
ABSTRACT
Th17 cells remain one of the most important subsets of T cells in numerous autoimmune and chronic inflammatory diseases. Posttranscriptional regulation (PTR), especially mRNA stability, has recently emerged as an important mechanism that controls the fate of Th17 cells. This review summarizes the current knowledge on RNA-binding proteins (RBPs), microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) that induce mRNA stability changes and their roles in mediating the differentiation, proliferation, function, and migration of Th17 cells. In addition, we summarize the role of RNA modifications and nonsense-mediated mRNA decay (NMD) in Th17 cells. Ongoing research will help to identify practical applications for the regulation of mRNA stability and provide potential targets to prevent and treat Th17-related autoimmune diseases.
Introduction
Interleukin (IL)-17-expressing T cells are now widely known as a third subset of effector T helper cells (Th17 cells), which are closely related to host defence against extracellular fungi and bacteria, including Mycobacterium tuberculosis, and homoeostasis maintenance in mucosal tissue [Citation1–5]. Differentiation factors (TGF-β plus IL-6 or IL-21), growth and stabilization factors (IL-23), and transcription factors (signal transducer and activator of transcription 3 (STAT3), retinoid-related orphan nuclear receptor γt (RORγt), and retinoid-related orphan nuclear receptor α (RORα)) are involved in the development of Th17 cells [Citation6–9]. In addition, IL-17, IL-17 F, IL-21, IL-22 and granulocyte-macrophage colony-stimulating factor (GM-CSF) are also secreted by Th17 cells, and these factors are mainly involved in tissue inflammation by cooperating with other immune cells, thus bridging the gap between innate and adaptive immunity [Citation10–12]. Recently, an increasing number of studies have demonstrated that Th17 cells are important in numerous autoimmune and chronic inflammatory diseases in both humans and experimental animals [Citation5,Citation13].
Accumulating studies have indicated that the regulation of key gene expression, including transcriptional and posttranscriptional regulation (PTR), is crucial in the differentiation, maturation, and function of Th17 cells [Citation14–16]. Although transcriptional regulation has been widely characterized in Th17 cell differentiation, PTR, especially RNA stability, has recently emerged as an important mechanism to control the fate of Th17 cells, and it provides rapid and flexible control of the maturation, destruction, and translation of mRNA [Citation15,Citation17–19]. mRNA decay is mainly caused by the interactions between RNA-binding proteins (RBPs) and RNA in the cytoplasm [Citation14]. These regulatory RBPs bind to elements in the 3′ untranslated region (UTR), such as AU-rich elements (AREs) or constitutive decay elements (CDEs).
There are three mechanisms of mRNA decay [Citation14]. The first mechanism of mRNA decay is exonucleolytic decay, which is mediated by RBPs binding to AREs or CDEs [Citation14]. RBPs, such as Roquin, recruit exonucleases, such as poly(A)-specific ribonuclease (PARN) or CCR4-NOT complex, which remove the poly (A) tail from the target mRNA. Following the decapping, the mRNA can be degraded from the 3′ end or the 5′ end. The degradation from the 3′ end is then initiated by the binding of exosomes containing exonucleolytic enzymes, which leads to the degradation of mRNA. The degradation from the 5′ end is initiated by the formation of a Lsm1–7 complex at the 3′ end. The complex recruits the decapping enzymes, such as Dcp1 and Dcp2, to remove the 5′ cap. The mRNA is then degraded by the 5′→3 exonuclease XRN1. The second mRNA decay pathway is endonucleolytic cleavage [Citation14]. Site-specific RNases, such as Reganse-1, induce internal cleavage to produce RNA fragments, which are then degraded by exonucleases. The third mechanism is nonsense-mediated mRNA decay (NMD), which degrades abnormal mRNAs and thus prevents the production of abnormal proteins [Citation14]. The presence of premature termination codons (PTCs), the improper recognition of the 5′CAP by the cap-binding protein (CBP) complex (CBC), or the generation of the exon junction complex (EJC) initiates the NMD pathway [Citation20–22]. The up-frameshift proteins 1, 2, and 3 (UPF1, UPF2, and UPF3) play a central role in NMD and enable exonuclease decay by XRN1-, SMG5/SMG7- or SMG6-mediated endonuclease decay [Citation22].
Herein, we summarize and discuss the effects of RBPs on Th17 cell differentiation, function, proliferation and migration and focus on the underlying mechanisms involving RBP target mRNAs, including STAT3, OX40, c-Rel, IκBζ, IκBNS, Interferon-regulatory factor 4 (IRF4), IL-6, Inducible T Cell Costimulator (ICOS), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), IL-17, cyclin A and cyclin B1, GM-CSF and chemokine receptor 6 (CCR6). In addition, the role of miRNAs and lncRNAs in mRNA stabilization is also discussed. Finally, we provide conclusions on the role of RNA modifications and NMD in the regulation of Th17 cells (). However, these detailed mechanisms require further investigation for clarification, and such work might provide potential therapeutic applications to treat various immune-related diseases, such as allergic inflammation and autoimmune diseases.
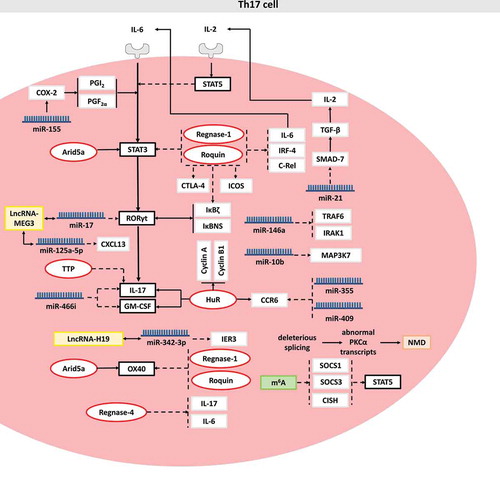
Figure 1. Posttranscriptional regulation in Th17 cell. RBPs, miRNAs, lncRNAs, mRNA modification and NMD are shown in red, blue, yellow, green and brown, respectively. For a clear description, these posttranscriptional regulators are not necessarily placed in the correct cell site. Solid arrows indicate RBP-mediated mRNA stabilization, miRNA-mediated mRNA stabilization or the promoting effect of the target. Dashed arrows indicate RBP-mediated mRNA decay, miRNA-mediated mRNA decay or the inhibiting effect of the target. Solid double-arrows indicate the interaction between lncRNA and miRNA or the cooperation between transcriptional factors. Molecular interactions are explained in the text
1. Effects of Regnase and Roquin on mRNA stability in Th17 cells
Regnase-1 (also known as Zc3h12a and MCPIP1) is a kind of cytoplasmic protein encoded by Zc3h12 [Citation23], and it contains a PilT N-terminus (PIN)-like RNase domain in the N-terminus followed by a CCCH-type zinc-finger domain [Citation23,Citation24]. The CCCH-type zinc finger domain of Regnase-1 is responsible for the recognition of the stem-loop structure [Citation23,Citation24]. Through the RNase domain, Reganse-1 induces internal cleavage to produce RNA fragments, which are then degraded by exonucleases, such as XRN1, and exosomes containing exonucleolytic enzymes [Citation14,Citation20,Citation23]. Regnase-1 functions as an endonuclease that prevents immune disorders by directly controlling the stability of a set of mRNAs, including IL-2, c-Rel, OX40 and the p40 subunit of IL-12, by directing the cleavage of the 3′UTR of these mRNA transcripts in macrophages and various T helper cells [Citation25].
Regnase-4 (also known as transformed follicular lymphoma (TFL) or ZC3H12D) contains a CCCH type zinc finger domain and PIN-like RNase domain [Citation26,Citation27]. However, the domain that is responsible for the recognition of target mRNA and the domain the mediates mRNA decay remain unknown. Further study is required to identify the mechanism underlying the Regnase-4-mRNA interaction.
The E3 ubiquitin ligase Roquin is encoded by the Rc3h1 gene and contains a ROQ domain and an adjacent CCCH-type zinc-finger domain in the N‐terminal region and a proline-rich domain in the C-terminal region [Citation28–30]. Roquin specifically recognizes the stem-loop motifs in the CDE of the 3′UTR through the ROQ domain and adjacent CCCH zinc finger domain [Citation31]. Subsequently, the C-terminal domain recruits a CCR4-NOT deadenylase complex that initiates the degradation of mRNA transcripts [Citation30,Citation32]. The exonuclease CCR4-NOT complex removes the poly (A) tail from the target mRNA at the 3′ end, and then the 3′ end is targeted by exosomes containing exonucleolytic enzymes, which leads to the degradation of mRNA [Citation14,Citation20].
Here, we provide a current overview of the immunoregulatory roles of Regnase-1, Regnase-4 and Roquin in Th17 cell differentiation and proliferation mediated by regulating the mRNA stability of target genes, including STAT3, OX40, IκBζ, IκBNS, c-Rel, IRF4, ICOS and IL-17 ().
1.1 STAT3
Studies have demonstrated that STAT3 activation is a key step in pathogenic Th17 cells, cancer and autoimmune diseases [Citation33,Citation34]. Hyperactive STAT3 protein in peripheral T cells has been shown to induce the accumulation of Th17 cells in the lung, which leads to thickened airway epithelium and increased mucus production [Citation35].
Masuda et al. found that in Th17 cells, Regnase-1 selectively decreases the stability of STAT3, thus contributing to the inhibition of Th17 cell differentiation [Citation36]. Moreover, it is physically bound to the stem-loop region in the STAT3 3′UTR (1738–1765), which competitively prevents the mRNA-stabilizing protein AT-rich interactive domain-containing 5a (Arid5a) from binding to the same region [Citation36]. The authors further found that the level of STAT3 was dramatically decreased in Arid5a-deficient T cells in an IL-6-dependent manner, which drove an impairment in Th17 cell differentiation [Citation36].
Jeltsch et. al. further found that the cooperation of Regnase-1 and Roquin destabilized STAT3 mRNA in Th17 cells, which suppressed the development of Th17 cells [Citation37]. More importantly, compared with the wild type (WT) mice, Roquin-deficient mice exhibited an increased half-life of STAT3 mRNA, which promoted Th17 differentiation and caused more severe spontaneous lung inflammation and pathology [Citation37]. However, whether Roquin and Arid5a competitively bind to STAT3 mRNA in Th17 cells remains unknown and requires further research.
1.2 OX40
In addition to STAT3, OX40, a T cell costimulatory molecule belonging to the tumour necrosis factor receptor (TNFR) superfamily, has broad impacts on the fate and function of activated T cells [Citation38–42]. Many studies have revealed that the OX40-OX40 ligand (OX40L) pathway plays a critical role in promoting the differentiation of Th17 cells, which may be partially dependent on local conditions [Citation43]. A study on Th17 cells showed that Regnase-1 and Roquin destabilized OX40 mRNA through binding to the ADE-like stem-loop (GGUG sequence in the stem region and the GUU sequence in the loop region) of the OX40 3′UTR, which is the same region that is competitively recognized by Arid5a [Citation44]. Moreover, Arid5a-deficient mice showed dramatically decreased mRNA expression of OX40 before experimental autoimmune encephalomyelitis (EAE) onset [Citation44]. These results suggest that the Arid5a/OX40 axis in CD4+ T cells may have potential implications on the pathogenesis of autoimmune diseases, which may be prevented by the induction of Regnase-1 and Roquin.
1.3 IκBζ and IκBNS
IκBζ is a member of the nuclear IκB protein family encoded by the Nfkbiz gene and harbours six ankyrin repeats at its carboxyl terminus [Citation45]. It interacts with the NF-κB subunit p50 through the ankyrin repeat domain (ARD) and has an important role in Th17 cell development, functioning in a T cell-intrinsic manner [Citation46,Citation47]. Okamoto et al. demonstrated that IκBζ acts together with the transcription factors RORα and RORγt to enhance IL-17 expression by binding directly to the regulatory region of the IL-17 gene [Citation46]. Nfkbid mRNA encodes the nuclear NF-κB inhibitor IκBNS, which contains two CDEs in its 3′UTR [Citation48]. Jeltsch et al. demonstrated that IκBNS is another transcriptional modulator, and its knockdown resulted in a remarkable decrease in IL-17 production, suggesting that IκBNS is as important as IκBζ in Th17 cell differentiation [Citation37].
Jeltsch et al. demonstrated that Regnase-1 and Roquin destabilize the target mRNAs IκBζ and IκBNS. The expression of IκBζ and IκBNS was significantly increased in Roquin- and Regnase-1-deficient mouse embryonic fibroblasts (MEFs), with the 3′UTRs of these transcripts shown to be specifically targeted by Roquin or Regnase-1 in reporter assays [Citation37]. Moreover, the increased differentiation of Th17 cells observed with Roquin-deficient T cells was at least partially mediated through the upregulation of IκBζ and IκBNS expression under Th17 cell-polarizing conditions [Citation37]. Furthermore, knockdown of Regnase-1 expression in Roquin-deficient T cells led to a greater abundance of IL-17-producing cells [Citation37]. These phenomena suggest that low expression of both Roquin and Regnase-1 may result in increased Th17 cell differentiation by regulating IκBζ and IκBNS mRNA stability. However, the molecular responses of IκBNS modulated in Th17 cell differentiation are not fully understood and need to be further investigated.
1.4 c-Rel, IRF-4, IL-6
In addition to IκBζ and IκBNS, many other sets of mRNAs have also been reported to be regulated by Roquin and Regnase-1 in Th17 cells simultaneously, and these target mRNAs include IL-6, c-Rel, and IRF4, which are critical and necessary in the differentiation of Th17 cells [Citation37,Citation49]. Studies have suggested that Roquin and Regnase-1 target the 3′UTR of c-Rel, IRF-4, and IL-6 via ICOS reporter assays with a substituted 3′UTR, which could downregulate the stability of these mRNAs [Citation37]. In addition, Minagawa et al. generated dual-luciferase reporter vectors with the 3′UTR of IL-6 mRNA and transfected them into T cells with Regnase-4, which led to destabilized IL-6 mRNA [Citation26]. However, the detailed mechanism of the Reganse-4-mediated mRNA decay needs to be further investigated.
1.5 ICOS
Although the differentiation of naive CD4+ T cells into Th17 cells does not require ICOS, ICOS has a critical role in Th17 cell proliferation and functions that are mediated by regulating the production of IL-21, which contributes to the expression and maintenance of the IL-23 receptor (IL-23 R) [Citation50]. Studies have demonstrated that ICOS mRNA is one of the targets of Roquin and Regnase-1, which bind to the ICOS 3′UTR, and that ICOS expression is dramatically increased in Roquin-deficient or Regnase-1-deficient mice under Th17 cell-polarizing conditions [Citation37].
1.6 CTLA-4
CTLA-4, also known as CD152, is a protein receptor that functions as an immune checkpoint and contributes to immune inhibitory functions [Citation51,Citation52]. It acts as an ‘off’ switch when bound to CD80 or CD86 on the surface of antigen-presenting cells, which results in protection against autoimmune disease-related deterioration [Citation53,Citation54]. A previous study showed that CTLA-4-B7 interaction inhibits Th17 cell differentiation in vitro and in vivo and suppresses Th17-mediated autoimmunity [Citation55]. Jeltsch et al. demonstrated that the expression of CTLA-4 was significantly increased in Roquin-deficient CD4+ T cells under Th17 cell-polarizing conditions [Citation37] and showed that the CTLA-4 mRNA 3′UTR was specifically targeted by Roquin and Regnase-1 in their specifically deficient MEF model [Citation37]. These phenomena suggest that increased CTLA-4 mRNA stability may play an inhibitory role in the differentiation of Th17 cells.
1.7 IL-17
IL-17 mRNA levels were markedly increased in Reganse-4 conditional knockout cells [Citation26]. Furthermore, Reganse-4-deficient mice exhibited more severe EAE compared with WT mice [Citation26]. These phenomena suggest that Regnase-4 may play an important role in attenuating local inflammation by mediating IL-17 mRNA decay.
2. Effects of HuR on mRNA stability in Th17 cells
HuR is a member of the Elav/Hu family and contains three RNA-recognition motifs (RRM) [Citation56]. The two tandem N-terminal RRM domains (RRM1 and RRM2) can selectively bind to the ARE [Citation57–59]. The third domain (RRM3) binds to the ARE and the poly(A) tail simultaneously, which protects the poly(A) tail from poly(A) exonuclease [Citation60]. An increasing number of studies have reported that HuR is emerging as a pivotal regulator in cell proliferation, differentiation, and immune responses and functions by binding to AREs located in the 3′UTR and mediating mRNA stabilization [Citation61]. Herein, we summarize the modulatory effects of HuR on Th17 cell differentiation mediated by the regulation of mRNA stability ().
2.1 IL-17
Chen et al. demonstrated that HuR normally binds to the 3′UTR of IL-17 and stabilizes IL-17 transcripts in CD4+ Th17 cells [Citation62]. HuR deficiency shortens the half-life of IL-17 mRNA in Th17-polarized cells, which impairs Th17 cell differentiation by activated T cells and delays disease onset, thereby reducing the severity of EAE in an adoptive transfer model [Citation62]. These results suggest that HuR may play important roles during the differentiation and immune function of Th17 cells via PTR and provide insights into the treatment of IL-17-mediated autoimmune diseases, such as EAE, by regulating the differentiation of Th17 cells [Citation62].
2.2 Cyclin A and cyclin B1
Previous studies demonstrated the involvement of the regulation of cyclin A and cyclin B1 mRNA stability in the cell cycle [Citation63–65]. Interestingly, Wang et al. reported that HuR-deficient mice exhibited decreased cyclin A and cyclin B1 mRNA stability and diminished proliferation rates of human colon adenocarcinoma (RKO) cells [Citation66]. Therefore, we hypothesized that HuR may regulate Th17 cell proliferation by specifically targeting cyclin A and cyclin B1 in a posttranscriptional manner. However, the detailed mechanism needs to be further investigated.
2.3 GM-CSF
In addition to IL-17, GM-CSF is essential for Th17 cells to induce autoimmune neuroinflammation [Citation67,Citation68]. Increased levels of GM-CSF have been reported in the cerebrospinal fluid of patients with relapsing-remitting multiple sclerosis (MS) [Citation69,Citation70]. Some trials targeting GM-CSF are still in progress for treating MS patients [Citation71]. A recent Phase I clinical trial showed that targeting human GM-CSF by MOR103 is safe for MS patients [Citation72]. Surprisingly, Chen et al. showed that HuR genetic ablation resulted in significant decreases in steady-state GM-CSF mRNA levels in Th17 cells [Citation73]. Further studies have indicated that HuR directly binds to the AREs in the 3′UTR of GM-CSF mRNA, suggesting a regulatory relationship. Thus, HuR binds to and stabilizes GM-CSF mRNA, which results in increased GM-CSF mRNA accumulation in Th17 cells [Citation73]. Given the importance of Th17 cells in MS [Citation74], the regulation of GM-CSF production requires further study to develop novel approaches for targeting this glycoprotein.
2.4 CCR6
CCR6 is a CC chemokine receptor protein that belongs to family A of the G protein-coupled receptor superfamily, and it is highly expressed in pathogenic Th17 cells [Citation75,Citation76]. Reboldi et al. revealed that CCR6 is necessary for the migration of pathogenic Th17 cells to sites of inflammation and that CCR6-deficient mice are resistant to the pathogenesis of EAE [Citation77]. Further studies have demonstrated that HuR moderately promotes CCR6 expression by binding to its mRNA transcript to prolong the transcript half-life [Citation61]. Moreover, HuR-deficient mice exhibited reduced expression of CCR6 in Th17 cells and impaired migration in response to the CCR6 ligand chemokine ligand 20 (CCL20), thereby ameliorating EAE [Citation27,Citation61,Citation77]. These results suggest that HuR plays an important role in the migration of Th17 cells via PTR, and it may represent a novel therapeutic intervention for autoimmune diseases.
3. Effects of tristetraprolin (TTP) on mRNA stability in Th17 cells
TTP (also known as TIS11, ZFP36, and Nup475) is a CCCH tandem zinc-finger protein (ZFP) coded by the gene Zfp36 and regulates inflammatory responses at the post-transcriptional level [Citation78]. The two zinc finger domains within TTP recognize specific ARE elements, such as UUAUUUAUU, in the 3′UTR of their target mRNA [Citation79]. TTP binds to the AREs and recruits the deadenylation complex CCR4-NOT via its C-terminal domain to remove the poly(A) tail at the 3′ end, which initiates mRNA decay [Citation14,Citation80]. Subsequently, TTP recruits decapping enzymes via its N-terminal domain, and they remove the cap at the 5′ end [Citation14,Citation81]. The 5′ end is then targeted by 5′→3′ exonuclease, such as XRN1, which leads to mRNA decay [Citation14,Citation20,Citation79,Citation82,Citation83].
Previous study showed that TTP controls the function of Th17 cells via promoting IL-17 mRNA decay [Citation81]. Peng’s group further found that TTP-KO mice showed more IL-17–producing effector T cells, which led to more severe colitis compared with WT mice. These phenomena support a pathogenic role for TTP in intestinal inflammation via destabilizing IL-17 mRNA in Th17 cells () [Citation84].
4. Effects of miRNAs and lncRNAs on mRNA stability in Th17 cells
In addition to RBPs, miRNAs also regulate gene expression in a sequence-specific manner, often through interplay with RBPs that associate with the same mRNAs [Citation85]. miRNAs are a class of noncoding RNAs that are approximately 21 to 24 nucleotides (nt) in length, and most miRNAs regulate the expression of numerous target genes by mediating mRNA decay and/or repressing translation [Citation85–87]. miRNA-mediated mRNA decay requires the RNA-induced silencing complex (RISC). The function of the RISC in mRNA decay is determined by two components, Argonaute (Ago) proteins and miRNA. miRNA binds to the 3′UTR of target mRNA via central pairing, which guides the RISC to the target mRNA. Ago proteins then mediate mRNA decay through their intrinsic RNase H-like activity [Citation14,Citation88,Citation89]. The mRNA decay process mediated by the RISC is determined by the subtype of Ago [Citation14,Citation89]. The RISC containing Ago1 recruit deadenylation complexes, such as CCR4-NOT and PARN, which remove the poly (A) tail from the target mRNA at the 3′ end. The 3′ end is then targeted by exosomes containing exonucleolytic enzymes, which leads to the degradation of mRNA [Citation14,Citation89]. The RISC containing the translation initiation factor Ago2 induces endonucleolytic cleavage to produce RNA fragments, which are then degraded by exonuclease [Citation14,Citation89]. miRNAs are critical in the immune response, and their knock-out and silencing may lead to immune disorders, such as autoimmune diseases and cancers [Citation85].
LncRNAs are non-coding RNAs that are more than 200 nucleotides long, and they are involved in the development of multiple diseases, especially in asthma [Citation90,Citation91]. One of these mechanisms can be explained by the theory of competing endogenous RNA (ceRNA). The theory supposes that through base complementation, lncRNA adsorbs miRNAs and consequently reduces the binding of these miRNAs to their target genes, which indirectly alters the expression of the target genes of these miRNAs [Citation92].
Herein, we summarize the modulatory effects of miRNAs on Th17 cell differentiation mediated via regulation of mRNA stability and the function of lncRNAs in Th17-cells-related diseases via regulating miRNAs ().
4.1 COX-2
The activation of COX-2 induced the production of prostaglandin I2 (PGI2) and prostaglandin F2α (PGF2α) [Citation93], which increased STAT3 phosphorylation in naive CD4+ T cells and thereby promoted Th17 differentiation [Citation94]. Furthermore, Qiu et al. showed that miR-155 upregulated COX-2 mRNA stability by binding to the 3′UTR of COX-2 mRNA and that knocking out miR-155 resulted in significantly decreased production of Th17 cytokines [Citation95]. These results suggest that the modulation of COX-2 in Th17 cells can be mediated by miR-155 via PTR. However, the molecular mechanism underlying miR-155-mediated mRNA stabilization is not fully understood, which requires further research.
4.2 GM-CSF and IL-17
Chen et al. demonstrated that miR-466i functions to mediate GM-CSF and IL-17 mRNA decay by binding to the 3′UTRs of these transcripts in Th17 cells [Citation73]. Interestingly, they also demonstrated that knocking out HuR could increase the expression of miR-466i by downregulating the expression of Mxi1, an important repressor of a miRNA expression activator [Citation73]. Therefore, in addition to HuR directly regulating GM-CSF and IL-17 mRNA stability via the previously described process, HuR can also work indirectly by downregulating the expression of certain miRNAs.
4.3 CCR6
Chen et al. demonstrated that the miR-335 and miR-409 levels were increased in HuR-KO Th17 cells, with these miRNAs degrading CCR6 mRNA by binding to the CCR6 3′UTR [Citation61]. This phenomenon suggested that in addition to the direct regulation of CCR6 mRNA stability by HuR via the previously described process, indirect regulation also occurs through downregulation of the expression of certain miRNAs [Citation96–98].
4.4 SMAD-7
miR-21 mediates SMAD-7 mRNA decay via binding to the 3′UTR in CD4+ T cells [Citation99]. After SMAD-7 mRNA decay, TGF-β production is increased, which limits the production of IL-2 [Citation100,Citation101]. IL-2 has been shown to inhibit Th17 differentiation via activating STAT5, and miR-21 promotes the differentiation of Th17 cells. Further study found that miR-21-deficient mice showed a reduced clinical severity of EAE [Citation99].
4.5 TRAF6 and IRAK1
Studies demonstrated that miR-146a represses TRAF6 and IRAK1 protein production, resulting in the blocking of IL-6 and IL-21 signals in CD4+ T cells, which are important for Th17 cell differentiation [Citation102–104]. However, whether miR-146a mediates TRAF6 and IRAK1 mRNA decay in CD4+ T cells remains unknown and requires further research.
4.6 MAP3K7
MAP3K7-deficient CD4+ T cells are defective in IL −17 production when undergoing differentiation into Th17 cells [Citation105]. Moreover, the over-expression of miR-10b causes decreased MAP3K7 gene expression, suggesting that miR-10b destabilizes MAP3K7 mRNA to inhibit the differentiation and function of Th17 cells [Citation106].
4.7 RORγt
LncRNA-maternally expressed gene 3 (MEG3) is a tumour suppressor lncRNA that functions as a tumour suppressor gene in many different kinds of cancer, such as gastric cancer [Citation107,Citation108]. After transfection with the miR-17 mimic, RORγt mRNA is destabilized in CD4 + T cells from asthma patients, which inhibited the development of Th17 cells [Citation92]. A bioinformatic analysis demonstrated that MEG3 regulates miR-17 through base complementation in CD4 + T cells [Citation92]. Furthermore, the mRNA and protein levels of RORγt are decreased after the knockdown of MEG3 [Citation92]. Taken together, MEG3 acts as a ceRNA to inhibit miR-17-mediated RORγt mRNA decay, which inhibits the development of Th17 cells and thereby influences the development of asthma.
Interestingly, a clear protein band of Ago2 was detected by Qiu et al. in biotin-labelled MEG3 [Citation92], suggesting that an interaction occurs between the Ago2 and MEG. We suppose that MEG3 indirectly inhibits Ago2-mediated RORγt mRNA decay by competitively binding to miR-17 and preventing miR-17 from binding to RORγt mRNA in CD4+ T cells.
4.8 IER3
Immediate early response gene (IER3), also called IEX-1, is rapidly activated during inflammation and functions as an inhibitor in TNF-induced apoptosis [Citation109,Citation110]. A previous study showed that the lack of IER3 promotes Th17 differentiation [Citation111].
Liu et al. found that the mRNA stability of IER3 was downregulated by miR-342-3p [Citation112]. LncRNA-H19 is one of the most widely studied lncRNAs in cancers, angiogenesis, diabetes mellitus, etc. [Citation113,Citation114]. A previous study revealed that H19 competitively binds to endogenous miR-342-3p in gallbladder cancer cells [Citation115]. However, whether H19 regulates IER3 expression by competitively binding to endogenous miR-342-3p in Th17 cells remains unknown and requires further research.
4.9 CXCL13
An imbalance of the Th17 cell ratio plays an important role in immune thrombocytopenic purpura (ITP) [Citation116]. CXC chemokine ligand-13 (CXCL13) is a small cytokine belonging to the CXC chemokine family and functions as a chemoattractant and influences the Th17 cell ratio in multiple immunological diseases, including ITP [Citation117–119].
A previous study discovered that miR-125a-5p mediates CXCL13 mRNA decay by targeting the 3′UTR and participates in the ITP process [Citation120]. Furthermore, Li et al. reported that the overexpression of MEG3 upregulates CXCL13 mRNA [Citation121]. However, whether MEG3 regulates CXCL13 expression by competitively binding to endogenous miR-125a-5p in Th17 cells remains unknown and requires further research.
Interestingly, a clear protein band of Ago2 can be detected in biotin-labelled MEG3, suggesting that MEG3 interacts with Ago2 [Citation121]. We suppose that MEG3 also indirectly inhibits Ago2-mediated CXCL13 mRNA decay by competitively binding to endogenous miR-125a-5p in CD4+ T cells and preventing miR-125a-5p from binding to CXCL13 mRNA.
5. Effects of RNA modification and NMD on mRNA stability in Th17 cells
PTR events include not only mRNA decay but also RNA splicing, mRNA export and RNA modifications [Citation14]. However, except for RNA modifications, few studies have discussed these aspects in Th17 cells [Citation122]. A previous study reported that STAT5 activation is crucial to suppressing the differentiation of Th17 cell [Citation123]. Methyltransferase-like 3 (Mettl3) is the ‘writer’ protein of N6-methyladenosine (m6A) modification, the knockout of which leads to the loss of the m6A modification [Citation124]. Li et al. demonstrated that the loss of the m6A modification in Mettl3-KO naive T cells leads to increased mRNA stability of SOCS1, SOCS3 and CISH, which ultimately suppresses STAT5 signalling pathway, thereby promoting the differentiation of Th17 cells () [Citation122]. However, the underlying mechanism of decreased SOCS1, SOCS3 and CISH mRNA stability caused by m6A is not fully understood and requires further research.
In addition, the NMD caused by deleterious splicing is actively involved in the regulation of Th17 cells by mediating the decay of abnormal PKCα transcripts () [Citation125]. PKCα is a Th17-cell-selective kinase, and mice deficient in this kinase exhibit decreased mRNA expression levels of Th17-related transcription factors, such as RORγt, RUNX1, AHR, and IRF4, indicating that PKCα promotes Th17 differentiation [Citation125]. Further study is required to identify the degradation mechanism of abnormal PKCα transcripts through the NMD pathway in Th17 cells.
6. Conclusions and perspectives
In conclusion, we mainly reviewed the key roles of regulatory RBPs and miRNAs in the differentiation, function, proliferation and migration of Th17 cells and focuses on the mechanism of miRNA-mRNA and RBP-mRNA interactions. We also summarized the function of lncRNAs in Th17 cell-related diseases via regulating miRNAs. Finally, we provided conclusions on the role of modifications and NMD in the regulation of Th17 cells.
Interestingly, Th17 cells appear to be particularly susceptible to alternations of RBPs. The RBPs discussed above are not expressed exclusively in Th17 cells; however, some target mRNAs of these RBPs are Th17-specific. For instance, RORγt is a Th17-specific transcription factor [Citation5], which might be due to some unknown Th17-specific factors that mediate RBP regulation of RORγt mRNA stability.
In addition to mRNA stability, the role of other regulatory PTR events mediated by RBPs in Th17 cells are not fully understood. For example, HuR is reported to affect many other aspects of mRNA processing in addition to mRNA stability, such as splicing, translation, miRNA repression modulation, and intracellular mRNA trafficking [Citation126]. However, recent studies have only shown that HuR could regulate mRNA stability in Th17 cells; thus, other post-transcriptional regulatory mechanisms mediated by HuR require further research.
Further investigations are required to identify other potential RBPs and miRNAs and their targets that are involved in the differentiation of Th17 cells. We suppose that other RBPs harbouring the same domain may also be involved in the regulation of Th17 cells. For example, Regnase-2 and Regnase-3 are reported to share the same CCCH-type zinc finger domain with Regnase-1; thus, they likely function in Th17 cells [Citation127]. The RBP poly(C)-binding protein-1 (PCBP-1) has been reported to mediate the mRNA decay of forkhead box protein P3 (FoxP3), which inhibits Treg differentiation [Citation128]. Since the expression of FoxP3 inhibits Th17 differentiation by inhibiting RORγt transcriptional activity [Citation129], PCBP-1 may regulate mRNA stability in Th17 cells, which may influence the development of Th17 cells.
Despite the important roles of RBPs and miRNAs in controlling the fate of Th17 cells, direct evidence for a link between the regulation of mRNA stability and the development of autoimmune disease must still be obtained in the future. For example, knocking out the binding site in the ARE or SL of the 3′UTR of a target mRNA could be performed.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 31670889 and 31200668) and the National Key Research and Development Project (No. 2016YFA0502203 and 2016YFA0502204). The funders had no role in the study design, data analysis, or decision to publish.
Disclosure statement
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Matsuzaki G, Umemura M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol Immunol. 2007;51(12):1139–1147.
- Kolls JK, Khader SA. The role of Th17 cytokines in primary mucosal immunity. Cytokine Growth Factor Rev. 2010;21:443–448.
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348.
- Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132.
- Korn T, Bettelli E, Oukka M, et al. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517.
- Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189.
- Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238.
- Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234.
- Yosef N, Shalek AK, Gaublomme JT, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468.
- Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30(5):646–655.
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489.
- Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. 2019;41:283–297.
- Wilke CM, Bishop K, Fox D, et al. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011;32:603–611.
- Kafasla P, Skliris A, Kontoyiannis DL. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat Immunol. 2014;15:492–502.
- Bjur E, Larsson O, Yurchenko E, et al. Distinct translational control in CD4+ T cell subsets. PLoS Genet. 2013;9:e1003494.
- Ciofani M, Madar A, Galan C, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303.
- Bhatt DM, Pandya-Jones A, Tong AJ, et al. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–290.
- Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. 2009;10:281–288.
- Hao S, Baltimore D. RNA splicing regulates the temporal order of TNF-induced gene expression. Proc Natl Acad Sci U S A. 2013;110:11934–11939.
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126.
- Le Hir H, Izaurralde E, Maquat LE, et al. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. Embo J. 2000;19:6860–6869.
- Diaz-Munoz MD, Turner M. Uncovering the role of RNA-Binding proteins in gene expression in the immune system. Front Immunol. 2018;9:1094.
- Matsushita K, Takeuchi O, Standley DM, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–1190.
- Yang C, Huang S, Wang X, et al. Emerging roles of CCCH-Type zinc finger proteins in destabilizing mRNA encoding inflammatory factors and regulating immune responses. Crit Rev Eukaryot Gene Expr. 2015;25:77–89.
- Uehata T, Iwasaki H, Vandenbon A, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell. 2013;153:1036–1049.
- Minagawa K, Wakahashi K, Kawano H, et al. Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J Iimmunol. 2014;192:1512–1524.
- Yoshinaga M, Takeuchi O. RNA binding proteins in the control of autoimmune diseases. Immunological medicine. 2019;42:53–64.
- Athanasopoulos V, Ramiscal RR, Vinuesa CG. ROQUIN signalling pathways in innate and adaptive immunity. Eur J Immunol. 2016;46:1082–1090.
- Lin AE, Mak TW. The role of E3 ligases in autoimmunity and the regulation of autoreactive T cells. Curr Opin Immunol. 2007;19:665–673.
- Leppek K, Schott J, Reitter S, et al. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153:869–881.
- Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. 2017;17:130–143.
- Sgromo A, Raisch T, Bawankar P, et al. A CAF40-binding motif facilitates recruitment of the CCR4-NOT complex to mRNAs targeted by Drosophila Roquin. Nat Commun. 2017;8:14307.
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
- Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46:812–814.
- Fogli LK, Sundrud MS, Goel S, et al. T cell-derived IL-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J Iimmunol. 2013;191:3100–3111.
- Masuda K, Ripley B, Nyati KK, et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J Exp Med. 2016;213:605–619.
- Jeltsch KM, Hu D, Brenner S, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. 2014;15:1079–1089.
- Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68.
- Xiao X, Shi X, Fan Y, et al. The costimulatory receptor OX40 inhibits Interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity. 2016;44:1271–1283.
- Demirci G, Amanullah F, Kewalaramani R, et al. Critical role of OX40 in CD28 and CD154-independent rejection. J Iimmunol. 2004;172:1691–1698.
- Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839.
- Xiao X, Balasubramanian S, Liu W, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. 2012;13:981–990.
- Fu Y, Lin Q, Zhang Z, et al. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B. 2020;10:414–433.
- Hanieh H, Masuda K, Metwally H, et al. Arid5a stabilizes OX40 mRNA in murine CD4(+) T cells by recognizing a stem-loop structure in its 3ʹUTR. Eur J Immunol. 2018;48:593–604.
- Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276:27657–27662.
- Okamoto K, Iwai Y, Oh-Hora M, et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature. 2010;464:1381–1385.
- Muta T. IkappaB-zeta: an inducible regulator of nuclear factor-kappaB. Vitam Horm. 2006;74:301–316.
- Essig K, Kronbeck N, Guimaraes JC, et al. Roquin targets mRNAs in a 3ʹ-UTR-specific manner by different modes of regulation. Nat Commun. 2018;9:3810.
- Wawro M, Wawro K, Kochan J, et al. ZC3H12B/MCPIP2, a new active member of the ZC3H12 family. Rna. 2019;25:840–856.
- Paulos CM, Carpenito C, Plesa G, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med. 2010;2:55ra78.
- Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol. 2003;3:544–556.
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328:267–270.
- Chikuma S, Bluestone JA. CTLA-4 and tolerance: the biochemical point of view. Immunol Res. 2003;28:241–253.
- Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547.
- Ying H, Yang L, Qiao G, et al. Cutting edge: CTLA-4–B7 interaction suppresses Th17 cell differentiation. J Immunol. 2010;185:1375–1378.
- Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65:3168–3181.
- Pabis M, Popowicz GM, Stehle R, et al. HuR biological function involves RRM3-mediated dimerization and RNA binding by all three RRMs. Nucleic Acids Res. 2019;47:1011–1029.
- Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J. 1998;17:3448–3460.
- Peng SS, Chen CY, Xu N, et al. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. Embo J. 1998;17:3461–3470.
- Ma WJ, Chung S, Furneaux H. The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 1997;25:3564–3569.
- Chen J, Martindale JL, Cramer C, et al. The RNA-binding protein HuR contributes to neuroinflammation by promoting C-C chemokine receptor 6 (CCR6) expression on Th17 cells. J Biol Chem. 2017;292:14532–14543.
- Chen J, Cascio J, Magee JD, et al. Posttranscriptional gene regulation of IL-17 by the RNA-binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. J Iimmunol. 2013;191:5441–5450.
- Maity A, McKenna WG, Muschel RJ. Cyclin A message stability varies with the cell cycle. Cell growth differentiation. 1997;8:311–318.
- Howe JA, Howell M, Hunt T, et al. Identification of a developmental timer regulating the stability of embryonic cyclin A and a new somatic A-type cyclin at gastrulation. Genes Dev. 1995;9:1164–1176.
- Trembley JH, Kren BT, Steer CJ. Posttranscriptional regulation of cyclin B messenger RNA expression in the regenerating rat liver. Cell growth differentiation. 1994;5:99–108.
- Wang W, Caldwell MC, Lin S, et al. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19:2340–2350.
- El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575.
- Codarri L, Gyulveszi G, Tosevski V, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567.
- Carrieri PB, Provitera V, De Rosa T, et al. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20:373–382.
- Perrella O, Carrieri PB, De Mercato R, et al. Markers of activated T lymphocytes and T cell receptor gamma/delta+ in patients with multiple sclerosis. Eur Neurol. 1993;33:152–155.
- Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm. 2015;2015:568543.
- Constantinescu CS, Asher A, Fryze W, et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2015;2:e117.
- Chen J, Adamiak W, Huang G, et al. Interaction of RNA-binding protein HuR and miR-466i regulates GM-CSF expression. Sci Rep. 2017;7:17233.
- Maddur MS, Miossec P, Kaveri SV, et al. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18.
- Zaballos A, Varona R, Gutierrez J, et al. Molecular cloning and RNA expression of two new human chemokine receptor-like genes. Biochem Biophys Res Commun. 1996;227:846–853.
- Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426.
- Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523.
- Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans. 2002;30:945–952.
- Lai WS, Carballo E, Thorn JM, et al. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J Biol Chem. 2000;275:17827–17837.
- Lai WS, Stumpo DJ, Wells ML, et al. Importance of the conserved carboxyl-terminal CNOT1 binding domain to tristetraprolin activity in vivo. Mol Cell Biol. 2019;39.
- Lee HH, Yoon NA, Vo MT, et al. Tristetraprolin down-regulates IL-17 through mRNA destabilization. FEBS Lett. 2012;586:41–46.
- Lykke-Andersen J, Wagner E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005;19:351–361.
- Fenger-Gron M, Fillman C, Norrild B, et al. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915.
- Peng H, Ning H, Wang Q, et al. Tristetraprolin regulates TH17 cell function and ameliorates DSS-induced colitis in mice. Front Immunol. 2020;11:1952.
- Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36.
- Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433.
- Iwakawa HO, Tomari Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25:651–665.
- Arribas-Hernandez L, Kielpinski LJ, Brodersen P. mRNA decay of most arabidopsis miRNA targets requires slicer activity of AGO1. Plant Physiol. 2016;171:2620–2632.
- Fukaya T, Tomari Y. MicroRNAs mediate gene silencing via multiple different pathways in drosophila. Mol Cell. 2012;48:825–836.
- Narozna B, Langwinski W, Szczepankiewicz A. Non-Coding RNAs in pediatric airway diseases. Genes (Basel). 2017;8.
- Zhu YJ, Mao D, Gao W, et al. Peripheral whole blood lncRNA expression analysis in patients with eosinophilic asthma. Medicine (Baltimore). 2018;97:e9817.
- Qiu YY, Wu Y, Lin MJ, et al. LncRNA-MEG3 functions as a competing endogenous RNA to regulate Treg/Th17 balance in patients with asthma by targeting microRNA-17/RORgammat. Biomed Pharmacothe. 2019;111:386–394.
- Russell-Puleri S, Dela Paz NG, Adams D, et al. Fluid shear stress induces upregulation of COX-2 and PGI2 release in endothelial cells via a pathway involving PECAM-1, PI3K, FAK, and p38. Am J Physiol Heart Circ Physiol. 2017;312:H485–H500.
- Li H, Bradbury JA, Dackor RT, et al. Cyclooxygenase-2 regulates Th17 cell differentiation during allergic lung inflammation. Am J Respir Crit Care Med. 2011;184:37–49.
- Qiu L, Zhang Y, Do DC, et al. miR-155 modulates cockroach allergen- and oxidative stress-induced cyclooxygenase-2 in asthma. J Iimmunol. 2018;201:916–929.
- Hedrick MN, Lonsdorf AS, Shirakawa AK, et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. 2009;119:2317–2329.
- Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812.
- Yamazaki T, Yang XO, Chung Y, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Iimmunol. 2008;181:8391–8401.
- Murugaiyan G, da Cunha AP, Ajay AK, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. 2015;125:1069–1080.
- Ross SH, Cantrell DA. Signaling and function of Interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36(1):411–433.
- Li Q, Zhang D, Wang Y, et al. MiR-21/Smad 7 signaling determines TGF-beta1-induced CAF formation. Sci Rep. 2013;3:2038.
- Yang L, Boldin MP, Yu Y, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012;209:1655–1670.
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708.
- Li B, Wang X, Choi IY, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest. 2017;127:3702–3716.
- Suddason T, Gallagher E. Genetic insights into Map3k-dependent proliferative expansion of T cells. Cell Cycle. 2016;15:1956–1960.
- Chen L, Al-Mossawi MH, Ridley A, et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann Rheum Dis. 2017;76:620–625.
- Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:3850–3856.
- Uroda T, Anastasakou E, Rossi A, et al. Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol Cell. 2019;75:982–95 e9.
- Wu MX, Ao Z, Prasad KV, et al. IEX-1L, an apoptosis inhibitor involved in NF-kappaB-mediated cell survival. Science. 1998;281:998–1001.
- Zhang Y, Schlossman SF, Edwards RA, et al. Impaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proc Natl Acad Sci U S A. 2002;99:878–883.
- Zhi L, Ustyugova IV, Chen X, et al. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Iimmunol. 2012;189:1639–1647.
- Liu Z, Liu L, Zhong Y, et al. LncRNA H19 over-expression inhibited Th17 cell differentiation to relieve endometriosis through miR-342-3p/IER3 pathway. Cell Biosci. 2019;9:84.
- Qi M, Zhou Q, Zeng W, et al. Analysis of long non-coding RNA expression of lymphatic endothelial cells in response to type 2 diabetes. Cell Physiol Biochem. 2017;41:466–474.
- Hou J, Wang L, Wu Q, et al. Long noncoding RNA H19 upregulates vascular endothelial growth factor A to enhance mesenchymal stem cells survival and angiogenic capacity by inhibiting miR-199a-5p. Stem Cell Res Ther. 2018;9:109.
- Wang SH, Ma F, Tang ZH, et al. Long non-coding RNA H19 regulates FOXM1 expression by competitively binding endogenous miR-342-3p in gallbladder cancer. J Exp Clin Cancer Res. 2016;35:160.
- Ji L, Zhan Y, Hua F, et al. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PloS One. 2012;7:e50909.
- Lee HT, Shiao YM, Wu TH, et al. Serum BLC/CXCL13 concentrations and renal expression of CXCL13/CXCR5 in patients with systemic lupus erythematosus and lupus nephritis. J Rheumatol. 2010;37:45–52.
- Burkle A, Niedermeier M, Schmitt-Graff A, et al. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110:3316–3325.
- Jernas M, Nookaew I, Wadenvik H, et al. MicroRNA regulate immunological pathways in T-cells in immune thrombocytopenia (ITP). Blood. 2013;121:2095–2098.
- Li JQ, Hu SY, Wang ZY, et al. MicroRNA-125-5p targeted CXCL13: a potential biomarker associated with immune thrombocytopenia. Am J Transl Res. 2015;7:772–780.
- Li JQ, Hu SY, Wang ZY, et al. Long non-coding RNA MEG3 inhibits microRNA-125a-5p expression and induces immune imbalance of Treg/Th17 in immune thrombocytopenic purpura. Biomed Pharmacothe. 2016;83:905–911.
- Li HB, Tong J, Zhu S, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–342.
- Yang XP, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254.
- Wang CX, Cui GS, Liu X, et al. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. 2018;16:e2004880.
- Meisel M, Hermann-Kleiter N, Hinterleitner R, et al. The kinase PKCalpha selectively upregulates interleukin-17A during Th17 cell immune responses. Immunity. 2013;38:41–52.
- Eberhardt W, Badawi A, Biyanee A, et al. Cytoskeleton-dependent transport as a potential target for interfering with post-transcriptional HuR mRNA regulons. Front Pharmacol. 2016;7:251.
- Liang J, Wang J, Azfer A, et al. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. 2008;283:6337–6346.
- Ansa-Addo EA, Huang HC, Riesenberg B, et al. RNA binding protein PCBP1 is an intracellular immune checkpoint for shaping T cell responses in cancer immunity. Sci Adv. 2020;6:eaaz3865.
- Ren J, Li B. The functional stability of FOXP3 and RORgammat in Treg and Th17 and their therapeutic applications. Adv Protein Chem Struct Biol. 2017;107:155–189.