-like receptor (TLR) ligands. While activation of the TLR pathway is normal, also the Il-6 messenger RNA decay is severely impaired in Regnase-1 deficient macrophages (see ). Overexpression of Regnase-1 enhancesIl-6 mRNA degradation and regulates further RNAs such as Il-12band calcitonin receptor (Calcr) mRNA [Citation13]. Also, macrophages and splenocytes from Regnase-1 deficient mice display an elevated expression of inflammatory genes and increased JNK and IkBkinase activation [Citation14]. A specific immunomodulatory role of Regnase-1 has been identified in alveolar macrophages. Elevated protein levels of Regnase-1 mediate prolonged survival of rats in a model of acute lung injury. Through activation of the JNK/c-Myc pathway, macrophages alter their polarization from pro-inflammatory M1-like to the anti-inflammatory M2-like phenotype in this setting [Citation15].
Further, Regnase-1 is expressed in airway epithelial cells, which are essential for the innate immune defence against inhaled pathogens. Regnase-1 coordinates innate and adaptive immune responses against inhaledPseudomonas aeruginosa infections. Deletion of Regnase-1 augments innate defence pathways by sustainingRegnase-1-dependent inflammatory genes. This enhances the secretion of pseudomonas-specific immunoglobulins and T cell accumulation in the lung, culminating in significant resistance against P. pseudomonas re-infection in vivo [Citation16]. Likewise, an immune-modulatory role of Regnase-1 has also been described in epidermal keratinocytes, where is mediates decreased skin inflammation. In this context, Regnase-1 limits the Il-36 mediated inflammation in epidermal cells [Citation17].
Regnase-1 in the adaptive immune system
The modulatory role of Regnase-1 in the adaptive immune response is primarily mediated by T cells. In these cells, Regnase-1 regulates sets of genes, including c-Rel, Ox40, and Interleukin-2(Il-2). Mechanistically, T cell receptor stimulation leads to cleavage of Regnase-1 by Malt1/paracaspase and thereby liberates T cells from Regnase-1 mediated suppression of inflammatory mRNA. The Malt1 protease activity is also critical for controlling the stability of mRNA of T-cell effector genes, thereby regulating their abundance in the cytosol [Citation18].
Roquin also represents an immune-related RNA-binding protein that coordinates the stability of inflammatory mRNA by recognizing stem-loop structures in target mRNA. Roquin mutation in mice is implicated in the development of systemic lupus erythematosus (SLE)-like pathology (reviewed by [Citation2]). Regnase-1 and Roquin are closely intertwined in regulating adaptive immune homoeostasis. Defects in both Regnase-1 and Roquin result in a substantial increase in their target mRNAs [Citation12]. Although deficiency of either Regnase-1 or Roquin leads to enhanced T-cell activation, their functional relationship has not been fully elucidated yet, because of lethality in mouse models with a double genetic knockdown. Cui and Takeuchi tackled this question by using a Regnase-1 conditional allele model, in which mutations of both Regnase-1 and Roquin in T-cells lead to strong lymphocyte activation. Mutation of either of them affected T cell activation to a lesser extent compared to the double mutation [Citation5]. These double mutants suffered from severe autoimmune inflammation and early fibrotic tissue turnover, especially in the heart. This is accompanied by an increased expression of Interferon γ, while Il-4 and Il-17a expression remain unaltered. Consistently, mutation of both Regnase-1 and Roquin lead to an increase in T helper cells (Th) 1, but not Th2 or Th17 populations, in the spleen compared to the single knockouts. Regnase-1 and Roquin repress the expression of mRNAs encoding for proteins involved in the Th1 differentiation, such as Furin(encoding for a proprotein convertase) and Il12rb1(encoding for the subunit of the IL-12 receptor). Regnase-1 is also capable of repressing Roquin mRNA, which may lead to a cross-regulated synergistic control of T cell activation. Recently, the role of Regnase-1 in adoptive cell therapy in effector T cells was elucidated by using an in vivo CRISPR platform [Citation19]. Regnase-1 deficient CD8+T cells display a new therapeutic approach with high efficacy against mouse melanoma and leukaemia. Further, the Basic leucine zipper ATF-like transcription factor (BATF) was discovered as a key target of Regnase-1 in T cells. Engineered Regnase-1 deficient T cells could, therefore, provide a new tool in cancer therapy.
Regnase-1 in viral infections
Regnase-1, apart from its immunomodulatory role in innate immune cells, exhibits direct effector functions in immune defence by degrading genomic nucleic acids of positive-sense, negative-sense RNA and DNA viruses [Citation20, Citation21]. Hereby, RNA-binding proteins act as sensors for viral RNA and DNA [Citation22, Citation23]. Regnase-1 expression can be induced by hepatitis C virus (HCV) infection in Huh7.5 hepatoma cells, and Regnase-3 expression is higher in liver tissue samples from patients with chronic HCV infection compared to those without chronic disease. The knockdown of Regnase-1 increases HCV replication and HCV-mediated expression of inflammatory cytokines. Overexpression of Regnase-1 leads to a significant reduction in HCV replication and pro-inflammatory cytokine expression.
This effect of Regnase-1 has also been observed in other viral infections, e.g., the Japanese encephalitis virus and dengue virus replication [Citation21]. Further mechanisticanalysisrevealedthat RNA-binding, RNA degradation (RNase), and oligomerization, but not deubiquitinases are required for proper functioning of Regnase-1 in this context. Noteworthy, even positive-sense RNA viruses (namely sindbis virus), encephalitis virus, negative-sense RNA virus (e.g., influenza virus), and DNA viruses (e.g., adenovirus), can be blocked by the RNase activity of Regnase-1 [Citation21].
Regnase-1 in haematopoietic homoeostasis
The role of Regnase-1 in haematopoietic homoeostasis involves modulation of Gata2 and Tal1 mRNA levels in haematopoietic stem- and progenitor cells [Citation24]. Dysfunction of Regnase-1 is associated with the accumulation of immature haematopoietic stem and progenitor cells, and therefore Regnase-1 may as well play a role as a suppressor of abnormal haematopoiesis, potentially leading to leukaemia.
Intracellular regulatory mechanisms in Regnase-1 biology
Mechanistically the IKK complex phosphorylates not only IkB α, thereby activating transcription, but also Regnase-1, and thus releases the `brake`on Il-6 mRNA expression [Citation25]. Furthermore, the IKK complex controls the phosphorylation of Regnase-1 that undergoes ubiquitination and degradation in the proteasome. Phosphorylated Regnase-1 is released from the endoplasmic reticulum into the cytosol, thereby losing its mRNA degrading function, which leads to expression of Il-17 associated genes [Citation26]. In the respective study, two knock-in mice were generated with blocked IKK phosphorylation sites in Regnase-1 as well as with deletion of the C-terminal portion of Regnase-1, resulting in a lack of phosphorylation of the RNase. The phosphorylation of Regnase-1 and its release from the endoplasmatic reticulum is sufficient to suppress the function of Regnase-1.Mechanistically, Regnase-1 mediated degradation of mRNA seems to be dependent on the IKK complex [Citation27].
Binding of various ubiquitin molecules to the same target protein is called ubiquitination. It represents a vital signal for degradation of respective protein in the proteasome. In addition to its post-transcriptional effects, Regnase-1 exhibits also post-translational potential through deubiquitination. Regnase-1 also promotes the removal of ubiquitin marker proteins from TNF receptor-associated factors (TRAF2, TRAF3, and TRAF6) and thereby interacts with JNK and NF-kB signalling pathways. It is still controversial if deubiquitination is performed by Regnase-1 itself or it merely acts as a scaffold for other deubiquitinases such as USP10 [Citation28]. Nevertheless, the interplay of cytokine production and degradation results in fatal inflammatory responses in Regnase-1 knockout mice [Citation14].
Regnase-2
The expression and function of Regnase-2 across tissues is incompletely understood. Regnase-2 is expressed in the brain, thymus, and testis [Citation29]. In a gene expression analysis, mRNA levels of Regnase-2 are highest in the human brain, and the neuroblastoma-derived cell line SH-SY5Y [Citation30].On structural analysis, Regnase-2 carries the conserved and membrane-associated as-like GTPase domain at the C-terminus, suggesting its potential involvement in signal transduction [Citation29]. Notably, Regnase-2 is not inducible by LPS and IFNγ stimulation in Raw264.7 cells, which is in contrast to Regnase-1 or Regnase-3, representing an unusual feature of this RNA-binding protein family. The biological function of Regnase-2 is depended on an intact NYN/PIN (PilT N-terminal) RNase domain for substrate recognition [Citation30]. Regnase-2 binds endogenous Il-6 mRNA and regulates its turnover upon stimulation with Il-1ß. Furthermore, Regnase-2 interacts with other mRNAs, namely the immediate early response 3 gene (IER3), possibly protecting from TNFα induced apoptosis. In summary, the current literature paintsan incomplete picture of gene expression, cellular distribution, and function of Regnase-2. Data on the characterization of this RNA-binding protein in the mice models are missing entirely. Thus, further research with global and conditional deficiency models in mammals are needed to understand the nature of Regnase-2.
Regnase-3
To date, only sparse data give insight into the modulatory role of Regnase-3 in endothelial and myeloid cell lines. In studies using human umbilical vein endothelial cells (HUVEC), Regnase-3 inhibits the endothelial inflammatory response [Citation31]. The overexpression of Regnase-3 significantly attenuates TNFα induced expression of chemokines and adhesive molecules, and reduces monocyte adherence to HUVECs. In the first publication of the myeloid Regnase-3of Liang et al.in 2008, LPS stimulated macrophages upregulatedRegnase-1 and Regnase-3, which is in line with the upregulation of macrophage expression patterns by inflammatory signals such as TNFα, MCP-1, Il-1β and Il-6 [Citation9].
After a decade of this first analysis of the expression pattern of myeloid Regnase-3, the first characterization of the function of the Regnase-3 in mouse modelshas been published using global and conditional Cre-induced knockouts. Global Regnase-3 deficiency in mice promotes systemic increased interferon (IFN) signalling and suppression of germinal centre formation. Nevertheless, deficient animals did not display manifestations towards autoimmune disease. Sequencing analysis revealed 12 significantly up-regulated genes that are part of the interferon response genes. This phenotype is observed in the absence of Regnase-3, as well as in mice with conditional Regnase-3 deficiency in myeloid cells. The abundance of Regnase-3 is increased explicitly in macrophages upon TLR3 activation and is regulated by IRF3/IRF7 signalling (see ). This stays in contrast to Regnase-1, which is highly expressed in the lymphoid cell line and interacts via NF-kB [Citation32].
Regnase-1 and Regnase-3 share a high homology (97%) for the PIN domain and zinc finger domain that are thought to act as RNA sensors. In vitro reporter experiments confirmed that Regnase-1 UTR is a target for Regnase-3 in macrophages. By now, it remains unclear which other RNAs are targeted by Regnase-3 and whether Regnase-3 degrades intracellular mRNAs or incoming RNA from cell debris or viruses.
Nevertheless, Regnase-3 is a new potent player in immune homoeostasis that warrants further studies on the mode of action. This first in mouse study supports observations from genome-wide associations identifying loci polymorphisms in Regnase-3 that are associated with human inflammatory diseases, such as psoriasis [Citation33].
Regnase-4
In the field of neuroinflammatory disorders of the central nervous system (CNS), a gene-ontology pathway analysis in white matter alterations (leukoaraiosis) revealed significant inflammatory changes to be associated with Regnase-4 gene- and mRNA expression. Thiscouldimplicatepotential molecular mechanisms in cerebro-inflammatory diseases [Citation34]. When tested in an autoimmune encephalitis animal model, Regnase-4 seems to play a role in T cell effectors function by inhibiting the synthesis of cytokines such as Il-2, Il-6, Il-10, TNFα, and Il-17a [Citation35].
Besides the neurological research, Liang et al. conducted a genome-wide survey and expression profiling of CCCH Zinc finger families that revealed high expression of Regnase-4 in organs with abundance of macrophages such as thymus, spleen, lung, intestine and adipose tissue [Citation9]. This distribution pattern is supported by expression analysis of Regnase-4 being enriched in the spleen, lung, and lymph nodes and, to a lesser extent, in the heart, liver, intestine, thymus, and kidney[Citation36]. Their experiments showed that Regnase-4 negatively regulates TLR signalling and macrophage activation. In a model of acute lung injury, Regnase-4 attenuated the inflammatory response by reducing mRNA stability of pro-inflammatory genes [Citation37]. Regnase-4 levels are decreased in airway immune cells in LPS-induced inflammation. The overexpression of the RNA-binding protein inhibits the expression of inflammatory genes, e.g., NF-kBsubunitRelA (p65) and cytokines. In line with these results, Regnase-4 reducesthe mRNA stability of cytokines (Il-6, Il-1ß, and TNFα), NF-kBp65, and c-fos.
In macrophages, the expression of Regnase-4 is induced by TLR ligands through JNK and NF-kB signal pathways. The overexpression of Regnase-4 inhibits TLR2 and TLR4 activation-induced JNK, ERK and NF-kB signalling as well as macrophage inflammation. Interestingly, similar to Regnase-1, Regnase-4 decreases the global cellular protein ubiquitination in vitro. Thereby, Regnase-4 could act as a novel negative feedback regulator of TLR signalling and macrophages activation in inflammation.
Furthermore, Regnase-4 interacts with Regnase-1 by forming a complex which regulates mRNA degradation [Citation38]. Regnase-4 is identified as an interaction partner of Regnase-1 by mass-spec analysis and co-immunoprecipitation. Immunofluorescence staining showed that Regnase-4 co-localized with Regnase-1 in the processing bodies, which play a fundamental role in mRNA decay. However, the localization of these RNA-binding proteins is still controversial and further characterization is necessary to clarify the cellular localization and interaction between Regnase-1 and Regnase-4 [Citation12,Citation35,Citation39,Citation40]. In vitro, both Regnase family members can degrade Il-6 mRNA, with an enhanced effect when both RBPs are present. Similarly to Regnase-1, the Regnase-4 participates in the 3ʹ UTR-dependent regulation of the turnover of Il-6, TNFα, and immediate early response 3 gene (IER3) in an NYN/PIN-like domain-dependent mechanism [Citation41]. In summary, deciphering the immune-modulatory role of Regnase-4 has only scratched the surface. The purpose of Regnase-4 in multiple inflammatory conditions and models remains to be elucidated by further experiments, which could lay the foundation for target-specific modulation of inflammatory diseases.
Role of Regnase family members in inflammatory diseases
The purpose of the Regnase family members in immune homoeostasis is evident for both adaptive and innate immune responses. Uncontrolled inflammation in autoimmune-related diseases such as psoriasis or Crohn’s disease are characterized by the breakdown of self-tolerance towards antigens, and the imbalance between activation and repression of immune cells. There are nowadays established treatment regimens harnessing antibodies as immune modifying agents, such as adalimumab or infliximab. Regarding intestinal inflammation, mutation of Regnase-1 is associated with the pathogenesis of ulcerative colitis [Citation42,Citation43]. Whole-exome genome sequencing data from human colon tissue identified mutations in Regnase-1 and genes related to IL-17 signalling in the inflamed epithelium. Regnase-1 deficiency in intestinal epithelial cells does not increase inflammation at steady state, but colitis induced by dextran sulphate sodium administration is blunted in the knockout mice [Citation44]. Even iron homoeostasis in duodenal enterocytes is affected by this RNA-binding protein [Citation45]. The research group of Osamu Takeuchi identified a destabilizing role of Regnase-1 for transferrin receptor 1 mRNA, and that lack of duodenal Regnase-1 leads to iron deficiency anaemia in vivo.
In cardiovascular diseases, inflammation plays a significant role not only in inflammatory conditions like myocarditis but also in atherosclerosis and even more so in cardiac remodelling after myocardial infarction. Here the inflammatory processes are a two-edged sword, on the one hand being essential for, e.g., clearing of dead cell debris, but on the other hand, throughovershooting inflammation, enhancing ischaemia-induced tissue damage. Novel therapeutic approaches concerning such inflammatory responses are already been tested. The CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial invested the interleukin-1ß targeting monoclonal antibody, which in the setting of human atherosclerosis reduced the rate of cardiovascular events such as myocardial infarction [Citation46]. Other studies are ongoing, that test the effect of anti-interleukin-6 receptor antibodies in the context of atherosclerosis [Citation47]. The ability of Regnase-1 to interact and modulate the amount of pro-inflammatory mRNA such as Il-6 and Il-12b could thereby lead to new treatment approaches. In viral-induced diseases, overexpression of Regnase-1 suppresses the replication of the hepatitis c virus, Japanese encephalitis virus, and dengue virus infection. Enhancing the function of Regnase-1 might, therefore, represent a new therapeutic strategy.
Conclusion
The Regnase protein family has been identified as a new player in mammalian immune homoeostasis. However, a significant gap of knowledge exists regarding their precise functions and modes of molecular actions, especially regarding Regnases-2, 3, and 4. Further research in animals and in vitro experiments is needed to validate their targeted mechanism of mRNA degradation and to address their interacting partners. Elucidating the inflammatory RNA modifying potential of Regnase family members could lay the foundation for modulating immune cell functions and shaping immune-related pathways.
Disclosure of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest; Friedrich-Baur-Stiftung; DZHK Standortprojekt [81Z0600204]; DZHK Shared Expertise [81X2600256]; DFG [project number MA 2186/14-1].
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15(12):829–845.
- Yoshinaga M, Takeuchi O. RNA binding proteins in the control of autoimmune diseases. ImmunolMed. 2019;42(2):53–64.
- de Bruin RG, Rabelink TJ, van Zonneveld AJ, et al. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. 2017;38(18):ehw567.
- Akira S. Regnase-1, a ribonuclease involved in the regulation of immune responses. Cold Spring Harb Symp Quant Biol. 2013;78(1):51–60.
- Cui X, Mino T, Yoshinaga M, et al. regnase-1 and roquin nonredundantly regulate Th1 differentiation causing cardiac inflammation and fibrosis. J Immunol. 2017;ji1701211. DOI:10.4049/jimmunol.1701211
- King KR, Aguirre AD, Ye YX, et al. IRF3 and type i interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23(12):1481–1487.
- Mino T, Takeuchi O. Post-transcriptional regulation of immune responses by RNA binding proteins. Proc Jpn Acad Ser B. 2018;94(6):248–258.
- Jura J, Skalniak L, Koj A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim Biophys Acta Mol Cell Res. 2012;1823(10):1905–1913.
- Liang J, Wang J, Azfer A, et al. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. 2008;283(10):6337–6346.
- Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9(4):353–359.
- Shyu A-B, Wilkinson MF, van Hoof A. Messenger RNA regulation: to translate or to degrade. Embo J. 2008;27(3):471–481.
- Mino T, Murakawa Y, Fukao A, et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. 2015;161(5):1058–1073.
- Matsushita K, Takeuchi O, Standley DM, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458(7242):1185–1190.
- Liang J, Saad Y, Lei T, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J Exp Med. 2010;207(13):2959–2973.
- Zhang Y, Huang T, Jiang L, et al. MCP-induced protein 1 attenuates sepsis-induced acute lung injury by modulating macrophage polarization via the JNK/c-Myc pathway. Int Immunopharmacol. 2019;75(April):105741.
- Nakatsuka Y, Vandenbon A, Mino T, et al. Pulmonary Regnase-1 orchestrates the interplay of epithelium and adaptive immune systems to protect against pneumonia. Mucosal Immunol. 2018;March:1–16. DOI:10.1038/s41385-018-0024-5
- Takaishi M, Satoh T, Akira S, et al. Regnase-1, an immunomodulator, limits the IL-36/IL-36R autostimulatory loop in keratinocytes to suppress skin inflammation. J Invest Dermatol. 2018;138(6):1439–1442.
- Uehata T, Iwasaki H, Vandenbon A, et al. XMalt1-induced cleavage of regnase-1 in CD4+ helper T cells regulates immune activation. Cell. 2013;153(5):1036.
- Wei J, Long L, Zheng W, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576(7787):471–476.
- Lin R-J, Chu J-S, Chien H-L, et al. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J Immunol. 2014;193(8):4159–4168.
- Lin RJ, Chien HL, Lin SY, et al. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013;41(5):3314–3326.
- Pichlmair A, Diebold SS, Gschmeer S, et al. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via toll-like receptor 9. J Virol. 2007;81(2):539–547.
- Pichlmair A, Reis E Sousa C. Innate recognition of viruses. Immunity. 2007;27(3):370–383.
- Kidoya H, Muramatsu F, Shimamura T, et al. Regnase-1-mediated post-transcriptional regulation is essential for hematopoietic stem and progenitor cell homeostasis. Nat Commun. 2019;10:1.
- Iwasaki H, Takeuchi O, Teraguchi S, et al. The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat Immunol. 2011;12(12):1167–1175.
- Tanaka H, Arima Y, Kamimura D, et al. Phosphorylation-dependent Regnase-1 release from endoplasmic reticulum is critical in IL-17 response. J Exp Med. 2019;216(6):1431–1449.
- Matsushita K, Tanaka H, Yasuda K, et al. Regnase-1 degradation is crucial for interleukin-33- and interleukin-25-mediated ILC2 activation. JCI Insight. 2020. DOI:10.1172/jci.insight.131480
- Niu J, Shi Y, Xue J, et al. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. Embo J. 2013;32(24):3206–3219.
- Liang J, Song W, Tromp G, et al. Genome-wide survey and expression profiling of CCCH-zinc finger family reveals a functional module in macrophage activation. PLoS ONE. 2008;3:8.
- Wawro M, Wawro K, Kochan J, et al. ZC3H12B/MCPIP2, a new active member of the ZC3H12 family. RNA. 2019;25(7):840–856.
- Liu L, Zhou Z, Huang S, et al. Zc3h12c inhibits vascular inflammation by repressing NF-κB activation and pro-inflammatory gene expression in endothelial cells. Biochem J. 2013;451(1):55–60.
- von Gamm M, Schaub A, Jones AN, et al. Immune homeostasis and regulation of the interferon pathway require myeloid-derived Regnase-3. J Exp Med. 2019;216(7):1700–1723.
- Tsoi LC, Spain SL, Trembath RC. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44(12):1341–1348. https://doi.org/10.1038/ng. 2467
- Huang W-Q, Yi K-H, Li Z, et al. DNA methylation profiling reveals the change of inflammation-associated ZC3H12D in leukoaraiosis. Front Aging Neurosci. 2018;10(MAY):1–18.
- Minagawa K, Wakahashi K, Kawano H, et al. Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J Immunol. 2014;192(4):1512–1524.
- Huang S, Qi D, Liang J, et al. The putative tumor suppressor Zc3h12d modulates toll-like receptor signaling in macrophages. Cell Signal. 2012;24(2):569–576.
- Zhang H, WC W, JK C, et al. ZC3H12D attenuated inflammation responses by reducing mRNA stability of proinflammatory genes. Mol Immunol. 2015;67(2):206–212.
- Huang S, Liu S, Fu JJ, et al. Monocyte chemotactic protein-induced protein 1 and 4 form a complex but act independently in regulation of interleukin-6 mRNA degradation. J Biol Chem. 2015;290(34):20782–20792.
- Bloch DB, Nobre R. p58TFL does not localize to messenger RNA processing bodies. Mol Cancer Res. 2010;8(1):131–132.
- Minagawa K, Yamamoto K, Nishikawa S, et al. Deregulation of a possible tumour suppressor gene, ZC3H12D, by translocation of IGK@ in transformed follicular lymphoma with t(2;6)(p12;q25). Br J Haematol. 2007;139(1):161–163.
- Wawro M, Kochan J, Krzanik S, et al. Intact NYN/PIN-like domain is crucial for the degradation of inflammation-related transcripts by ZC3H12D. J Cell Biochem. 2017;118(3):487–498.
- Kakiuchi N, Yoshida K, Uchino M, et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature. 2020;577(7789):260–265.
- Nanki K, Fujii M, Shimokawa M, et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature. 2020;577(7789):254–259.
- Nagahama Y, Shimoda M, Mao G, et al. Regnase-1 controls colon epithelial regeneration via regulation of mTOR and purine metabolism. Proc Nat Acad Sci. 2018;115(43):11036–11041.
- Yoshinaga M, Nakatsuka Y, Vandenbon A, et al. (2017). Regnase-1 maintains iron homeostasis via the degradation of transferrin receptor 1 and Prolyl-hydroxylase-domain-containing protein 3 mRNAs. Cell Rep. 1614–1630;19(8). DOI:10.1016/j.celrep.2017.05.009
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131.
- Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207.e15.