
ABSTRACT
Eight-segmented, negative-sense, single-stranded genomic RNAs of influenza A virus are terminated with 5′ and 3′ untranslated regions (UTRs). All segments have highly conserved extremities of 13 and 12 nucleotides at the 5′ and 3′ UTRs, respectively, constructing the viral RNA (vRNA) promoter. Adjacent to the duplex stem of 3 base pairs (bps) between the two conserved strands, additional 1–4 bps are existing in a segment-specific manner. We investigated the roles of the matrix (M) segment-specific base pair between the 14th nucleotide uridine (U14′) of the 5′ UTR and the 13th nucleotide adenosine (A13) of the 3′ UTR by preparing possible vRNA promoters, named vXY, as well as cRNA promoters, named cYX. We analysed their RNA-dependent RNA replication efficiency using the minigenome replicon system and an enzyme assay system in vitro with synthetic RNA promoters. Notably, in contrast to vAC(s) that is a synthetic vRNA promoter with A14′ and C13, base-pair disruption at the complementary RNA (cRNA) promoter in cAC(s), which has A13′ and C14, not only reduced viral RNA replication in cells but also impaired de novo initiation of unprimed vRNA synthesis. Reverse genetics experiments confirmatively exhibited that this breakage in the cRNA promoter affected the rescue of infectious virus. The present study suggests that the first segment-specific base pair plays an essential role in generating infectious viruses by regulating the promoter activity of cRNA rather than vRNA. It could provide insights into the role of the segment-specific nucleotides in viral genome replication for sustainable infection.
Introduction
Influenza A virus belonging to the family Orthomyxoviridae is one of the human respiratory pathogens that causes seasonal or zoonotic epidemics and unpredictable global pandemics. Its genome is composed of eight single-stranded negative-sense RNA segments. Within a viral particle, they are individually encapsidated with nucleoprotein (NP) to form viral ribonucleoproteins (vRNPs) complexed with the three polymerase subunits, PB2, PB1, and PA. When the virus enters the cell, these vRNP complexes are released into the cytoplasm through the endocytic pathway[Citation1]. Depending on the nuclear localization signals (NLS) within NP, they are delivered into the nucleus where transcription and RNA replication take place[Citation2].
Influenza A viral RNA (vRNA) has a unique genetic characteristic that all eight fragments terminate with 13 and 12 highly conserved nucleotides at the ends of the 5′ and 3′ untranslated regions (UTRs)[Citation3]. These UTR extremities called the universal sequence are partially complementary to each other, constructing an RNA promoter with a panhandle structure when naked, or a 5′ stem-loop (‘hook’) structure in the presence of polymerase components () [Citation4–7].The compact stem-loop of the 5′ end is stabilized by the binding pocket formed by PB1 and PA residues, while the single-stranded 3′ end enters the polymerase active site[Citation7]. Both ends of the universal sequence become spatially close on account of hybridization between nucleotides 11–13 of the 5′ UTR and nucleotides 10–12 of the 3′ UTR, achieving the proximal stem. This structural characteristic is conversed in a complementary RNA (cRNA) promoter[Citation8]. In the nuclei of infected cells, a viral polymerase complex initiates de novo synthesis of cRNA or vRNA from each relevant promoter [Citation9–12].
Figure 1. Mutations at the M segment-specific non-coding nucleotides of the influenza A vRNA and cRNA promoters. Mutations at the X14′ and Y13 nucleotides of the vRNA promoter were introduced by site-directed mutagenesis, named vX14′Y13 or vXY (left, black). Position 1′ means the starting point of the 5′ UTR, while position 1 does the terminating point of the 3′ UTR. Their complementary RNA (cRNA) cassettes were designed and named cY′13′X′14 or cYX (right, red). According to the hybridization ability of the target nucleotides within the vRNA and cRNA promoters, they are classified into the three groups. wt, wild-type. Paired or unpaired nucleotides at the target site are highlighted in red or blue boxes
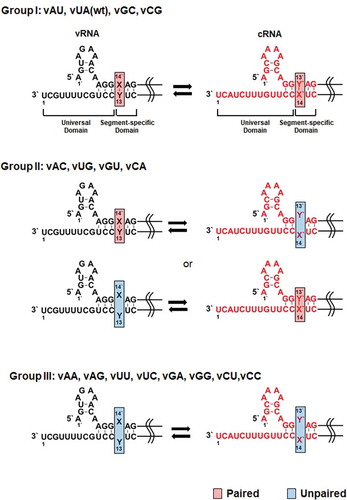
The entire non-coding regions of influenza A viral genome also include non-conserved sequences of 5 to 45 nucleotides along with the universal sequences at both termini[Citation13]. Many previous studies have focused on the roles of the highly conserved universal sequences, whereas functions of the variable domain have not been well elucidated [Citation10,Citation14,Citation15]. Segment-wide sequence analysis of different influenza A viral subtypes or isolates showed that the variable regions have intra-segmental homology at individual genome segments[Citation16]. It is implying that they could be actively participated either in viral RNA replication or transcription or in viral genome packaging together with the universal domains. One of the underlying characteristics of the non-conserved regions is a perfect complementarity between initial 1–4 bases. They are directly adjacent to the universal domains in 5′ and 3′ UTRs and complete the proximal stem of the vRNA and cRNA promoters (, Group I) [Citation3,Citation17]. Particularly, conservation of the first segment-specific base pair appears in every segmented RNA of all types of influenza A viruses[Citation18]. We targeted the M segment as a representative genome to investigate whether and how this base pair regulates RNA replication, protein expression, and viral growth. The present study suggests pivotal roles of hybridization of the two nucleotides for initiation of vRNA synthesis from the cRNA promoter and discovery of a recombinant mutant virus with an alternative nucleotide pair that allows sustainable infection, eventually unveiling viral preferences for evolutionary selection.
Results
Hybridization of the first segment-specific non-coding nucleotide pair is indispensable to the cRNA promoter for RNA-dependent RNA replication
We explored the effect of the first segment-specific pair composed of U14′ and A13 of the M genome UTRs on influenza A viral RNA replication. The antisense strand of the enhanced green fluorescent protein (EGFP) open reading frame flanked by the 5′ and 3′ UTRs was cloned under the control of the human RNA polymerase I (Pol I) promoter. The mimetic vRNA [vRNA(EGFP)] with U14′·A13 was systematically mutated to generate all 15 mutations, named vX14′Y13, or simply vXY (, left column). They were classified into three groups based on their hybridization ability in the vRNA and cRNA promoters. Group I includes vAU, vGC, and vCG with Watson-Crick base pairs in both promoters like the wild-type vUA. Group II consists of vAC, vUG, vGU, and vCA, which can form non-canonical G-U base pairs either in the vRNA promoter or in the cRNA promoter. Group III includes vAA, vAG, vUU, vUC, vGA, vGG, vCU, and vCC, which lack the ability to accomplish hybridization in any promoters. The reporter plasmids were individually co-transfected with the four plasmids pVP-PB2, -PB1, -PA, and -NP, that express the three viral polymerase subunits and NP [Citation19,Citation20]. At 48 h post-transfection, cell lysates were harvested to quantify the amounts of recombinant vRNA, cRNA, and mRNA by primer extension experiments. The result showed that the efficiency of RNA replication and transcription initiated from the vRNA templates varied depending on mutations (). Three mutants of Group I, vAU, vGC, and vCG, were amplified at similar levels to the wild-type vUA. Within Group II, RNA replication from the two mutant promoters, vAC and vCA, was more active, compared to the others, vUG and vGU. Statistically significant differences were found between vUG and vUA in the mRNA level and between vGU and vUA in the vRNA level. The Group III set exhibited unfavourable promoter activities overall. Notably, RNA replication and transcription were most significantly suppressed in vGA and vGG, indicating that the vRNA promoter activity is negatively regulated when X14′ is G. Expression of EGFP from the transfected live cells enabled visualization of the final products from cap-snatching-mediated mRNA transcripts (), being correlated with the mRNA levels in the primer extension experiments (). The data particularly from Group II suggested that base-pairing between X14′ and Y13 of the vRNA promoter has little relationship with its competence in RNA-dependent RNA synthesis.
Figure 2. RNA-dependent RNA replication initiated from the vRNA promoter in cells. (A) Primer extension for detecting vRNA(EGFP) [138 nucleotides (nt)] and its cRNA (162 nt) or mRNA (>162 nt) transcripts. At 24 h after transfection of 293 T cells with individual vX14′Y13 reporter constructs [pHH21-vRNA(EGFP) series] together with pVP-PB2, -PB1, -PA, and -NP, total RNA was purified from the cell lysates. Cellular 5S rRNA (100 nt) was used as an internal control. The primer extension products are indicated with arrows at the right side. The wild-type vUA sample was loaded in each panel (underlined). (B) Quantification of band intensities of (A) using a phosphorimage analyser. Values were normalized against 5S rRNA. Error bars represent standard error of the mean (SEM) from three independent experiments. Relative changes are given as percentages of the vUA sample (100%). Statistical analysis was performed by two-way ANOVA with Dunnett’s multiple comparison test against the control sample, vUA, in vRNA (black asterisks), mRNA (red asterisks) and cRNA (green asterisks) levels. *P < 0.05, **P < 0.01, and ***P < 0.001. (C) At 24 h post-transfection, EGFP expression was captured by fluorescent microscopy. Original magnification, 10 ×. (D) The fluorescent images were captured every 12 h for 48 h using a live cell imaging system. Fluorescent spots were counted 16 nonoverlapped areas per well and compared with the spot number from vUA at 48 h (100%). Error bars represent SEM from four independent experiments
![Figure 2. RNA-dependent RNA replication initiated from the vRNA promoter in cells. (A) Primer extension for detecting vRNA(EGFP) [138 nucleotides (nt)] and its cRNA (162 nt) or mRNA (>162 nt) transcripts. At 24 h after transfection of 293 T cells with individual vX14′Y13 reporter constructs [pHH21-vRNA(EGFP) series] together with pVP-PB2, -PB1, -PA, and -NP, total RNA was purified from the cell lysates. Cellular 5S rRNA (100 nt) was used as an internal control. The primer extension products are indicated with arrows at the right side. The wild-type vUA sample was loaded in each panel (underlined). (B) Quantification of band intensities of (A) using a phosphorimage analyser. Values were normalized against 5S rRNA. Error bars represent standard error of the mean (SEM) from three independent experiments. Relative changes are given as percentages of the vUA sample (100%). Statistical analysis was performed by two-way ANOVA with Dunnett’s multiple comparison test against the control sample, vUA, in vRNA (black asterisks), mRNA (red asterisks) and cRNA (green asterisks) levels. *P < 0.05, **P < 0.01, and ***P < 0.001. (C) At 24 h post-transfection, EGFP expression was captured by fluorescent microscopy. Original magnification, 10 ×. (D) The fluorescent images were captured every 12 h for 48 h using a live cell imaging system. Fluorescent spots were counted 16 nonoverlapped areas per well and compared with the spot number from vUA at 48 h (100%). Error bars represent SEM from four independent experiments](/cms/asset/782cf8e0-5eaa-45fa-8b48-460b954e687a/krnb_a_1864182_f0002_c.jpg)
In the vRNA promoter constructs, we could not define whether the vRNA level accounts for plasmid-derived transcripts mainly or replication products amplified by the viral polymerase complex. It was needed to verify the reliability of the result by using another de novo synthesis cassette. We prepared a series of cRNA promoters in a pairwise manner. The wild-type cRNA promoter was designed to encode the sense strand of the EGFP coding region flanked by the 5′ and 3′ UTRs of cRNA, transcribing cRNA(EGFP) from the RNA pol I promoter as a template for vRNA(EGFP) (, right column). Various mutants at the segment-specific pair U13′∙A14 were created and named cY′13′X′14, simply cYX. Using these constructs, viral RNA replication initiated from the cRNA promoter was analysed. As expected, primer extension experiments showed that RNA replication and transcription occurred efficiently in Group I, but marginally in Group III (, B). Consistent with the vRNA promoters (, B), RNA synthesis was more dramatically inhibited in cCA and cAC rather than in cGU and cUG within Group II. The mRNA expression was visualized again by monitoring reporter gene expression, confirming the reliability of the primer extension (, D). Given the relationship between the promoter activity and complementarity, these results suggested that hybridization between the first M segment-specific non-coding nucleotides is more critical in the cRNA promoter rather than the vRNA promoter for RNA-dependent RNA replication.
Figure 3. RNA replication initiated from the cRNA promoter in cells. (A) Primer extension for detecting cRNA(EGFP) [162 nucleotides (nt)] and its vRNA (138 nt) or mRNA (>162 nt) transcripts. HEK 293 T cell were transfected as mentioned in but by substitution with individual cY′13′X′14 reporter constructs [pHH21-cRNA(EGFP) series]. The primer extension products are indicated with arrows at the right side of the gels. The wild-type cUA sample was loaded in each panel (underlined). (B) Quantification of band intensities of (A). Values were normalized against 5S rRNA. Error bars represent SEM from three independent experiments. Relative fold changes are given as percentages of the cUA sample (100%). Statistical analysis was performed by two-way ANOVA with Dunnett’s multiple comparison test against the control sample, cUA, in vRNA (black asterisks), mRNA (red asterisks) and cRNA (green asterisks) levels. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (C) At 24 h post-transfection, EGFP expression was captured by fluorescent microscopy. Original magnification, 10 ×. (D) Fluorescent spots were counted from 16 nonoverlapped areas per well every 12 h and compared with the spot number from cUA at 48 h (100%). Error bars represent SEM from four independent experiments
![Figure 3. RNA replication initiated from the cRNA promoter in cells. (A) Primer extension for detecting cRNA(EGFP) [162 nucleotides (nt)] and its vRNA (138 nt) or mRNA (>162 nt) transcripts. HEK 293 T cell were transfected as mentioned in Fig. 2 but by substitution with individual cY′13′X′14 reporter constructs [pHH21-cRNA(EGFP) series]. The primer extension products are indicated with arrows at the right side of the gels. The wild-type cUA sample was loaded in each panel (underlined). (B) Quantification of band intensities of (A). Values were normalized against 5S rRNA. Error bars represent SEM from three independent experiments. Relative fold changes are given as percentages of the cUA sample (100%). Statistical analysis was performed by two-way ANOVA with Dunnett’s multiple comparison test against the control sample, cUA, in vRNA (black asterisks), mRNA (red asterisks) and cRNA (green asterisks) levels. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (C) At 24 h post-transfection, EGFP expression was captured by fluorescent microscopy. Original magnification, 10 ×. (D) Fluorescent spots were counted from 16 nonoverlapped areas per well every 12 h and compared with the spot number from cUA at 48 h (100%). Error bars represent SEM from four independent experiments](/cms/asset/7ffab22f-86ae-4e99-aa80-4a68ffefd3f3/krnb_a_1864182_f0003_c.jpg)
Hybridization between the segment-specific nucleotides is essential for efficient initiation of unprimed vRNA synthesis from the cRNA promoter
It was intriguing that the replication pair, vAC and cGU, within Group II displayed promoter activity as potently as their wild-type ones, in contrast to the other pair, vGU and cAC (, B and 3A, B). We hypothesized that distinct from the vRNA promoter, the cRNA promoter could be more sensitive to hybridization of the two nucleotides. To explore this in vitro, viral polymerase complex was purified (Fig. S1A) according to previous reports [Citation21,Citation22] and the vRNA and cRNA promoters of 35 nucleotides in length were chemically synthesized, named vXY(s) or cYX(s) (Fig. S1B and ). They were subjected to both unprimed de novo and primed synthesis. Under the unprimed condition, cRNA synthesis by RdRp revealed increased or similar accumulation of the dinucleotide AG in the presence of the mutant vRNA promoters, vAC(s) and vGU(s) (). However, initiation of vRNA synthesis decreased in cAC(s) with a statistical significance, but not in cGU(s), compared to cUA(s) (). When in vitro RNA synthesis was initiated with the AG primer, no inhibition of the trinucleotide RNA products was observed in any of the vRNA and cRNA promoters (–H). This data informed that reduced intracellular RNA replication between the complementary templates, vGU and cAC, as observed in , is mainly responsible for a defect at the dinucleotide AG synthesis step from the cAC promoter.
Figure 4. Effects of hybridization between the segment-specific nucleotides at the vRNA or cRNA promoters on the unprimed or dinucleotide AG-primed RNA synthesis. Dinucleotide pppApG synthesis on the vRNA promoters (A, B) or on the cRNA promoter (C, D). De novo synthesis of pppApG (red in A or black in C) on the 5′-hooked synthetic vRNA promoter (black in A) and the cRNA promoter (red in C) is schematized. Each mutation site is highlighted in a box. Synthetic promoters were subjected to the RdRp reaction as templates in the presence of ATP and [α-32P]GTP (ppp*G). vUA(S) or cUA(s) without RdRp [(-)RdRp] was loaded as a negative control. (B, D) Quantification as percentage of isotope intensity from pppApG normalized to the input RNA amount. Extension of a 5′-radiolabeled pApG with CTP on the vRNA promoters (E, F) and the cRNA promoters (G, H). Primer-dependent cRNA synthesis (red in E) from the vRNA promoters (black in E) and primer-dependent vRNA synthesis (black in G) from the cRNA promoters (red in G) are schematized. The template RNAs were individually incubated with the radio-labelled dinucleotide AG primer (*pApG), and RdRp in the presence of CTP (E) or UTP (G). vUA(s) or cUA(s) without RdRp [(-)RdRp] was used as a negative control. The dinucleotide primer and the trinucleotide product are marked on the right of the denaturing gels. Quantification as percentage of isotope intensity from trimer pApGpC (F) or pApGpU (H). In B, D, F and H, data are representative of three independent experiments. Error bars represent SEM. *P < 0.05, ***P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. n.s., non-significant
![Figure 4. Effects of hybridization between the segment-specific nucleotides at the vRNA or cRNA promoters on the unprimed or dinucleotide AG-primed RNA synthesis. Dinucleotide pppApG synthesis on the vRNA promoters (A, B) or on the cRNA promoter (C, D). De novo synthesis of pppApG (red in A or black in C) on the 5′-hooked synthetic vRNA promoter (black in A) and the cRNA promoter (red in C) is schematized. Each mutation site is highlighted in a box. Synthetic promoters were subjected to the RdRp reaction as templates in the presence of ATP and [α-32P]GTP (ppp*G). vUA(S) or cUA(s) without RdRp [(-)RdRp] was loaded as a negative control. (B, D) Quantification as percentage of isotope intensity from pppApG normalized to the input RNA amount. Extension of a 5′-radiolabeled pApG with CTP on the vRNA promoters (E, F) and the cRNA promoters (G, H). Primer-dependent cRNA synthesis (red in E) from the vRNA promoters (black in E) and primer-dependent vRNA synthesis (black in G) from the cRNA promoters (red in G) are schematized. The template RNAs were individually incubated with the radio-labelled dinucleotide AG primer (*pApG), and RdRp in the presence of CTP (E) or UTP (G). vUA(s) or cUA(s) without RdRp [(-)RdRp] was used as a negative control. The dinucleotide primer and the trinucleotide product are marked on the right of the denaturing gels. Quantification as percentage of isotope intensity from trimer pApGpC (F) or pApGpU (H). In B, D, F and H, data are representative of three independent experiments. Error bars represent SEM. *P < 0.05, ***P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. n.s., non-significant](/cms/asset/cfedf40c-ae46-4d52-b918-aba2303f2eb8/krnb_a_1864182_f0004_c.jpg)
The first segment-specific nucleotides determine a rescue of infectious virus
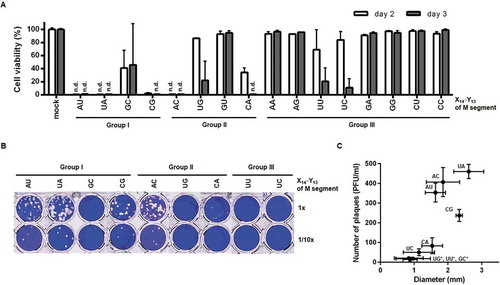
Next, we introduced the eight-plasmid-based reverse genetics technique to investigate whether these mutations affect the generation of infectious virus or protein expression[Citation23]. The Influenza A/Puerto Rico/8/34 (PR8) virus-derived M genome was subjected to site-directed mutagenesis at X14′·Y13 to generate 15 modified plasmids, named pVP-M(X14′Y13) or M(XY). Viral yield and infectivity were assessed by inoculating Madin-Darby canine kidney (MDCK) cells with culture supernatants from the transfected human embryonic kidney (HEK) 293 T cells (designated passage 0 or P0). Cell viability assays revealed that in Groups I and II, M(AU), M(CG), and M(AC) led to complete cytopathic effect (CPE) to the same extent as the wild-type segment, M(UA), on days 2 and 3 (). With reduced infectivity, M(GC), M(UG), M(GU), and M(CA) mutants resulted in partial or little cytopathogenesis. In Group III, CPE was induced marginally with a reduction of cell viability by less than 10% except for M(UU) and M(UC), suggesting that U14′ of the M segment 5′ UTR is a preferential element for maintaining viral infectivity. In parallel, plaque titration was performed by infection with the nine CPE-inducible P0 stocks (). The three main mutants, M(AU), M(CG), and M(AC), exhibited obvious viral plaques with the number ranging from 236.7 to 406.7 plaque forming units (PFU)/ml and sizes of 1.63 to 2.34 mm in diameter. Those were similar to the M(UA) virus with plaques of 460.0 ± 36.1 PFU/ml and sizes of 2.62 ± 0.45 mm in diameter. Two other mutants, M(CA) and M(UC), formed viral plaques (80.3 and 50.0 PFU/ml, respectively), but far less productively than the four dominant rescued isolates. Plaque sizes from M(GC), M(UG), and M(UU) were too small to be counted with naked eyes (13.3 to 20.0 PFU/ml and 0.84 to 0.98 mm in diameter under a microscope). Both CPE and plaque assays suggested that M(AU), M(CG), and M(AC) are able to sustain the entire viral life cycle as the wild-type M(UA).
Figure 5. Rescue of recombinant viruses by eight-plasmid-based reverse genetics. (A) CPE assay to compare virus rescue efficiency with variable pVP-M series. HEK293T cells were transfected with the eight pVP plasmids for reverse genetics by substituting pVP-M(UA) with one of other 15 mutant plasmids listed at the bottom of the graph. On day 2 after transfection, 2-fold diluted culture supernatants of P0 were loaded onto MDCK cells. On days 2 and 3 post-infection, cell viability (%) was calculated by treatment with MTT, in which the mock-transfected cells were used as a control (100%). n.d., not detected. Error bars represent SEM of four independent experiments. mock, inoculated with culture supernatants after empty-vector transfection. (B) Plaque titration. The culture supernatants of P0 which led to CPE in (A) were infected into MDCK cells in 48-well plates (upper, undiluted; lower, 10-fold diluted). On day 3 post-infection, plaques were visualized by crystal violet staining. (C) Plaque numbers (y-axis) and sizes (x-axis) from four independent experiments of (B) are represented as means ± SEM. Mutations at X14′·Y13 of the M segment are described on the xy-coordinate plane. *, Plaque numbers and sizes were determined using a microscope when they were too small to measure with the naked eye
For multicycle growth curve analysis at the same MOI, recombinant viruses (P0) were amplified by infection of MDCK cells. On day 2 post-infection, titre of the resultant viral stocks (P1) was determined by plaque assay (). The result exhibited that the M(UG) virus has the lowest titre, thus not being available for further investigations on a common condition. We wondered whether the mutations affect expressions of M segment-derived proteins, M1 and M2. MDCK cells were infected with individual viruses (P1) at the same multiplicity of infection (MOI) of 10−4. Western blot analysis presented that both proteins were strongly expressed from the four dominant viruses of M(AU), M(UA), M(CG), and M(AC), with the greatest abundance in M(AC), on day 1 (, left). NP levels were relatively consistent, with an exception in M(CA) that showed little NP. On day 2, M1 and M2 levels were enhanced in most samples, but still not in M(CA) (, right). Subsequently, we analysed the growth rates of the wild-type virus, M(UA), and the seven infectious mutants of P1 by infecting MDCK cells at an equal MOI of 10−4 (–E). Plaque titration with culture supernatants harvested at 12-h intervals revealed that the main mutant viruses, including M(AU) and M(CG) of Group I together with M(AC) of Group II, replicated as rapidly as M(UA), while the remaining mutants, M(GC), M(CA), M(UU), and M(UC), grew slowly, resulting in 2-log titre reduction at most at 60 h post-infection. Taken together, these results show that the first segment-specific nucleotides ultimately influence the rescue and growth of infectious viruses. As with the effect on RNA replication, the maintenance of the base pair between first segment-specific nucleotides on the cRNA is vital for viral growth. In addition, the facts that both M(GC) and M(AC) have ability to achieve base paring at the cRNA promoter but grow relatively slow indicate a potential preference to specific nucleotide sequences to maintain viral infectivity.
Figure 6. Effect of the X14′·Y13 mutation on the viral protein expression and growth. (A) Plaque titration of the amplified recombinant viruses at P1. An equal volume of P0 supernatants was inoculated in MDCK cells (100 µl per well in 12-well plates) for 2 days. The number of viral particles of P1 was quantified by plaque assay and expressed as means ± SEM (log10 PFU/ml) (n = 3). (B) Western blot analysis for detecting M1 and M2 expression. MDCK cells were infected with the different recombinant viruses (P1) at an MOI of 10−4. Cell lysates harvested on days 1 (left) and 2 (right) were subjected to SDS-PAGE for immunoblotting. Viral proteins M1, M2, and NP were detected with their specific primary antibodies and subsequently HRP-conjugated secondary antibodies. Cellular β-actin was used as a loading control. Proteins are indicated on the right side of the gels. (C-E) Viral growth curve of reverse-genetically rescued recombinant viruses. Wild-type and mutant viruses of Groups I (C), II (D), and III (E) of P1 were treated in MDCK cells at an MOI of 10−4 at 35°C. Culture supernatants were harvested every 12 h for 60 h for plaque titration (log10 PFU/ml). The graphs show means ± SEM (n = 4). In all infection experiments, influenza viruses were amplified in the presence of 2 µg/ml TPCK-trypsin
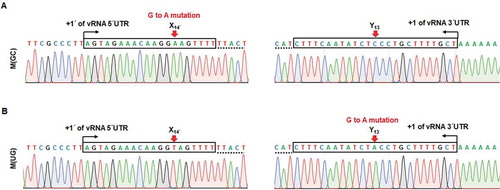
Recombinant viruses, M(GC) and M(UG), undergo G to A mutation at the target sites
To examine the genetic stability of the non-coding nucleotides, we investigated whether additional or reverse mutations are created during viral propagation. A series of successive generations was acquired by infecting wild-type or each mutant virus of P1 into MDCK cells at an MOI of 10−4, except for the slowest-growing virus, M(UG), which was used at a 5 × 10−6 MOI due to its limited amount (). At 48 h post-infection, cell culture supernatants were harvested for sequence analysis. Unexpectedly, plaque titration of the resulting passage 2 (P2) exhibited that all recombinant viruses were enriched at high titres over 106 PFU/ml (Fig. S2). Most prominently, the plaque titre of M(UG) that was steadily grown at P1 () suddenly increased, being comparable to the wild-type titre: 4.0 × 107 PFU/ml of M(UG) versus 5.2 × 107 PFU/ml of M(UA). Using the purified viral RNAs from P2, rapid amplification of cDNA ends (RACE) and sequence analysis proceeded. The result displayed G to A mutations at the target sites of both M(GC) and M(UG) (), but no mutations in other recombinant viruses (Fig. S3A). Quantitatively, based on the results from the analysis of individual 10 clones, 90% of M(GC)-derived clones and 100% of M(UG) clones were changed to M(AC) and the wild-type M(UA), respectively. To follow when the reverse mutations were created, we analysed again their 5′ and 3′ UTR nucleotides with a pool of the earlier passage, P1 (). We found no mutation in M(GC) of P1 (Fig. S3B). Unfortunately, it was not available for M(UG) of P1 because its vRNA amount was not enough for RACE. The data indicated that the mutant viruses, M(GC) and M(UG), are genetically unfavourable and evolved to acquire non-coding nucleotides optimized for continuous viral infection by using the error-prone genome replication machinery. However, it remains to be elucidated whether these mutations were mediated intrinsically by G to A substitution at the vRNA promoter or indirectly by C to U substitution at its cRNA promoter.
Figure 7. Genetic preference and evolution of the M segment-specific non-coding nucleotides. Identification of acquired mutations within the M genome segment of recombinant viruses, M(GC) (A) and M(UG) (B), of P2. Terminal sequences of vRNA were determined by 5′ (left) and 3′ RACE (right). Both 5′ and 3′ UTR regions of the M segments are marked in boxes with terminal ends indicated by black arrows. Coding sequences are underlined with dotted lines. Red arrows point to the target sequences, X14′ and Y13. The sites with ‘G to A mutation’ are denoted at the top of the sequencing chromatograms
Discussion
In the present study, we investigated the regulatory function of the influenza A viral M segment-specific non-coding sequences by focusing on the first base pair directly adjacent to the universal non-coding domain (). Through systematic mutation of the target sites, we discovered that RNA-dependent RNA replication between vRNA and cRNA relies on the hybridization of the segment-specific nucleotides, Y′13′ and X′14, of the cRNA promoter, rather than their counterpart nucleotides of the vRNA promoter (). Confirmatively, in vitro activity assay with synthetic RNA promoters proved that complementarity between Y′13′ and X′14 of the cRNA promoter is required for initiation of unprimed vRNA synthesis (). It is predicted that this breakage could render the G11 and G12 nucleotides at 5′ UTR of the cRNA promoter to participate in the temporary or non-fixed hybridization with any nucleotides of the pyridine-rich template of the 3′ UTR extremity (). The structural intolerability of the cRNA promoter might be associated with the length of its proximal stem, which is one base-pair shorter than that of the vRNA promoter. Thus, it is not difficult to deduce that its wobble state could hamper the initiation step of vRNA synthesis at the correct site, the 4th nucleotide U, from the 3′ UTR terminus as demonstrated in the internal initiation and realignment model[Citation10].
It is interesting that the mutant virus M(AC) exhibited the desirable properties in all comparison assays, including RNA replication from vRNA to cRNA and vice versa, virus rescue or protein expression, multicycle growth, and genetic stability. The primary finding to be addressed is about the increased expression of M1 and M2 at an early time of infection (). It could be attributed to the modification of viral mRNA splicing machinery. It has been reported that influenza A viral M genome has four different mRNA transcripts, such as unspliced M1 mRNA transcript and spliced M2 mRNA, mRNA3, and mRNA4 transcripts[Citation24]. Particularly, mRNA3 is produced by splicing from the 5′ splicing donor site of mRNA (the 11th nucleotide) to the 3′ splicing acceptor (the 740th nucleotide). Within the highly conserved splicing donor nucleotides, G12 and U13, the 13th intronic nucleotide U corresponds to one of our target nucleotides, Y′13′ of the cRNA promoter or Y13 of the vRNA promoter (). Accordingly, it is expected that during M(AC) virus replication, A13 C mutation at the 3′ UTR of the vRNA promoter is transcribed to create the M1 mRNA transcript with U13G mutation, accumulating the unspliced precursor M1 mRNA and the alternatively spliced transcript, M2 mRNA.
Considering the sustainable growth of dominant mutant viruses including M(AU), M(CG), and M(AC) (), we wondered whether influenza A viral isolates with any of these mutations are existing in nature and retrieved full genome sequences of the M segment from the NCBI GenBank database. After sequence alignment analysis, we couldn’t find any isolates to possess the double substitutions in the M segment. Regardless of dominance in M1 and M2 expression level in the M(AC)-infected cells (), influenza virus seems to evolve to another way by choosing segment-specific non-coding sequences ultimately to maintain a balanced ratio among the eight different segments and harmonized functions of the viral proteins.
To the best of our knowledge, this is the first report to show that hybridization, either canonical or non-canonical, between the segment-specific non-coding nucleotides plays a pivotal role within the cRNA promoter for de novo synthesis of vRNA, eventually being responsible for sustainable viral growth (). It has been identified that all influenza A viral genome segments have their own specific nucleotides at X14′ and Y13: the PB2, PA and M segments have a U-A pair; the HA, NP and NS segments with a G-C pair; the PB1 segment with a C-G pair; and the NA segment with any of possible pairs depending on the viral subtype[Citation18]. To generalize our finding, we are going to extend this research to another influenza A viral segment that has a discriminative pair from the U-A pair. In summary, the present study through systematic mutational analysis and reverse genetics provides a fundamental understanding of the roles of the M segment-specific nucleotides X14′ and Y13 during the entire viral life cycle.
Materials and methods
Cells and viruses
MDCK and HEK 293 T cells were purchased from the American Type Culture Collection (ATCC). Cells were grown in minimum essential medium (MEM; Invitrogen) or Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen), respectively, supplemented with 10% foetal bovine serum (FBS; Invitrogen). Influenza A virus strain PR8 (H1N1) was purchased from ATCC and amplified in 10-day-old chicken embryos for 3 days at 37°C. Recombinant PR8 and its mutants rescued by reverse genetics were inoculated in MDCK cells in the presence of 2 µg/ml TPCK-trypsin (Sigma Aldrich) at 35°C[Citation25].
Plasmids
To establish the systems for reverse genetics and viral polymerase assay, full-length viral genomes of PR8 were individually amplified from vRNAs using universal and segment-specific primers [Citation25,Citation26]. PCR products were cloned between human RNA Pol I and cytomegalovirus RNA Pol II promoters which are oppositely located within the pVP vector, generating pVP-PB2 (Genbank accession no. AB671295), -PB1 (accession no. KC866596.1), -PA (accession no. KC866595.1), -HA (accession no. AB671289.1), -NP (accession no. KC866598.1), -NA (accession no. CY033579.1), -M [or -M(UA); accession no. EF467824.1], and -NS (accession no. EF467817) as described previously [Citation23,Citation27]. The plasmid pHH21-vRNA(EGFP) was prepared by cloning the negative-sense EGFP coding region flanked by the 5′ and 3′ UTRs of the M vRNA under the Pol I promoter [Citation28,Citation29]. In parallel, the plasmid pHH21-cRNA(EGFP) was obtained by cloning its inverted insert at the BsmBI site in the mock vector, pHH21. The primers for amplifying the inserts, vRNA(EGFP) (named vUA) and cRNA(EGFP) (named cUA), by PCR are listed in Table S1. A series of mutants was prepared by site-directed mutagenesis by targeting X14′·Y13 of pHH21-vRNA(EGFP) or pVP-M or targeting Y′13′·X′14 of pHH21-cRNA(EGFP) using the GeneArt Site-Directed Mutagenesis System (Thermo Fisher Scientific). The primer sets used are summarized in Table S2. After mutagenesis, all plasmids were identified by sequencing analysis using EGFP- or M-specific primers. DNA concentration was determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific) and by densitometric analysis of an agarose gel. Plasmids expressing viral polymerase complex, pcDNA3-PB2-TAB, pcDNA3-PB1 and pcDNA3-PA, derived from influenza A/WSN/33 virus, were kindly provided by Prof. Ervin Fodor (Oxford University, UK).
Polymerase activity assay in cells
The polymerase activity assay was performed according to our previous report with some modifications[Citation27]. In brief, 293 T cells seeded onto 12-well plates (5 × 105 cells per well) were transfected with pVP-PB2, -PB1, -PA, and -NP, together with any of the pHH21-vRNA(EGFP) or -cRNA(EGFP) constructs (each 0.25 µg) using Lipofectamine 2000 (Invitrogen). The next day, total RNA was extracted for primer extension experiments using Trizol (Invitrogen). At six hours post-transfection, the plates were loaded into a live cell imaging system (IncuCyte FLR; Essen BioScience) to capture fluorescent images every 12 h for 48 h. Fluorescent objects from 16 dissected areas per well were counted using the built-in software.
Primer extension
The 5′-ends of DNA primers hybridizing the negative-sense EGFP coding region [5′-CCTGAGCACCCAGTCCGC-3′; for detecting the vRNA(EGFP)], its positive sense [5′-TTCAGGGTCAGCTTGCCG-3′; for detecting the cRNA(EGFP) and mRNA transcripts], or cellular 5S rRNA (5′-TCCCAGGCGGTCTCCCATCC-3′) as a loading control were labelled with [γ-32P]-ATP (PerkinElmer)[Citation30]. The mixture of labelled primer and RNA extracts was heated at 95°C for 3 min followed by rapid cooling at 4°C for 10 min and stabilization at 55°C for 1 min. Reverse transcription reaction was performed at 55°C using the SuperScript III reverse transcriptase kit (Invitrogen). After 1 h, it was stopped by addition of an equal volume of 2× RNA loading dye (Invitrogen). The samples were separated on 6% polyacrylamide gel with 7 M urea. The images were scanned using an FLA3000 image analyser (Fujifilm). Band intensities were quantified using ImageJ-Fuji Software.
In vitro dinucleotide synthesis and pApG extension assays
Purification of influenza virus polymerase was performed by transfection of 293 T cells with pcDNA3-PB2-TAP, pcDNA3-PB1 and pcDNA3-PA according to previous reports [Citation21,Citation22]. For analysing dinucleotide pppApG synthesis, 0.5 µM of each synthetic vRNA or cRNA promoter was incubated with 10 ng of the purified polymerase complex in a 4 µl reaction mixture containing 0.05 µM [α-32P]GTP (3,000 Ci/mmol; Perkin-Elmer), 1 mM ATP, 2 U/µl RNase inhibitor (Enzynomics), 5 mM MgCl2 and 1 mM DTT at 30°C for 12 h[Citation31]. The samples were heated at 95°C for 3 min and treated with 1 U of shrimp alkaline phosphatase (SAP) (New England BioLabs) for 1 h at 37°C. The reaction was stopped by addition of 5 µl of 2× RNA loading dye (Invitrogen) and analysed on an 8 M urea/20% denaturing PAGE gel.
For in vitro pApG-primed synthesis assay, the dinucleotide primer was prepared by labelling of 1 µM ApG (Jena Bioscience) with 0.5 μM [γ-32P]ATP (6,000 Ci/mmole, Perkin-Elmer) and 1 U/µl T4 polynucleotide kinase (T4 PNK) (Enzynomics) in 10 μl 1× T4 PNK reaction buffer (Enzynomics) at 37°C for 1 h[Citation31]. The RdRp assay was performed as mentioned above but with some key modifications in which ATP and [α-32P]GTP were replaced with radiolabeled pApG and 1 mM CTP (for cRNA synthesis) or UTP (for vRNA synthesis) without SAP treatment.
Reverse genetics and virus amplification
For rescue of PR8 virus, 293 T cells were seeded on 6-well plates (1 × 106 cells per well) and, on the next day, transfected with the eight pVP plasmids (each 0.5 µg)[Citation23]. For mutant virus rescue, the plasmid pVP-M(UA) among the eight plasmids mixture was substituted with one of the 15 mutant plasmids, pVP-M(X14′Y13). On day 2 after transfection, the culture supernatants (passage 0 or P0; 300 µl) were loaded into MDCK cells in 6-well plates at 35°C for 1 h. Unabsorbed virus was removed by washing with PBS, followed by the addition of FBS-free MEM with 2 µg/ml TPCK-treated trypsin. On day 2 post-infection, viral titres of the MDCK inoculum (P1) were determined by plaque assay. Fresh MDCK cells were infected with the P1 viruses individually at an MOI of 10−4 [but at an MOI of 5 × 10−6 for the mutant virus of M(GU) due to its limited titre] for 2 days to produce the P2 recombinant viruses.
Virus titration
For the CPE assay, MDCK cells (5 × 104 cells per well) seeded in 96-well plates were allowed to reach 100% confluency. They were then incubated with 50 µl of 2-fold diluted P0 supernatants for 1 h. After removing unabsorbed virus, the cells were incubated at 35°C for 2 or 3 days. Cell viability was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide assay (MTT; Sigma Aldrich) as described previously[Citation32]. For plaque assay, confluently grown MDCK cells in 48-well plates were infected with 100 µl of 10-fold serially diluted viruses at 35°C for 1 h. After washing with PBS, cells were grown in FBS-free MEM with 2 µg/ml TPCK-trypsin and 0.6% CMC (Sigma Aldrich) for 3 days[Citation25]. Plaques were visualized by crystal violet staining to count their numbers (plaque forming units per ml, PFU/ml) and sizes. In the case of mutant viruses producing too small-sized plaques, they were measured using a microscope.
Western blot analysis
Cell lysates collected on day 1 or 2 after virus infection were loaded onto 10% (for NP and β-actin) or 15% (for M1 and M2) SDS-PAGE (40 µg per well). After electrotransfer to PVDF membrane, viral proteins were probed with anti-M1 (cat. no. sc-57,881; Santa Cruz Biotechnology), -M2 (cat. no. sc-32,238; Santa Cruz Biotechnology), or -NP (cat. no. 11,675-RP02; Sino Biological) antibodies and then HRP-conjugated anti-mouse (for M1 and M2; Thermo Fisher Scientific) or -rabbit IgG (for NP; Thermo Fisher Scientific). β-Actin was used as a loading control using anti-β-actin antibody (cat. no. A1978; Sigma Aldrich) and HRP-conjugated anti-mouse secondary antibody.
RACE
To identify the 5′ terminal sequence of viral RNAs, template switch-based 5′ RACE was performed [Citation33,Citation34]. Influenza viral RNA was extracted from culture supernatants using QIAamp Viral RNA mini kit (Qiagen). 5′ Ends of extracted RNAs (15 µl in a 20 µl reaction volume) were capped with 7-methylguanylate using the Vaccinia Capping System (New England Biolabs). All primer sequences used for RACE experiments are listed in Table S3. Capped RNAs (5.5 µl in a 10 µl reaction volume) were reverse-transcribed with the M5RACE-out(fwd) primer using the SuperScript III kit (Invitrogen). The template switching reaction was initiated by addition of an equal volume of 1 nM template switching oligonucleotide (TSO) dissolved in the RT reaction buffer with 0.1% BSA and 4 mM MnCl2. Then, nested PCR was performed to amplify the 5′ terminal sequences: outer primer set, M5RACE-out(fwd) and TSO out; inner primer set, M5RACE-in(fwd) and M5RACE-in(rev). With the same RNA extracts, the 3′ RACE experiment was performed as previously described[Citation34]. Poly(A) tail was added to the vRNAs with E. coli Poly(A) Polymerase (1 U) in 10 µl of E-PAP buffer (Thermo Fisher Scientific) at 37°C for 1 h. The RT reaction was performed by incubation of tailed RNAs (5.5 µl) and 5 pmoles of the M3RACE dT primer using the SuperScript III kit (Invitrogen). The 3′ terminal sequences were determined by nested PCR: the first round PCR primers were M3RACE-out(fwd) and M3RACE(rev); the second round PCR primers were M3RACE-in(fwd) and M3RACE(rev). The amplified products were purified from an agarose gel using MEGA quick-spin Plus Total Fragment DNA Purification Kit (iNtRON Biotechnology). They were cloned into the T-Blunt vector (SolGent) for subsequent sequencing analysis using a conventional T7 primer.
Statistical analysis
Data were analysed with Prism 8 Software (GraphPad Software). Unless otherwise stated, P values were calculated from three independent experiments. Statistical analysis was conducted using unpaired two-way ANOVA with a control group. If the P values were less than 0.05, they were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplemental Material
Download PDF (442.5 KB)Acknowledgments
We thank Prof. H.R. Lee and Prof. B.S. Choi for careful reading and constructive comments.
Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary materials
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Chu VC, Whittaker GR. Influenza virus entry and infection require host cell N-linked glycoprotein. Proc Natl Acad Sci U S A. 2004;101:18153–18158.
- Ozawa M, Fujii K, Muramoto Y, et al. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J Virol. 2007;81:30–41.
- Desselberger U, Racaniello VR, Zazra JJ, et al. The 3ʹ and 5ʹ-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene. 1980;8:315–328.
- Bae SH, Cheong HK, Lee JH, et al. Structural features of an influenza virus promoter and their implications for viral RNA synthesis. Proc Natl Acad Sci U S A. 2001;98:10602–10607.
- Flick R, Neumann G, Hoffmann E, et al. Promoter elements in the influenza vRNA terminal structure. RNA. 1996;2:1046–1057.
- Fodor E, Pritlove DC, Brownlee GG. Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J Virol. 1995;69:4012–4019.
- Pflug A, Guilligay D, Reich S, et al. Structure of influenza A polymerase bound to the viral RNA promoter. Nature. 2014;516:355–360.
- Fan H, Walker AP, Carrique L, et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature. 2019;573:287–290.
- Robb NC, Te Velthuis AJ, Wieneke R, et al. Single-molecule FRET reveals the pre-initiation and initiation conformations of influenza virus promoter RNA. Nucleic Acids Res. 2016;44:10304–10315.
- Deng T, Vreede FT, Brownlee GG. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J Virol. 2006;80:2337–2348.
- Honda A, Endo A, Mizumoto K, et al. Differential roles of viral RNA and cRNA in functional modulation of the influenza virus RNA polymerase. J Biol Chem. 2001;276:31179–31185.
- Zhang S, Wang J, Wang Q, et al. Internal initiation of influenza virus replication of viral RNA and complementary RNA in vitro. J Biol Chem. 2010;285:41194–41201.
- Zhao L, Peng Y, Zhou K, et al. New insights into the nonconserved noncoding region of the subtype-determinant hemagglutinin and neuraminidase segments of influenza A viruses. J Virol. 2014;88:11493–11503.
- Crow M, Deng T, Addley M, et al. Mutational analysis of the influenza virus cRNA promoter and identification of nucleotides critical for replication. J Virol. 2004;78:6263–6270.
- Kim HJ, Fodor E, Brownlee GG, et al. Mutational analysis of the RNA-fork model of the influenza A virus vRNA promoter in vivo. J Gen Virol. 1997;78(Pt 2):353–357.
- Benkaroun J, Robertson GJ, Whitney H, et al. Analysis of the variability in the non-coding regions of Influenza A viruses. Vet Sci. 2018;5(3):76.
- Robertson JS. 5ʹ and 3ʹ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res. 1979;6:3745–3757.
- Wang J, Li J, Zhao L, et al. Dual roles of the hemagglutinin segment-specific noncoding nucleotides in the extended duplex region of the Influenza A Virus RNA promoter. J Virol. 2017;91(1):e01931-16.
- Bussey KA, Bousse TL, Desmet EA, et al. PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. J Virol. 2010;84:4395–4406.
- Lutz A, Dyall J, Olivo PD, et al. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods. 2005;126:13–20.
- Fodor E, Crow M, Mingay LJ, et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol. 2002;76:8989–9001.
- Deng T, Sharps J, Fodor E, et al. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J Virol. 2005;79:8669–8674.
- Hoffmann E, Neumann G, Kawaoka Y, et al. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000;97:6108–6113.
- Wise HM, Hutchinson EC, Jagger BW, et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012;8:e1002998.
- Jang Y, Shin JS, Yoon YS, et al. Salinomycin inhibits influenza virus infection by disrupting endosomal acidification and viral matrix protein 2 function. J Virol. 2018;92(24):e01441-18.
- Hoffmann E, Stech J, Guan Y, et al. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289.
- Kim M, Kim SY, Lee HW, et al. Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antiviral Res. 2013;100:460–472.
- Fujii K, Fujii Y, Noda T, et al. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J Virol. 2005;79:3766–3774.
- Pleschka S, Jaskunas R, Engelhardt OG, et al. A plasmid-based reverse genetics system for influenza A virus. J Virol. 1996;70:4188–4192.
- Turrell L, Lyall JW, Tiley LS, et al. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat Commun. 2013;4:1591.
- Te Velthuis AJ, Robb NC, Kapanidis AN, et al. The role of the priming loop in Influenza A virus RNA synthesis. Nat Microbiol. 2016;1(5):16029.
- Jang YJ, Achary R, Lee HW, et al. Synthesis and anti-influenza virus activity of 4-oxo- or thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-ones. Antiviral Res. 2014;107:66–75.
- Liu F, Zheng K, Chen HC, et al. Capping-RACE: a simple, accurate, and sensitive 5ʹ RACE method for use in prokaryotes. Nucleic Acids Res. 2018;46:e129.
- Wang R, Xiao Y, Taubenberger JK. Rapid sequencing of influenza A virus vRNA, cRNA and mRNA non-coding regions. J Virol Methods. 2014;195:26–33.