
ABSTRACT
After infection by flaviviruses like Zika and West Nile virus, eukaryotic hosts employ the well-conserved endoribonuclease Xrn1 to degrade the viral genomic RNA. Within the 3ʹ untranslated regions, this enzyme encounters intricate Xrn1-resistant structures. This results in the accumulation of subgenomic flaviviral RNAs, an event that improves viral growth and aggravates viral pathogenicity. Xrn1-resistant RNAs have been established throughout the flaviviral genus, but not yet throughout the entire Flaviviridae family. In this work, we use previously determined characteristics of these structures to identify homologous sequences in many members of the genera pegivirus, hepacivirus and pestivirus. We used structural alignment and mutational analyses to establish that these sequences indeed represent Xrn1-resistant RNA and that they employ the general features of the flaviviral xrRNAs, consisting of a double pseudoknot formed by five base-paired regions stitched together by a crucial triple base interaction. Furthermore, we demonstrate that the pestivirus Bungowannah virus produces subgenomic RNA in vivo. Altogether, these results indicate that viruses make use of a universal Xrn1-resistant RNA throughout the Flaviviridae family.
Introduction
The genetically diverse Flaviviridae family encompasses a large and rapidly expanding selection of enveloped, positive sense RNA viruses. Their genomes are generally comprised of around 10 to 12.5 kilobases, containing a single open reading frame that encodes the viral polyprotein. Within the Flaviviridae family, viruses are subdivided into the four genera of flavi-, pegi-, hepaci- and pestiviruses [Citation1–3]. Of these, the flavivirus genus has received the most attention due to several member viruses, such as Dengue virus and Yellow fever virus, being globally distributed and responsible for recent outbreaks causing concerns for human health [Citation4–6]. However, the impact of viruses that belong to the other Flaviviridae genera pegi-, hepaci- and pestiviruses, cannot be understated. Hepaciviruses such as hepatitis C virus (HCV) are responsible for hepatitis and hepatocellular carcinoma in humans, while other hepaciviruses target monkeys, livestock and rodents [Citation7]. Pegiviruses like hepatitis GB virus A (GBV-A) and equine pegivirus 1 (EPgV) were originally classified as hepaciviruses, and are therefore closely related. However, pegiviruses generally differ in the fact that they do not appear to encode a core protein and often cause persistent infections without clinical symptoms [Citation8–10]. Originally, the genus pestivirus comprised four species of viruses pestivirus A through D that include bovine viral diarrhoea virus 1 (BVDV-1) and classical swine fever virus (CSFV). These viruses have a severe impact on the dairy and meat industry, as they infect mainly ruminants and pigs [Citation11–13]. However, recently proposed changes in taxonomy of the pestiviruses have expanded this genus to include the recently identified Bungowannah virus (BuPV) as a member of pestivirus F and atypical porcine pestivirus (APPV) as a member of pestivirus K [Citation3].
Members of the Flaviviridae family usually contain 5ʹ- and 3ʹ-untranslated regions (UTRs) that harbour a variety of highly structured elements. The 5ʹ UTR of flaviviruses are capped and generally make use of cap-dependent translation [Citation14–16], although recently cap-independent translation has been shown for Dengue and Zika virus as well [Citation17]. In contrast, pegi-, hepaci- and pestiviruses mostly control translation initiation through the use of internal ribosome entry site (IRES) elements [Citation18–21]. In the non-polyadenylated 3ʹ UTR, all Flaviviridae harbour conserved pseudoknot and stem–loop interactions [Citation22]. Flaviviruses have been demonstrated to employ an especially intricate structure within their 3ʹ UTR that enables them to resist degradation by Xrn1 after infection of a host. This well-conserved 5ʹ→3ʹ exoribunuclease is responsible in the host for cellular RNA homoeostasis through degradation of mono-phosphorylated RNA, and has been shown to colocalize with subgenomic flaviviral RNA (sfRNA) within cytoplasmic processing bodies after infection with Kunjin virus [Citation23]. As Xrn1 progresses through the viral genome, it will encounter the flaviviral Xrn1-resistant RNA (xrRNA), where it stalls. This leads to the accumulation of large amounts of sfRNA corresponding with the 3ʹ end of the genome. A current hypothesis for why the virus profits from this action, while losing their entire protein-coding genome, is that sfRNAs may function in sequestering of RNA-binding proteins involved in the RNA interference or interferon pathways, resulting in a delayed or impaired immune response [Citation24–27]. Furthermore, Xrn1 is sequestered or slowly released by the sfRNA and thus, cannot perform its canonical cellular function of dealing with aberrant RNA, causing dysregulation that may improve viral growth [Citation28,Citation29].
X-ray crystallography has elucidated the secondary and tertiary interactions required for stalling Xrn1 by RNA structures employed by Zika virus and Murray Valley Encephalitis virus [Citation30,Citation31]. This revealed the formation of intricate structural motifs, including five well-conserved stem-interactions, including a double pseudoknot and a base-triple interaction at a fixed location. Altogether, these interactions form a ring-like fold with a specific topology that disables progression of Xrn1 through the genomic RNA. Recently, we demonstrated that these xrRNA structures are present throughout the flavivirus genus, in which they employ the same interactions in order to stall Xrn1, regardless of variations in sequence and stem-lengths [Citation32,Citation33].
Until recently, research on xrRNAs within the Flaviviridae family has been focused mostly on the flavivirus genus. The hepacivirus HCV and pestivirus BVDV are notable exceptions, as structures in their 5ʹ UTRs have also been demonstrated to stall Xrn1 degradation in vitro [Citation28], and in the case of HCV in vivo, through the additional recruitment of miR-122 [Citation34]. Pegi-, hepaci- and pestiviruses do possess 3ʹ UTRs that harbour pseudoknot and stem-loop structures that appear to share structural homology to flaviviral xrRNA, and several of such structures have recently been demonstrated to be capable of stalling Xrn1 in vitro [Citation35]. In this study, we investigate the 3ʹ UTRs of pegi-, hepaci- and pestiviruses for additional, flavivirus-like Xrn1-resistant structures. Through structural alignment and in vitro Xrn1 degradation assays, we show that most of these viruses carry a structure equivalent to xrRNA found within flaviviruses and that these conserved structures are able to stall Xrn1. In addition, we demonstrate how one of these structures from the pestivirus BuPV, is able to stall Xrn1 in vivo as well. As such, it seems that throughout the Flaviviridae family, a universal structure is responsible for resisting host RNA degradation machinery.
Materials and methods
Identification of putative xrRNA motifs
3ʹ UTRs of pegi-, hepaci- and pestiviruses were retrieved from GenBank and fed to MFOLD [Citation36]. The output of MFOLD was manually checked for features that were previously found in other flaviviral xrRNA motifs. These features included, for example, the presence of tetraloop hairpins upstream of a GC-rich hairpin, representing γ and δ hairpins, respectively, and the presence of a CAGG or CAAGG sequence upstream of a putative γ hairpin and downstream of a G-rich sequence that could form a putative α stem with a complementary sequence directly downstream of the δ hairpin. Pseudoknots (β and ε interactions) were subsequently identified manually as well. Putative xrRNA motifs were placed in structural alignment manually, using these features as anchor points.
Design and production of DNA templates for in vitro RNA transcription
Flaviviridae xrRNA constructs within this study were amplified through oligonucleotide templates carrying reverse complementary sequences on the 3ʹ ends. These were purchased from Sigma Aldrich and Eurogentec in desalted form. Forward primers carried a T7 promoter sequence (GTAATACGACTCACTATA), followed by an AU-rich 12 nt leader sequence. A list of oligonucleotides is available on request. PCR and subsequent purification were performed as described in Dilweg et al. [Citation33].
In vitro Xrn1 degradation assay
RNA was produced in vitro using T7 RiboMAX™ Large Scale RNA Production System (Promega) as described in Dilweg et al. [Citation33]. The reaction mixture was treated with 1 unit RQ1 RNase-Free DNase, after which transcript concentration was checked on agarose gel. Per reaction, about 200 ng RNA was treated either with or without RppH and Xrn1, as described in Dilweg et al. [Citation33]. After the addition of an equal volume of denaturing loading buffer (8 M urea, 20 mM Tris-HCl, 20 mM EDTA, trace amounts of bromophenol blue and xylene cyanol FF), RNA was denatured for 5 min at 75 °C. These samples were run on 8 M urea 10% polyacrylamide gels in TBE buffer, equilibrated at 60–65 °C. Gels were stained with EtBr and each construct was subjected to this assay at least twice. Bands were quantified using the Quantity One 1-D analysis software.
Virus growth, isolation of intracellular RNA and analysis of sfRNA production
SK-6 cells were propagated in Minimum Essential Medium (MEM). Monolayers of SK-6 cells were infected with BuPV at m.o.i. 1.0. At 48 h post infection the medium was aspirated and discarded. Total intracellular RNA of infected and mock-infected cells was isolated using Trizol (Invitrogen) as described by the supplier. RNA was dissolved in H2O. Samples containing 7.5 μg of total RNA from infected and mock-infected cells were analysed for the production of BuPV sfRNA by agarose gel electrophoresis under denaturing conditions and northern blotting as described previously [Citation33]. 5ʹ 32P labelled oligonucleotide (Pesti004) that is complementary to positions 12,645 to 12,674 of the BuPV genome was used as a probe to detect viral xrRNA.
Results
Recent discoveries regarding the presence of xrRNA structures in the 3ʹ UTR of flaviviruses inspired our search for homologous motifs within all other genera of Flaviviridae. The previously elucidated characteristics of such structures [Citation31,Citation33] served as parameters which were used to search GenBank for similar structures in pegi-, hepaci- and pestiviruses. This yielded a large collection of sequences within each of these genera, of RNA sequence and/or structure fingerprints that strongly resembled the Xrn1 stalling sites of known flaviviral xrRNA sequences. Through structural alignment of these sequences, we modelled the presence of five putative stem interactions that correspond with stems α through ε, including double pseudoknots in a well-defined order ().
Figure 1. Structural alignment of putative xrRNA sequences found within pegi-, hepaci- and pestiviruses. For particular viruses, different strains or isolates were found carrying divergent motifs, which are shown here with corresponding accession numbers. Multiple sequences listed at a single isolate indicate tandem xrRNA motifs found within that 3ʹ UTR. The first nucleotide within the sequence corresponds with the given position downstream of the genome coding sequence. The proposed stem-interactions α, β, γ, δ and ε are colour-coded in blue, green, magenta, red and orange, respectively. Three grey background columns depict the nucleotides involved in the base triple interaction. Sequences that were tested in this study are tagged with a ‘*’
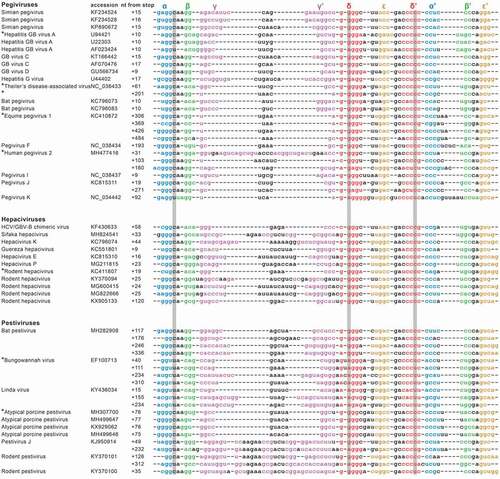
From the structural alignment, we can conclude that of all stem elements, stem γ appears the most variable, ranging from one base pair in EPgV, to 13 base pairs in human pegivirus 2 (HPgV-2). The other modelled interactions show smaller variations in stem lengths, within ranges that have previously been determined to be present in flaviviral xrRNA [Citation33]. While certain base positions within these sequences are highly conserved, covariation is prevalent throughout the five stems, which provides an evolutionary argument for their structural conservation. Previous work on the flaviviral xrRNAs has identified a crucial base triple interaction formed by the first nucleotide downstream of the 5ʹ side of stem α, together with the second base pair within stem δ [Citation30–33]. Our alignment shows that, similar to the flaviviruses, the potential for such a base triple interaction is well-conserved in pegi-, hepaci- and pestivirus. Either a C•G-C or isomorphic U•A-U base triple was predicted for all these viruses, except for hepatitis GB virus A, GB virus D, rodent hepacivirus isolates 05VZ and B349 and rodent pestivirus isolate RtAp-PestV/JL2014. These viruses can form a C•G-U base triple using nucleotides at equivalent positions. Such a base triple is also predicted to be formed in the flaviviral xrRNA motifs within the 3ʹ UTRs of Quang Binh, Ochlerotatus caspius, and Culex theileri flaviviruses (not shown). The Rodent pestivirus isolate RtNn-PestV/HuB2014 even appears to possess differing base triples between its two subsequent xrRNA motifs. Such covariations point towards the conservation and requirement of this interaction in the putative xrRNA motifs, similar to flaviviral xrRNAs.
In order to determine whether the predicted structures indeed represented Xrn1-resistant RNA, a selection of sequences from each genus was subjected to an in vitro Xrn1 degradation assay. These included the most upstream motifs of EPgV and human pegivirus 2 (HPgV-2), both motifs from Theiler’s disease associated virus (TDAV), the single motifs found in GBV-A, rodent hepacivirus (RHV), APPV and the most upstream motif of BuPV. For these selected viruses, we modelled the three-dimensional structure of their xrRNA motifs to better illustrate their predicted conformation and stem orientation (). The Xrn1 degradation assay involved an AU-rich leader sequence that was placed 5ʹ of the RNA corresponding with the sequences as depicted in . This sequence was predicted not to interfere with the formation of the tested xrRNA structures, and served as a landing spot for Xrn1, as it can only initiate degradation of RNA if the 5ʹ end remains unpaired. RNA derived from these constructs was treated with the pyrophosphatase RppH, subjected to Xrn1 and analysed on denaturing gels (). The bands of shortened RNA that remain after this treatment indicate that all candidate xrRNA motifs were capable of resisting Xrn1-mediated degradation in vitro.
Figure 2. (A) Representation of the proposed structure formed by the putative xrRNA sequences carrying stem-interactions α through ε, predicted by structural alignment of putative xrRNA sequences. The stem-colours match those used in . Nucleotides involved in the base triple interaction are given in bold. (B) In vitro Xrn1 degradation assays probing Xrn1-resistance of the constructs corresponding with the sequences as shown in (A). RNA was treated with or without Xrn1 and loaded on denaturing polyacrylamide gels. Data below the gels indicate the average percentage (± SD) of Xrn1-resistant RNA
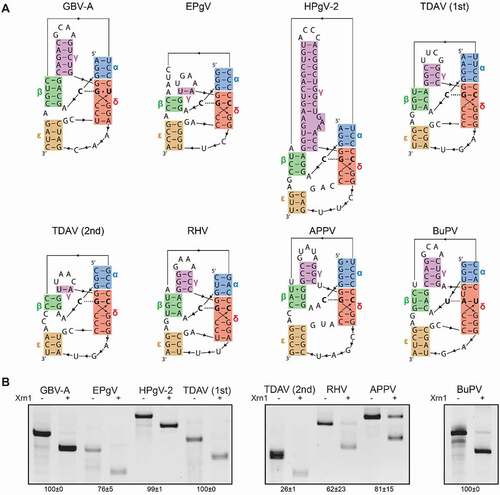
The pegi-, hepaci- and pestivirus xrRNA models all uncover the putative β stem interaction, long-range pseudoknot ε and base triple interaction, equivalent to those found to be crucial in previously tested flaviviruses [Citation30–33]. In order to supplement the phylogenetic evidence for the occurrence of such elements for at least one species within each genus, constructs were made carrying mutations that would impair these interactions (). These were subjected to in vitro Xrn1 degradation assays, together with constructs carrying complementary mutations that would putatively restore such interactions. Disruption of the pegiviruses GBV-A and EPgV β stem at the 5ʹ side lead to the complete loss Xrn1 resistance, which could be restored by compensatory changes at the 3ʹ side (). For the hepacivirus RHV, and the pestiviruses APPV and BuPV, this approach provided similar results. This pestivirus APPV wild-type construct has a relatively low Xrn1-stalling capacity, which is not completely lost by mutation of its 5ʹ side β stem. However, this construct harbours a uracil base between α and β, which may shift this interaction to form an alternative β stem made up of one U-G and one U-A base pair, potentially safeguarding some residual Xrn1-resistance. The BuPV-construct is able to compensate for the complete loss of Xrn1 resistance that occurs when its 5ʹ side of the β stem is mutated through complementary mutations at the 3ʹ side.
Figure 3. In vitro Xrn1 degradation assays probing Xrn1-resistance of (A) β stem mutants, (B) ε stem mutants and (C) triple base interaction mutants. A schematic depiction above each column shows the location of the respective element of interest within the xrRNA model. RNA was treated with or without Xrn1 and loaded on denaturing polyacrylamide gels. Construct mutations are shown above the corresponding lanes, in the same structural orientation as depicted in . Data below the gels indicate the average percentage (± SD) of Xrn1-resistant RNA
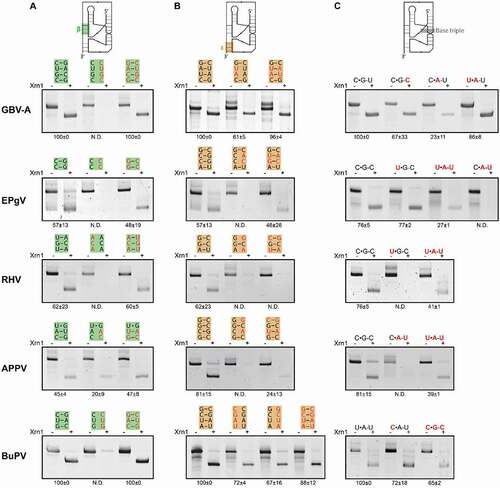
In order to further validate whether our xrRNA models are overall similar to previously determined flaviviral xrRNA structures, we tested whether the pseudoknot interaction ε is required for the xrRNA motifs to stall Xrn1-mediated degradation in vitro (). For GBV-A, we disrupted two base pairs through mutation of the 3ʹ side of the ε stem, which resulted in a partial loss of Xrn1 resistance, which was restored completely by complementary mutations at the 5ʹ side. The partial loss can likely be explained by the fact that shifting of the 5ʹ side nucleotides in the 3ʹ side mutant could yield a G-U and U-A base pair. Mutants disrupting and restoring the middle two base pairs of the EPgV ε interaction reveal that these changes more drastically disrupt and restore resistance towards Xrn1. For RHV, mutations at the 5ʹ side of the ε stem proved detrimental for Xrn1-resistance. However, we were not able to restore this interaction through complementary mutations at the 3ʹ side, which is likely due to the fact that the top G-C base pair was flipped. This pair is well conserved throughout the motifs modelled in this study, which suggests that this interaction performs a crucial structural function. Furthermore, we tested two mutants of the APPV xrRNA motif, changing two base pairs and subsequently restoring them. The disturbed ε stem yielded a construct unable to stall Xrn1, while restoring these base pairs restored Xrn1 resistance only slightly. Similar to the RHV restoration mutant, the fact that this restored mutant does not return to a wildtype level of resistance, is likely due to the flipping of the top G-C base pair. For the BuPV construct, we tested mutants from both sides of the ε stem, which reduced Xrn1 resistance only slightly. However, through shifting of the paired nucleotides, the 3ʹ mutant would still be able to pair C-G and G-C base pairs, while the 5ʹ mutant could still form a stem of three base pairs. As expected, the combination of these complementary mutations did not result in a level of Xrn1-resistance significantly different from that of the wildtype.
Modelling of the GBV-A xrRNA suggested that a non-canonical C•G-U triple may be formed in the position of the conserved base triple that has been demonstrated in flaviviruses to be essential for the formation of a functional Xrn1-resistant RNA motif. Mutating this triple to a C•G-C or U•A-U triple did not appear to disturb Xrn1 resistance suggesting that the model is correct (). However, mutation to A-U of the δ stem base pair involved in the triple base pair formation – yielding a more unstable C•A-U interaction – resulted in a strong reduction in the level of Xrn1 resistance for the motif. Within the EPgV motif, a mutation of its C•G-C base triple to an isomorphic U•A-U was not tolerated very well, while a U•G-C actually retained a level of RNA close to that of the wild type. Here again, employing a C•A-U triple proved detrimental for Xrn1 resistance. For RHV and APPV, Xrn1-resistance was lost entirely due to mutations of their modelled C•G-C triples to a U•G-C or C•A-U, respectively. These effects could both be rescued by compensatory mutations changing them into U•A-U triples. Finally, the BuPV xrRNA appears to undergo a slight reduction in Xrn1 resistance levels when its modelled U•A-U triple was changed into either a C•A-U or C•G-C. This is probably correlated with the presence of the unique U-bulge in the BuPV xrRNA, which may result in more rotational freedom allowing a nearby G-C pair to form the C•G-C triple.
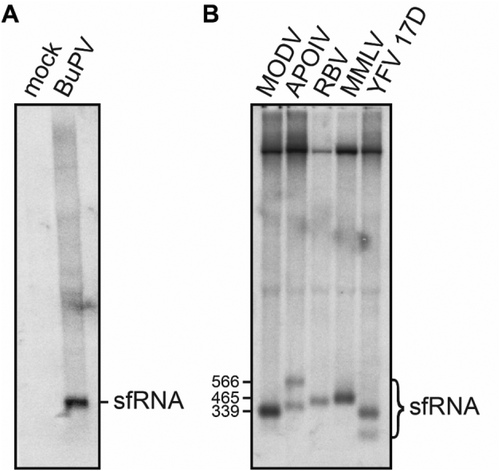
While the structural alignments and mutational analyses given above strongly indicate that pegi, hepaci- and pestiviral xrRNA motifs form a structure comparable to that of flaviviral xrRNA, this does not necessarily mean that these structures are able to stall Xrn1 in vivo and thus produce sfRNA. In order to obtain in vivo support for the functionality of these motifs, we sought to demonstrate formation of sfRNA in swine kidney cells infected by BuPV. A Northern blot analysis using a probe targeted to the 3ʹ end of this construct, revealed the presence of an intense band that we estimated to be roughly 450 nucleotides by comparison to a simultaneously produced Northern blot of YFV sfRNA and other flaviviral sfRNAs (). As the 5ʹ end of the most-upstream BuPV xrRNA motif is located 464 nucleotides from the 3ʹ end of the probe, this band likely represents subgenomic RNA that has been partially degraded by Xrn1, indicating that this BuPV xrRNA motif constitutes a genuine Xrn1-stalling site.
Figure 4. Northern blot analyses of total RNA isolated from (A) SK-6 cells after infection with BuPV, or (B) from BHK-21 J cells transfected with infectious cDNA clones carrying the 3ʹ UTRs of Modoc virus (MODV), Apoi virus (APOIV), Rio Bravo virus (RBV), Montana myotis leukoencephalitis virus (MMLV) or the Yellow fever virus (YFV) 17D variant. Size markers correspond in ascending order with the MODV, MMLV and larger APOIV sfRNA
Discussion
In previous work, we scrutinized xrRNA motifs within the tick-borne and no-known vector flavivirus clades, comparing them to mosquito-borne and insect-specific flaviviruses and concluded that within the flavivirus genus all the Xrn1 stalling sites share a single, similar structural organization [Citation33]. In this study, we extend this important observation to the entire Flaviviridae family, as we demonstrate that pegi-, hepaci- and pestiviruses contain equivalent xrRNA motifs which require the same interactions that characterize these Xrn1-resistant structures. In we depicted an overview of the common elements that we identified, and variability occurring within the Flaviviridae xrRNAs examined in this, and our previous work [Citation33]. Notable observations include the fact that stems α and δ are present in a range of sizes, but that they likely require a stacked conformation totalling 8–10 base pairs. Furthermore, the loop sequence linking stems α and β is variably present throughout the motifs in different genera. However, we demonstrated for tick-borne and no-known vector flaviviruses that this sequence is redundant for conferring Xrn1-resistance. As discussed before, the junction between stems β and ε is formed by mismatches or non-Watson-Crick base pairs, often involving an A-G pair. The tick-borne encephalitis virus xrRNA structure did not allow for the substitution of these junction nucleotides into Watson-Crick base pairs. Recently novel pegi-, pesti- and hepaciviral xrRNAs were also identified by Szucs et al [Citation35]., who classified them into a new subclass based on minor stem-size and sequence differences. However, their study did not include a mutational analysis of the proposed structures to support their models. Although indeed, stem sizes and number of nucleotides between junctions differ, we do not think this justifies a division into structural subclasses.
Figure 5. Overview of the universal Flaviviridae xrRNA structure. Characteristics of stem elements and junctions or loops are based on mutational analyses and structural alignments from this, and our previous work [Citation33]
![Figure 5. Overview of the universal Flaviviridae xrRNA structure. Characteristics of stem elements and junctions or loops are based on mutational analyses and structural alignments from this, and our previous work [Citation33]](/cms/asset/4e110364-a8ac-4a53-ac76-a612f822f389/krnb_a_1907044_f0005_oc.jpg)
The biogenesis of sfRNA by flaviviruses, due to stalling of Xrn1 on xrRNA structures, appears to fulfil a key role in the infection cycle and pathogenesis of flaviviruses [Citation29]. The identification of Xrn1-resistant RNAs in vitro within all genera of Flaviviridae, raises the question of whether these viruses produce sfRNA during their life cycle. We have demonstrated that this does indeed occur in vivo in the case of an infection with BuPV RNA. Interestingly, BuPV is one of several pesti- and pegiviruses within which we have identified multiple subsequent xrRNA motifs. Only a single sfRNA was produced post-infection (), which begs into question whether the other three sites that we identified are redundant. In vitro degradation of the two xrRNAs that were identified in TDAV indicates that the second structure is not as stable as the first (). This may be caused by the slight deviations it manifests from the motif consensus, notably carrying three instead of two nucleotides linking the 3ʹ sides of stems β and ε, and having an A-U instead of a G-C base pair on top of the ε stem. These factors indicate that the second stalling site may have deteriorated in structure over time due to redundancy. Of course, whether these structures stall Xrn1 in vitro may not equally reflect their function during a viral infection as cellular conditions may affect structural stability. Notably, we have identified only a single flavivirus-like xrRNA within hepacivirus species (). Several mosquito- and tick-borne flaviviruses have shown formation of more sfRNA species than the number of flavivirus-like xrRNAs demonstrated in their 3ʹ UTR [Citation30,Citation32,Citation37,Citation38,Citation39]. Furthermore, we have demonstrated before that after infection of BHK-21 cells with infectious clones carrying mutated versions of the Modoc virus xrRNA, an additional sfRNA appears. These viruses do harbour additional, highly structured RNA formations in their 3ʹ UTRs [Citation40], which likely represent other types of structures that also stall Xrn1.
Since their discovery over ten years ago, research on Xrn1-resistant RNAs within Flaviviridae has been focused almost primarily on those found within the flavivirus genus. In this work, we were able to find such structures in members of all currently classified species of pegivirus, including pegivirus A through K [Citation2]. In hepacivirus and pestivirus genera, we found putative xrRNA motifs in isolates in almost every virus, with notable exceptions being HCV, BVDV and CSFV. HCV has evolved a different strategy for circumventing Xrn1 degradation by employing genomic RNA that halts Xrn1 by binding of miR-122 to two sites in the proximal end of its 5ʹ UTR [Citation34]. This highly structured RNA, and an equivalent structure within the 5ʹ UTR of BVDV, have also been shown to stall Xrn1 successfully in vitro, without the aid of miR-122 [Citation28]. As such, HCV appears to compensate for not having a flavivirus-like xrRNA motif in its 3ʹ UTR. Members of the pegi-, hepaci- and pestiviruses are assumed to not possess a methylated 5ʹ-cap due to presence of an IRES in order to initiate ribosomal scanning preceding translation [Citation19,Citation41,Citation42]. This would allow for continued translation of the viral genome after Xrn1 stalls at the 5ʹ side. In contrast, the flavivirus-like xrRNAs investigated in this study are all located in 3ʹ UTRs. While the role of miR-122 is well-established for HCV, more research will be required to establish the interplay between xrRNA structures, their genomic location and whether binding of miRNA-species occurs.
All putative xrRNA structures treated in this work were also predicted using a novel algorithm developed recently by Zammit et al [Citation43]. In addition, they identified putative motifs in several viruses isolated from marine hosts [44], with among them a sequence from the Crangon crangon flavivirus that can be folded into the typical flavivirus xrRNA (Supplementary figure S1). When tested, we found this sequence capable of stalling Xrn1 as well. The presence of Xrn1-resistant structures in a large majority of Flaviviridae viruses suggests their importance as a general mechanism for improving infection efficiency through generation of subgenomic RNA and/or sequestering of Xrn1, even beyond the currently proposed genera of flavi-, hepaci-, pesti- and pegiviruses. However, in order to assign specifically which viruses make use of this system, and to determine whether other potential functions can be assigned to these structures, more extensive, in vivo investigations will be necessary.
Acknowledgments
We acknowledge Jeffrey Kieft for acknowledging R.C.O. and P.J.B. in his previous manuscript on novel xrRNA motifs in the Flaviviridae.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Calisher CH, Gould EA. Taxonomy of the virus family Flaviviridae. Adv Virus Res. 2003;59:1–19.
- Smith DB, Becher P, Bukh J, et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J. Gen. Virol. 2016;97(11):2894–2907.
- Smith DB, Meyers G, Bukh J, et al. Proposed revision to the taxonomy of the genus Pestivirus, family Flaviviridae. J. Gen. Virol. 2017;98(8):2106–2112.
- Bhatt S, Gething PW, Brady OJ, et al. The global distribution and burden of dengue. Nature. 2013;496:504–507.
- Faria NR, Kraemer MUG, Hill SC, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science. 2018;361:894–899.
- Pierson TC, Diamond MS. The continued threat of emerging flaviviruses. Nat Microbiol. 2020;5:796–812.
- Hartlage AS, Cullen JM, Kapoor A. The strange, expanding world of animal hepaciviruses. Annu Rev Virol. 2016;3:53–75.
- Schlauder G, Pilot-Matias T, Gabriel G, et al. Origin of GB-hepatitis viruses. Lancet. 1995;346:447–448.
- Alter HJ. G-pers creepers, where’d you get those papers? A reassessment of the literature on the hepatitis G virus. Transfusion. 1997;37:569–572.
- Stapleton JT, Foung S, Muerhoff AS, et al. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011;92(Pt 2):233–246.
- Houe H. Economic impact of BVDV infection in dairies. In: Biologicals. Vol. 31. Academic Press; 2003. p. 137–143.
- Becher P, Ramirez RA, Orlich M, et al. Genetic and antigenic characterization of novel pestivirus genotypes: implications for classification. Virology. 2003;311(1):96–104.
- Pinior B, Garcia S, Minviel JJ, et al. Epidemiological factors and mitigation measures influencing production losses in cattle due to bovine viral diarrhoea virus infection: a meta‐analysis. Transbound. Emerg. Dis. 2019;66(6):2426–2439.
- Brinton MA, Dispoto JH. Sequence and secondary structure analysis of the 5′-terminal region of flavivirus genome RNA. Virology. 1988;162(2):290–299.
- Ray D, Shah A, Tilgner M, et al. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 2006;80(17):8362–8370.
- Issur M, Geiss BJ, Bougie I, et al. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15:2340–2350.
- Song Y, Mugavero J, Stauft CB, et al. Dengue and Zika virus 5ʹ untranslated regions harbor internal ribosomal entry site functions. 2019;10(2):e00459-19
- Yoo BJ, Spaete RR, Geballe AP, et al. 5′ end-dependent translation initiation of hepatitis C viral RNA and the presence of putative positive and negative translational control elements within the 5′ untranslated region. Virology. 1992;191:889–899.
- Simons JN, Desai SM, Schultz DE, et al. Translation initiation in GB viruses A and C: evidence for internal ribosome entry and implications for genome organization. J Virol. 1996;70(9):6126-6135.
- Lemon SM, Honda M. Internal ribosome entry sites within the RNA genomes of hepatitis C virus and other flaviviruses. Semin. Virol 1997;8:274–288.
- Pestova TV, Shatsky IN, Fletcher SP, et al. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 1998;12(1):67–83.
- Thurner C, Witwer C, Hofacker IL, et al. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004;85(Pt 5):1113–1124.
- Peltier C, Hleibieh K, Thiel H, et al. Molecular biology of the Beet necrotic yellow vein virus. Plant Viruses. 2008;2:14–24.
- Schuessler A, Funk A, Lazear HM, et al. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012;86(10):5708–5718.
- Schnettler E, Sterken MG, Leung JY, et al. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and Mammalian cells. J. Virol. 2012;86(24):13486–13500.
- Schnettler E, Tykalová H, Watson M, et al. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Research. 2014;42(14):9436–9446.
- Moon SL, Dodd BJT, Brackney DE, et al. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology. 2015;485:322–329.
- Moon SL, Blackinton JG, Anderson JR, et al. XRN1 stalling in the 5ʹ UTR of hepatitis C virus and bovine viral diarrhea virus is associated with dysregulated host mRNA stability. PLoS Pathog. 2015;11:e1004708.
- Moon SL, Anderson JR, Kumagai Y, et al. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. Rna. 2012;18(11):2029–2040.
- Chapman EG, Costantino DA, Rabe JL, et al. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science. 2014;344:307–310.
- Akiyama BM, Laurence HM, Massey AR, et al. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science. 2016;354:1148–1152.
- MacFadden A, Òdonoghue Z, Silva PAGC, et al. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018;9(1):1–11.
- Dilweg IW, Bouabda A, Dalebout T, et al. Xrn1-resistant RNA structures are well-conserved within the genus flavivirus. RNA Biol. 2020.DOI:https://doi.org/10.1080/15476286.2020.1830238.
- Thibault PA, Huys A, Amador-Cañizares Y, et al. Regulation of hepatitis C virus genome replication by Xrn1 and microRNA-122 binding to individual sites in the 5′ untranslated region. J Virol. 2015;89:6294–6311.
- Szucs MJ, Nichols PJ, Jones RA, et al. A new subclass of exoribonuclease-resistant RNA found in multiple genera of flaviviridae. MBio. 2020;11:1–15.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research. 2003;31(13):3406–3415.
- Chapman EG, Moon SL, Wilusz J, et al. RNA structures that resist degradation by Xrn1 produce a pathogenic dengue virus RNA. Elife. 2014;3:e01892.
- Göertz GP, Fros JJ, Miesen P, et al. Noncoding subgenomic flavivirus RNA is processed by the mosquito RNA interference machinery and determines West Nile virus transmission by Culex pipiens mosquitoes. J Virol. 2016;90:10145–10159.
- Villordo SM, Carballeda JM, Filomatori CV, et al. RNA structure duplications and flavivirus host adaptation. Trends Microbiol. 2016;24:270–283.
- Brock KV, Deng R, Riblet SM. Nucleotide sequencing of 5′ and 3′ termini of bovine viral diarrhea virus by RNA ligation and PCR. J Virol Methods. 1992;38:39–46.
- Arhab Y, Bulakhov AG, Pestova TV, et al. Dissemination of internal ribosomal entry sites (IRES) between viruses by horizontal gene transfer. Viruses. 2020;12:612.
- Zammit A, Helwerda L, Olsthoorn RCL, et al. A database of flavivirus RNA structures with a search algorithm for pseudoknots and triple base interactions. Bioinformatics. 2020;DOI:https://doi.org/10.1093/bioinformatics/btaa759.
- Parry R, Asgari S. Discovery of novel crustacean and cephalopod flaviviruses: insights into the evolution and circulation of flaviviruses between marine invertebrate and vertebrate hosts. J Virol. 2019;93:432–451.