
ABSTRACT
The Androgen Receptor (AR), transcriptionally activated by its ligands, testosterone and dihydrotestosterone (DHT), is widely expressed in cells and tissues, influencing normal biology and disease states. The protein product of the AR gene is involved in the regulation of numerous biological functions, including the development and maintenance of the normal prostate gland and of the cardiovascular, musculoskeletal and immune systems. Androgen signalling, mediated by AR protein, plays a crucial role in the development of prostate cancer (PCa), and is presumed to be involved in other cancers including those of the breast, bladder, liver and kidney. Significant research and reviews have focused on AR protein function; however, inadequate research and literature exist to define the function of AR mRNA in normal and cancer cells. The AR mRNA transcript is nearly 11 Kb long and contains a long 3’ untranslated region (UTR), suggesting its biological role in post-transcriptional regulation, consequently affecting the overall functions of both normal and cancer cells. Research has demonstrated that many biological activities, including RNA stability, translation, cellular trafficking and localization, are associated with the 3’ UTRs of mRNAs. In this review, we describe the potential role of the AR 3’ UTR and summarize RNA-binding proteins (RBPs) that interact with the AR mRNA to regulate post-transcriptional metabolism. We highlight the importance of AR mRNA as a critical modulator of carcinogenesis and its important role in developing therapy-resistant prostate cancer.
Introduction
AR is a ligand-dependent transcription factor that binds to Androgen Response Elements (AREs) in the promoter and enhancer regions of its numerous target genes and exerts its biological effects through histone modifications and chromatin remodelling [Citation1,Citation2]. AR plays a crucial role in the normal growth and maintenance of prostate epithelial cells by transactivating regulators of cell growth, cell differentiation and apoptosis [Citation3]. One of the most well-documented AR target genes is prostate-specific antigen (PSA), a member of the Kallikrein family of serine proteases that functions in liquefying seminal coagulum [Citation4]. However, PSA is more widely known for its diagnostic potential in PCa indication and progression [Citation5], since proteolytic cleavage of pro-PSA to PSA is often ineffective in PCa. Thus, high serum levels of total PSA usually indicate advanced disease.
PCa initially begins as a localized malignancy. The 5-year survival rate at this stage is approximately 98% and is typically cured by tumour removal through surgery or radiation [Citation6]. However, androgen deprivation therapy (ADT) is customarily the primary treatment method if these procedures fail or if the cancer metastasizes. Various ADT options are available and may be combined to prevent AR signalling pathways from fuelling PCa cells. Such methods include surgical castration, antiandrogens and androgen signalling inhibitors. One of the most frequently prescribed ADT therapies is the antiandrogen, enzalutamide (Enz). Enz is an AR antagonist that binds the ligand-binding pocket of the AR protein, competes with its ligand and interferes with its ability to localize to the nucleus and transcriptionally activate its numerous target genes involved in cellular proliferation [Citation7].
Because androgens are synthesized from cholesterol, therapies such as the CYP17A inhibitor, Abiraterone Acetate, were designed to inhibit intratumoral androgen synthesis. These drugs effectively mimic castration and have high therapeutic efficacy [Citation8]. However, despite low levels of serum androgens, AR signalling is maintained through activation by cytokines or growth factors or by atypical steroids, such as progesterone, oestrogen and glucocorticoids [Citation9–11]. Consequently, after approximately 24 months of androgen ablation, many patients are likely to develop castration-resistant disease. Thus, there is a pressing need for novel and effective AR- or androgen signalling-targeted therapies.
While plentiful evidence suggests therapeutically targeting androgen synthesis and AR-mediated signalling at the protein level is efficacious, our knowledge regarding the mechanisms that regulate AR expression and function at the mRNA level is entirely lacking. Following gene transcription, precursor-messenger RNA (mRNA) is subjected to several levels of post-transcriptional regulation and modifications that affect various aspects of the pre-mRNA including RNA splicing, mRNA stability, translocation and translation. Post-transcriptional regulation of gene expression often involves cis-acting RNA elements typically found within the untranslated regions (UTRs) of mRNAs. Such cis elements include binding sites for miRNAs, long noncoding RNAs (lncRNAs) and RNA-binding proteins. Interestingly, longer 3’ UTRs are typically associated with downregulated levels of gene expression, presumably due to the abundance of binding sites for trans-acting factors, such as RNA-binding proteins and lncRNAs.
The 11 Kb long AR mRNA contains an unusually lengthy ~6.8 Kb 3’ UTR (), suggesting that this region harbours many sites and motifs potentially involved in post-transcriptional regulation that govern differential gene expression and, thus, affects overall cell function. Strong evidence shows that the shortening of mRNA 3’ UTRs is often observed in various cancers [Citation12–14]. Thus, various cis-acting elements and trans-acting factors, including RBPs that interact with the AR mRNA, can potentially control post-transcriptional regulation during therapy resistance and may present novel strategies for CRPC therapies. This review summarizes the current knowledge of cis-acting elements and RBPs that interact with the AR mRNA to presumably affect the central cellular processes that become differentially perturbed in castration-sensitive and castration-resistant PCa.
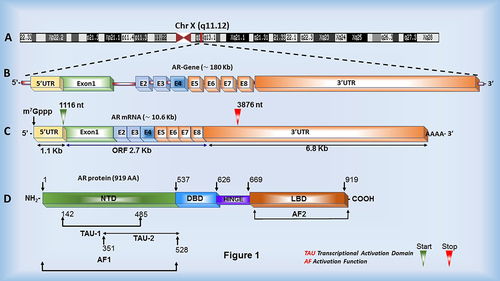
Figure 1. Androgen Receptor gene, mRNA and protein structure. (a, b) The AR gene is located on the X chromosome and consists of eight exons (E1-8) and seven variable size introns. (c) The AR mRNA is nearly 10.7 kb long, containing a 1.126 kb long 5’ UTR, a 6.778 kb long 3’ UTR and a 2.7 kb long coding region. (d) The AR protein consists of multiple functional domains: N-terminal domain (NTD), DNA-binding domain (DBD), Hinge region and C-terminal domain (CTD)/Ligand-binding domain (LBD). The N-terminal domain contains two transcriptional activation units (TAU) imperative for AR function.
Androgen receptor gene, mRNA and protein structure and function
The Androgen Receptor (AR) gene is located on the X chromosome and encodes the AR protein consisting of 919 amino acids [Citation15,Citation16]. The AR mRNA consists of two regulatory regions, a 1.1 Kb 5’ UTR and a ~6.8 Kb 3’ UTR, flanking a 2.7 Kb open reading frame (ORF) (). UTRs of mRNAs play a crucial role in mRNA localization, stability and overall mRNA function, typically through interactions with various trans-acting factors, reviewed extensively elsewhere [Citation17]. Therefore, it is logical to speculate that trans-acting factors such as miRNAs and RBPs interact with the untranslated regions of the AR mRNA and modulate AR mRNA properties such as mRNA stability, translatability, export and subcellular localization, thus ultimately affecting AR protein translation and subsequent function. However, the regulatory roles of the 5’ and 3’ UTRs of the AR mRNA remain an unexplored aspect of prostate cancer biology.
The AR protein consists of three functional domains encoded by eight exons (). The N-terminal domain (NTD), encoded by exon 1, makes up a large portion of the AR protein and is required for total AR activity [Citation18,Citation19]. The relatively sizable amino-terminal domain is also the least conserved among the human population and comprises varying lengths of polyglutamine (CAG) and polyglycine (GGC) repeats [Citation20]. Interestingly, polymorphic GGC and CAG tract lengths have been demonstrated to affect the folding and alpha-helix structure of the NTD and negatively correlate with AR transactivation activity and PCa incidence rates [Citation20,Citation21]. Within the NTD, the activation function 1 (AF1) region contains two transcription activation units (Tau-1 and Tau-5). These Tau units mediate protein–protein interactions with coactivators and other basal transcription factors and interactions between the amino and carboxyl terminal domains of AR, essential for dimerization and ligand association, thus affecting overall AR activity [Citation18,Citation19].
The DNA binding domain (DBD) of AR, encoded by exons 2 and 3, is a region essentially made up of cysteine residues and plays an important role in target sequence recognition and homodimer binding [Citation22,Citation23]. Like other nuclear receptor family members, the DBD of AR contains two zinc-finger motifs regulating α-helical structure, which bind AREs in promoter regions of AR target genes. The first zinc-finger contains an α-helix that interacts with the major groove of the consensus sequence, while the second zinc-finger plays an essential role in AR protein dimerization, necessary for binding AREs [Citation16,Citation24]. The DBD also has a carboxy-terminal extension (CTE) believed to play a role in ARE recognition and transactivation [Citation25].
Following the DBD, a flexible linker known as the hinge region connects the DBD and ligand-binding domains (LBD). The hinge region precedes the LBD in all steroid receptors and has proven to be involved in the transcriptional activation of AR [Citation26]. However, this region is better known for comprising most of the nuclear localization signal (NLS), which is revealed upon ligand binding and allows AR to localize to the nucleus and exert its effects on target genes [Citation27]. Finally, the crucial ligand-binding domain (LBD), located at the C-terminus of AR, is encoded by exons 4 through 8. This region functions in binding and stabilizing androgens by interacting with motifs in the N-terminus and plays an important role in promoting interactions between AR and chaperone proteins [Citation28].
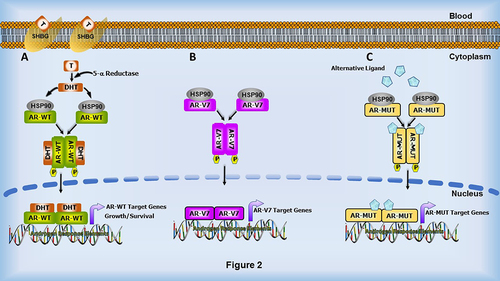
Mechanistically, AR resides in the cytoplasm until activated by binding androgenic hormones, testosterone or dihydrotestosterone (DHT), with DHT being the more potent activator. Testosterone is converted to DHT in the cytoplasm by 5-α reductase. Upon ligand binding, AR dimerizes, releases its heat shock proteins (HSPs) and localizes to the nucleus where it binds promoter and enhancer regions of target DNA via its DBD (). AR target genes often include those involved in cell proliferation via cell cycle regulation and other cellular events critical for the normal growth and function of the prostate [Citation29]. However, in PCa, these events often become perturbed due to AR modifications, such as AR mutations, AR splice variants and AR gene amplification, which contribute to PCa progression and ADT resistance [Citation30].
Figure 2. Androgen and cellular signalling alterations observed in AR-mediated transcriptional pathways during therapy resistance. (a) In the classical pathway, androgens such as Testosterone (T) present in the cytoplasm are converted to the highly potent Dihydrotestosterone (DHT) in the presence of 5-α reductase. DHT then binds to AR, inducing conformational changes, which results in the release of heat-shock proteins, followed by dimerization and translocation to the nucleus. In the nucleus, AR binds to Androgen response elements (AREs) and regulates the expression of AR target genes, and consequently the growth and survival of the cell. During PCa progression, changes in AR, such as AR splice variants, especially AR-V7 (b), and AR mutations (c), influence AR’s response to alternative ligands which can activate the AR signalling cascade, promoting the development of castration-resistant prostate cancer.
While AR function is largely dependent on ligand binding through the LBD, a plethora of research has revealed that all three domains are critical for full AR activity. Thus, the NTD and DBD have recently become targets for emerging PCa treatments. Although most androgen deprivation therapies rely on ligand binding of AR, castration resistance often occurs due to the presence of constitutively active splice variants that lack the LBD or other AR mutations [Citation30]. Therefore, novel therapies are currently being developed to interfere with the functions of other domains, such as those required for AR transactivation and DNA binding. It has been demonstrated that a small-molecule inhibitor of the AR NTD covalently binds the AF1 region and blocks AR transactivation without affecting the transactivation of other steroid hormone receptors [Citation31]. Another small molecule appears to inhibit the interaction of the DBD with DNA [Citation32]. Overall, the myriad activities of AR proteins have been extensively explored and targeted for PCa therapies. However, much remains to be learned about AR mRNA metabolism in normal cells as well as in PCa. Thus, investigating the mechanisms behind AR mRNA regulation would be of value in developing novel therapies.
Post-transcriptional regulation of AR mRNA metabolism
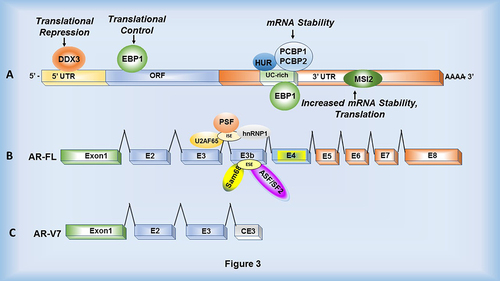
The AR gene consists of eight exons and seven introns transcribed into an mRNA with a 1.1 Kb 5’ UTR, a 2.7 Kb open reading frame (ORF) and a ~6.8 Kb 3’ UTR (). Numerous experimentally validated miRNAs and lncRNAs interact with the AR 3’ UTR and modulate AR gene expression [Citation33–37]. However, not much is known about the trans-acting RBPs that regulate AR mRNA metabolism. RBPs elicit a multitude of effects on cell function by regulating mRNA splicing, polyadenylation, translation, decay and localization, mediated by several RNA-binding domains [Citation38]. Thus, alterations in RBP expression can substantially affect central cellular processes controlled by mRNAs. Such alterations in RBP expression and function have been shown to contribute to diseases such as cancer by altering cell division, invasion, migration and proliferation [Citation39,Citation40]. A compilation of the currently known RBPs that may be involved in AR mRNA metabolism is illustrated in .
Figure 3. RNA-binding proteins that interact with the Androgen Receptor mRNA. (a) Various RNA-binding proteins including DDX3, EBP1, HuR, PCBP1/2 and MSI2 interact with AR mRNA within the 5ʹUTR, ORF and 3ʹUTR to control translation and stability. (b) Splicing factors including U2AF65 and hnRNP1 interact with an intronic splicing enhancer (ISE) while Sam68 and ASF/SF2 interact with an exonic splicing enhancer (ESE) located within the 3’ splice site of exon 3b. This results in cryptic exon splicing and generation of AR-V7 variant. PSF is predicted to bind within intron 3. (c) mRNA structure of AR-V7 lacking exons 5 through 8 (the ligand binding domain).
RNA-binding proteins targeting the androgen receptor
HuR and poly C-binding proteins 1 and 2
The earliest discovered RNA-binding proteins that bind AR mRNA include HuR and poly(C)-binding proteins 1 and 2 (PCBP1 and PCBP2). HuR is a multifaceted RNA-binding protein that is involved in various RNA processing events that are critical for regulating gene expression [Citation41]. Three RNA recognition motifs (RRMs) assist in the binding of HuR to various mRNAs, eliciting various effects on cell function [Citation42]. HuR was first identified for its role in mRNA stabilization [Citation43] but has recently been determined to have additional functions within the cell, such as modulating mRNA translation and nuclear shuttling [Citation44]. Because HuR has such a diverse array of roles in RNA metabolism within the cell, its role in cancer biology has been extensively scrutinized. HuR typically interacts with AU- and U-rich sequences within UTRs to stabilize target mRNAs [Citation43,Citation45]. Incidentally, in many cancers, HuR is overexpressed and stabilizes mRNAs that contribute to the over-proliferative properties of cancer cells [Citation46,Citation47]. Furthermore, HuR is also known to localize to the nucleus more frequently in malignant versus healthy cells [Citation48,Citation49]. Thus, HuR seems to participate in many aspects of carcinogenesis. PCBP1 and 2 share similar functions with HuR by modulating mRNA turnover and translation; however, these RBPs typically bind C-rich sequences within the UTRs of mRNAs [Citation50,Citation51].
The AR mRNA contains a well-conserved, 51 nucleotide UC-rich region located within the 3’ UTR, 153–156 nucleotides downstream from the stop codon [Citation52]. This UC-rich region was shown to reduce luciferase expression by 30% when cloned downstream of the transgene, luciferase, compared to luciferase vector alone in the AR-positive cell line, LNCaP [Citation52]. UC-rich motifs are often targets of RNA-binding proteins (RBP); thus, an RNA electrophoretic mobility shift assays (REMSA) discovered RBPs associated with this motif and revealed interactions with several cytoplasmic and nuclear proteins in human prostate and breast cancer cells [Citation52]. Experiments using anti-HuR and anti-PCBP1 or PCBP2 antibodies led to the discovery that both HuR and PCBP1 are associated with the AR mRNA UC-rich motif [Citation52]. Mutational analyses of the potential-binding sites in the AR mRNA 3’ UTR region indicated that a composite of multiple U-rich sequences within the UC-rich motif enables the optimal binding of HuR, while both CCC triplets flanking a central U in the CCCUCCC motif are critical for PCBP1 and PCBP2 binding. It is likely that these RBPs are potentially contributing to continued AR function in CRPC by stabilizing the AR mRNA, therefore affecting its translatability and oncogenic functions.
Since the discovery that HuR interacts with and stabilizes AR mRNA, several studies have investigated the implications of this RBP in PCa progression. HuR is highly expressed in human prostate cancer tissues as well as in LNCaP and PC3 PCa cell lines compared to non-cancerous samples [Citation53]. The interaction of HuR with AR mRNA affects cellular properties such as cell proliferation, invasion and wound healing, contributing to carcinogenesis in castration-sensitive and -resistant cells [Citation53]. Interestingly, several PCa-related genes have been suggested to be regulated by HuR. For example, the enzyme that converts arachidonic acid to prostaglandins, COX-2, is highly expressed in PCa, stabilized by HuR, and is strongly correlated with cytoplasmic HuR expression (P = 0.0005) [Citation54,Citation55]. Similarly, the receptor tyrosine kinase ERBB-2, which is also overexpressed in PCa, exhibits increased levels that are lined with HuR-mediated events in PCa cells. HuR appears to antagonize miR-133-3p-mediated downregulation of ERBB-2 in PCa cells, presumably by occupying the miR-133-3p binding site and affecting the formation of the RNA-protein complex for miRNA-mediated repression of mRNA translation [Citation56]. HuR and PCBP1 likely become dysregulated during PCa progression, leading to dysregulation of target mRNAs, contributing to disease progression. However, only a handful of studies have dissected the mechanisms encompassing the HuR and PCBP1-mediated modulation of AR mRNA in PCa. Because the UC-rich motif to which HuR and PCBP1 and 2 bind is highly conserved, this motif has the potential to be an ideal target for novel PCa therapies. The aspects of HuR and PCBPs illustrated above suggest that the interplay between HuR, PCBP and other RBPs or miRNAs can contribute to mRNA stabilization or destabilization of target genes, potentially producing undesirable, heterogeneous patterns of gene expression in prostate carcinogenesis.
Musashi 2
Musashi2 (MSI2) is a member of the Musashi family of RNA-binding proteins involved in translational control and regulation of stem cell fate [Citation57]. Two conserved RNA recognition motifs located at the N-terminus of MSI proteins promote binding to motifs within the 3’ UTR of target mRNA [Citation58]. Numerous results indicate a unique role for mammalian MSI2 in the regulation of haematopoietic stem cell repopulation and differentiation while also playing an important role in the progression of leukaemia [Citation57,Citation59–62]. In chronic myeloid leukaemia (CML), disease progression from the chronic phase to the blast crisis is thought to be partially regulated by the Musashi2-Numb signalling pathway, which governs differentiation of CML cells [Citation62]. Interestingly, the increased MSI2 protein levels are correlated with decreased patient survival [Citation59,Citation63], thus suggesting a role for Musashi2 as a prognostic marker for the development and progression of CML.
While MSI2 has been extensively investigated for its role in leukaemia, MSI2 expression has also been shown to be altered or thought to play a role in the progression of gastric [Citation64], pancreatic [Citation65] and hepatocellular cancers [Citation66]. Recently, a potential role for MSI2 in prostate cancer progression has been described [Citation67]. First, MSI2 expression is upregulated in human PCa tumours, and various PCa cell lines compared to normal tissue [Citation67]. Moreover, MSI2 immunohistochemistry staining of prostate tumour samples with various Gleason scores revealed that MSI2 expression was significantly higher in samples with a Gleason score ≥7 compared to those with Gleason scores <7 (P < 0.001), suggesting a role for MSI2 during the progression of localized cancer to metastasis [Citation67].
Additionally, in vitro knockdown experiments using lentivirus-mediated shMSI2 indicated that MSI2 knockdown in both castration-sensitive and resistant cell lines led to decreased AR mRNA and protein expression, cell proliferation and colony formation [Citation67]. Similar results were reflected in vivo using a nude mouse model. Mice overexpressing MSI2 exhibited 30% tumour incidence rates compared to 60% in MSI2-downregulated animals. In the MSI2 downregulated phenotype, decreased tumour growth, tumour weight and AR protein expression were observed, suggesting the function of MSI2 in conjunction with AR expression in PCa [Citation67]. Furthermore, the role of MSI2 in mRNA decay and protein stability helped linked its direct role in the regulation of AR stability. MSI2 overexpression appears to play a role in AR mRNA stability, demonstrated by RNAi knockdown experiments in which reduced levels of MSI2 lead to expedited mRNA decay in multiple PCa cell lines. RNA-IP and RNA pull down assays elucidated direct interactions between MSI2 and AR 3’ UTR [Citation67]. Results of a cycloheximide assay demonstrated that AR protein degrades more quickly in MSI2 knockdown conditions, supporting a role for MSI2 in AR mRNA stabilization and expression. Therefore, mechanistically, MSI2 may play a vital role in cancer development by binding to the AR 3’ UTR and mediating AR stability. Together, these data strongly indicate a role for MSI2-mediated control of AR mRNA metabolism during prostate tumorigenesis.
ErbB3-binding protein 1 (EBP1)
In CRPC, ADT is no longer effective due to progression to a castration-resistant stage. Importantly, AR protein often remains present and active during this stage, but the mechanisms of PCa progression and the roles of androgens and AR during this stage remain poorly understood. Currently, it is accepted that AR becomes activated by compensating signal transduction pathways, such as cytokines and growth factors involved in protein kinase pathways, and that members of the ErbB receptor family play a role during CRPC development [Citation68,Citation69]. Members of this receptor family, such as ErbB2, are expressed at higher levels in castration-resistant cell lines compared to castration-sensitive cell lines and can activate ligand-independent AR signalling, evidenced by increases in transcriptional activation of the AR target gene, PSA [Citation70]. Moreover, ErbB2 expression levels increase accordingly during progression to castration-resistance, are amplified in PCa patients and negatively correlate with patient survival [Citation71–73]. More recently, HER2/ErbB3 signalling has been suggested to mediate AR function in which HER2 kinase stabilizes AR protein and enhances the binding of AR to AR-regulated promoters [Citation74]. Thus, cross-talk between growth factor and AR signalling pathways likely occurs during carcinogenesis.
Recently, a role for EBP1, an ErbB3 binding protein, in PCa progression has been described. EBP1 was originally identified as a repressor of AR-mediated transcription and transactivation [Citation72] and binds to nuclear receptors via its LXXLL motif [Citation75]. EBP1 has been shown to directly bind AR in vitro and in vivo [Citation72]. Further studies indicated that ectopic expression of EBP1 led to a 40% or more reduction of AR-responsive gene expression, a 90% decrease in colony formation, significantly decreased growth rate (P < 0.05), increased doubling time in vitro and decreased tumorigenesis of PCa xenografts [Citation76]. Interestingly only in the presence of the antiandrogen, bicalutamide is EBP1 recruited to the PSA promoter. Furthermore, EBP1 expression is decreased in castration-resistant xenografts, together delineating a potential role for EBP1 in combating drug resistance by acting as an intermediary between antiandrogens and AR [Citation76].
Ectopic expression of EBP1 has been shown to cause a significant decrease in AR protein in vitro [Citation76]. Thus, it has been postulated that EBP1 is participating in post-transcriptional regulation of AR mRNA stability, therefore affecting translation. Indeed, a study demonstrated that EBP1 interacts with AR mRNA through a UC-rich region within the AR 3’ UTR and stimulates AR mRNA decay [Citation77]. This interaction allows EBP1 to regulate AR mRNA turnover, indicated by significantly decreased luciferase activity and accelerated mRNA decay of the reporter gene, firefly luciferase containing the AR UC-rich region in the 3’ UTR [Citation77]. This same AR UC-rich region is a binding site for HuR and PCBPs, both RBPs known for their involvement in destabilizing AR. Therefore, it is likely that this AR UC-rich region within the 3’ UTR plays a pivotal role in regulating AR mRNA stability [Citation78].
Another binding site for EBP1 was discovered within the AR mRNA coding region. This region contains the widely studied CAG repeats that are located near the 5’ end of the open reading frame and vary in length among the human population. These CAG repeats have been implicated in PCa and repeat length has been shown to negatively correlate with PCa incidence rates and to reduce the transactivation function of AR [Citation79–81]. A biotin pulldown assay utilizing His-tagged EBP1 demonstrated that EBP1 is associated with CAG9, CAG20 and CAG42 repeat lengths. Furthermore, an in vivo pulldown assay indicated that endogenous EBP1 is associated with CAG20 repeat length in cell lysates expressing EBP1, but not in EBP1-silenced lysates [Citation77]. Paradoxically, the domain of EBP1 that associated with AR mRNA was mapped to the C-terminal end of EBP1 and was found to bind both the mRNA CAG repeats and the AR mRNA 3’ UTR. Thus, this domain is critical for EBP1 association with AR mRNA and affects AR expression and function. Lastly, EBP1 knockout cells exhibited a shift in the association of AR mRNA with more translationally active polysomes, suggesting that EBP1 may also enhance the translation of AR mRNA [Citation77]. Together, these data define the importance of EBP1 during PCa progression and suggest that decreases in EBP1 expression observed during later stages of PCa may contribute to dysregulation and anomalous expression of AR.
Dead-box helicase 3 X-linked (DDX3)
While AR-positive CRPC phenotypes continuously express AR protein, other CRPC classifications include AR-low and AR-negative prostate cancers, which bypass their dependency on AR by relying on other signalling pathways, such as MAPK and FGF [Citation82]. One CRPC cell line in particular, PC-3, was isolated from an AR-null bone metastasis and is often used to study novel therapies targeting AR-null PCa. Once CRPC cells experience a phenotypic shift and become resistant to AR-targeting therapies, such as Enz, treatment options become limited.
To further define the mechanisms contributing to the loss of AR expression and ADT responsiveness in AR-low or AR-negative (ARL/-) subtypes, the ability of the RBP, DEAD-box helicase 3 (DDX3), to suppress AR translation in ARL/- CRPC cells was investigated. DDX3 is an RNA helicase with a wide variety of functions in multiple cellular compartments [Citation83]. DDX3 modulates translation initiation by interacting with translation initiation factors and unwinding RNA secondary structures [Citation84]. DDX3 also participates in directing mRNAs to stress granules (SGs) during conditions that evoke cellular stress [Citation85]. Incidentally, DDX3 is overexpressed in cancers including PCa [Citation86]. Staining of patient samples of various PCa subtypes with anti-DDX3 antibody revealed that cytoplasmic DDX3 expression is increased in AR-low/negative CRPC LuCaP PDX subtype samples compared to AR-positive and in castration-resistant compared to castration-sensitive samples, with the highest expression found in AR-low/negative CRPC LuCaP PDX subtypes [Citation87]. These data indicate a correlation between AR depletion and enhanced DDX3 expression in CRPC. Furthermore, to elucidate potential interactions between DDX3 and AR, RIP-qPCR analysis was conducted, revealing DDX3 interacts with AR mRNA and that this interaction is enhanced in the CRPC cell lines, BCaP MT10 and LNCaP-C42, compared to parental cell lines, BCaP NT1 and LNCaP [Citation87]. RNAi-mediated knockdown and pharmacological inhibition of DDX3 activity using the small-molecule inhibitor, RK33, upregulated AR protein expression, but not AR mRNA in the AR-low/negative CRPC subtype, suggesting that DDX3 modulates AR mRNA metabolism [Citation87]. Furthermore, siDDX3 and RK33 also increased AR transactivation function, evidenced by an increase in PSA mRNA and protein expression [Citation87].
In CRPC xenografts, co-staining of DDX3 and the stress granule marker, PABP1, revealed that DDX3 and PABP1 colocalize to the cytoplasmic puncta and that this colocalization was found to have an inverse correlation with AR protein [Citation87]. Under hypoxic conditions, sodium azide-treated AR-positive cells displayed increased DDX3 expression and localization of DDX3 to cytoplasmic puncta, decreased AR expression and increased binding of AR mRNA to DDX3 [Citation87]. Together, these data suggest that cellular stress mechanisms evoked during CRPC may induce DDX3-mediated regulation of AR mRNA metabolism and translation, leading to decreased AR expression and responsiveness to ADT in AR-null phenotypes. Therapeutically targeting DDX3 may sensitize the PCa cells to ADT by inhibiting its interaction with the AR mRNA, affecting its stability and translation.
RNA-binding proteins that function as splicing factors
One of the most notable mechanisms by which AR alterations engender therapy resistance occurs through the generation of constitutively active AR splice variants that bypass reliance on their cognate ligands and androgens. Multiple AR splice variants (AR Vs) have been reported in CRPC samples, many of which contain a truncated C-terminal domain [Citation88–91]. The most well-studied and abundant of these variants is the variant, AR-V7, which lacks the LBD of AR and thus, is unaffected by therapeutics that interfere with ligand binding [Citation92–94]. Although AR splice variants lack various exons present in WT AR, they remain fully capable of translocating to the nucleus and activating gene transcription of AR-regulated genes, promoting cancer cell growth even under androgen ablation [Citation95]. In this way, AR becomes aberrantly reactivated and cells are no longer responsive to androgen deprivation therapies such as Enz. Rather, fully functional AR-Vs are highly expressed in castration-resistant PCa and are capable of continuously activating AR target genes, contributing to carcinogenesis. However, although many of these splice variants have been identified, few studies have investigated the factors that participate in the splicing event that induces exon skipping and generates these AR splice variants.
To identify splicing factors (SFs) that potentially contribute to the generation of AR-V7, a recent study investigated the RBPs, U2AF65 and ASF/SF2, which seem to generate AR-V7 by interacting with AR pre-mRNA. Initially, various SFs imperative for normal gene splicing were examined, including hnRNP1, U1A, PSF, p54nrb, U2AF65 and ASF/SF2 [Citation96]. While none of these proteins experienced alterations in expression following dihydrotestosterone (androgen) or enzalutamide treatment, ChIP analysis utilizing primers designed against the 5’ and 3’ splice sites of WT AR and AR-V7 indicated that ADT enhanced the association of all the SFs mentioned to AR mRNA, excluding PSF and hnRNP1 [Citation96]. To identify the exonic splicing enhancers (ESEs) contributing to AR-V7 generation, an AR-V7 minigene under the control of the CMV promoter was created to test the interaction of a putative ISE and an ESE at the 3’ splice site of cryptic exon 3b with the SFs hnRNP1 and ASF/SF2, respectively. Transfection of this minigene construct (AR-V7 WT) and constructs containing point mutations within the splicing enhancers (ESEm and ISEm) revealed that ESEm and ISEm led to decreased AR-V7 mRNA expression compared to AR-V7 WT, but not WT AR in PCa cells. Furthermore, co-transfection of the AR-V7 minigene (AR-V7 WT) and siRNAs targeting the three splicing factors significantly reduced AR-V7 levels. This effect was rescued in samples co-transfected with those constructs containing point mutations within the splicing enhancers (ESEm and ISEm), verifying that the SFs are responsible for this reduction in AR-V7 isoform generation. Thus, interactions between U2AF65, hnRNP1 and ASF/SF2, and the ESEs and ISEs they bind to play a paramount role in alternative splicing of AR mRNA to generate the AR-V7 isoform [Citation96]. RNA pulldown experiments using RNA oligonucleotides led to the discovery that hnRNP1 and U2AF65 interact with the AR pre-mRNA region containing the ISE and ESE, respectively. These interactions were disrupted using mutant ISEm and ESEm constructs, confirming specific interactions of RBPs with the AR pre-mRNA. Moreover, anti-ASF/SF2, hnRNP1, and U2AF65 all precipitated AR pre-mRNA in the region containing both the ESE and the ISE [Citation96]. Enzalutamide treatment increased the association of all three SFs to the 3’ splice site of the exon 3b. However, not all SFs were recruited to the 5’ splice site of AR and AR-V7 or to the 3’ splice site of AR, indicating ADT-regulated enhanced recruitment or binding of SFs to the ESE and ISE may contribute to generation of AR-V7 isoform [Citation96]. Knockdown of all three SFs using RNA silencing methods led to decreased AR-V7 levels, but hnRNP1 also affected WT AR levels, suggesting that this SF does not exclusively regulate alternative splicing of AR-V7. On the other hand, U2AF65 and ASF/SF2 knockdown did not alter AR mRNA or protein levels, indicating their specificity in participating in AR-V7 pre-mRNA splicing only [Citation96].
In another study, siRNA-mediated knockdown of the SFs hnRNPA1 and hnRNPA2 led to decreases of AR-V7 mRNA and protein in 22Rv1 and VCaP cell lines [Citation97]. Interestingly, RIP experiments utilizing anti-hnRNPA1 and hnRNPA2 antibodies indicated that the association of the two SFs to the AR-V7 splice sites was increased in enzalutamide-resistant 22Rv1 and C4-2B cell lines, compared to the parental cell lines, a trend not observed with WT AR. Moreover, siRNA knockdown of hnRNPA1 and 2 in the Enz-resistant 22Rv1 cells led to regained responsiveness of these cells to ADT [Citation97]. Another RBP, Sam68 was also found to interact with both WT AR and AR-V7 proteins through the NTD, regulate exon 3b expression and promote AR-V7 transcriptional activity [Citation98]. Lastly, an SF known to interact with various components of the spliceosome, PSF, has also been shown to be a prominent factor in AR and AR-V7 splicing [Citation99]. Taken together, these data suggest that the interactions between various SFs and AR pre-mRNA require further dissection to understand the mechanism by which SFs differentially promote AR-V7 mRNA in CRPC and contribute to the survival of cancer cells and development of therapy resistant PCa.
Discussion
CRPC is the aggressive form of PCa in which no viable treatment options are currently available. Mechanisms that contribute to castration-resistance involve alterations to AR, such as AR mutations, leading to reduced ligand specificity, AR gene amplification/overexpression, the generation of constitutively active AR splice variants, dysregulation of AR coregulators, intratumoral androgen biosynthesis and activation by other ligands and factors. While all these mechanisms are attributed to AR protein function, to what extent AR mRNA metabolism contributes to one or all of these mechanisms is unknown. In this review, we have discussed several RBPs associated with AR mRNA during PCa and modulate AR mRNA features, such as stability, localization to stress granules and alterations of AR pre-mRNA splicing. RBPs are frequently dysregulated in cancers, leading to dysregulation of their target mRNA and the central cellular processes that the protein products of these mRNAs control. Therefore, differential gene regulation can influence various processes involved in cancer development and progression. The overexpression of RBPs that regulate stability, such as HuR, in PCa leads to modulation of target mRNA stability and expression. Furthermore, RBPs that function as splicing factors seem to play a critical role in promoting AR-splice variants for the survival of ADT-treated cells. RBPs that suppress AR function by directing AR mRNA to stress granules, such as DDX3, allow for the development of ADT-insensitivity. Overall, RBPs are critical players in modulating various cell functions.
While RBPs modulate various aspects of mRNA metabolism, post-transcriptional regulation of gene expression is a multi-component process that includes other elements interacting through various axes. Given the central importance of AR in prostate carcinogenesis, several longstanding questions still need to be answered, including splicing of AR pre-mRNA, AR mRNA epigenetic modifications and AR mRNA cellular trafficking, to name a few. Thus, much remains to be learned about the post-transcriptional regulatory mechanisms modulating AR and the interplay between cis-acting elements within AR mRNA and other trans-factors, including RBPs, miRNAs and long noncoding RNAs. Since aberrant androgen signalling is a direct consequence of dysregulated AR expression and transactivation, understanding the factors and mechanisms that contribute to the regulation of AR during CRPC could aid in the discovery of novel therapeutic targets. Learning about AR mRNA metabolism is significant from basic biology and clinical perspectives and will help identify disease recurrence mechanisms and, consequently, lay a foundation for novel therapeutic interventions.
Author Contributions
EL and GCS wrote and edited the manuscript. AB and CMW contributed intellectually and provided the feedback. GCS finalized the manuscript.
Acknowledgments
Research in GCS lab is supported by NIH 1R15CA252997-01A1.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Denayer S, Helsen C, Thorrez L, et al. The rules of DNA recognition by the androgen receptor. Mol Endocrinol. 2010;24(5):898–913.
- Cai C, Yuan X, Balk SP. Androgen receptor epigenetics. Transl Androl Urol. 2013;2(3):148–157.
- Vickman RE, Franco OE, Moline DC, et al. The role of the androgen receptor in prostate development and benign prostatic hyperplasia: a review. Asian J Urol. 2020;7(3):191–202.
- Lilja H, Oldbring J, Rannevik G, et al. Seminal vesicle-secreted proteins and their reactions during gelation and liquefaction of human semen. J Clin Invest. 1987;80(2):281–285.
- Stamey TA, Yang N, Hay AR, et al. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med. 1987;317(15):909–916.
- Rawla P. Epidemiology of prostate cancer. World J Oncol. 2019;10(2):63–89.
- Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790.
- Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27(23):3742–3748.
- Nadiminty N, Gao AC. Mechanisms of persistent activation of the androgen receptor in CRPC: recent advances and future perspectives. World J Urol. 2012;30(3):287–295.
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34–45.
- Zhao XY, Malloy PJ, Krishnan AV, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6(6):703–706.
- Mayr C, Bartel DP. Widespread shortening of 3ʹUTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684.
- Sandberg R, Neilson JR, Sarma A, et al. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008;320(5883):1643–1647.
- Chang JW, Zhang W, Yeh HS, et al. mRNA 3’-UTR shortening is a molecular signature of mTORC1 activation. Nat Commun. 2015;6(1):7218.
- Brown CJ, Goss SJ, Lubahn DB, et al. Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet. 1989;44(2):264–269.
- Tan MH, Li J, Xu HE, et al. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36(1):3–23.
- Mayr C. What are 3’ UTRs doing? Cold Spring Harb Perspect Biol. 2019;11(10):a034728.
- Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006;66(1):543–553.
- Jenster G, van der Korput HA, Trapman J, et al. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270(13):7341–7346.
- Irvine RA, Yu MC, Ross RK, et al. The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer. Cancer Res. 1995;55(9):1937–1940.
- Giovannucci E, Stampfer MJ, Krithivas K, et al. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A. 1997;94(7):3320–3323.
- Verrijdt G, Schoenmakers E, Haelens A, et al. Change of specificity mutations in androgen-selective enhancers. Evidence for a role of differential DNA binding by the androgen receptor. J Biol Chem. 2000;275(16):12298–12305.
- Shaffer PL, Jivan A, Dollins DE, et al. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A. 2004;101(14):4758–4763.
- Claessens F, Denayer S, Van Tilborgh N, et al. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6(1):e008.
- Schoenmakers E, Alen P, Verrijdt G, et al. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem J. 1999;341(Pt 3):515–521.
- Haelens A, Tanner T, Denayer S, et al. The hinge region regulates DNA binding, nuclear translocation, and transactivation of the androgen receptor. Cancer Res. 2007;67(9):4514–4523.
- Cutress ML, Whitaker HC, Mills IG, et al. Structural basis for the nuclear import of the human androgen receptor. J Cell Sci. 2008;121(7):957–968.
- Dubbink HJ, Hersmus R, Pike AC, et al. Androgen receptor ligand-binding domain interaction and nuclear receptor specificity of FXXLF and LXXLL motifs as determined by L/F swapping. Mol Endocrinol. 2006;20(8):1742–1755.
- Kaarbo M, Klokk TI, Saatcioglu F. Androgen signaling and its interactions with other signaling pathways in prostate cancer. Bioessays. 2007;29(12):1227–1238.
- Jernberg E, Bergh A, Wikstrom P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr Connect. 2017;6(8):R146–R61.
- Andersen RJ, Mawji NR, Wang J, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–546.
- Dalal K, Roshan-Moniri M, Sharma A, et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289(38):26417–26429.
- Ebron JS, Shankar E, Singh J, et al. MiR-644a disrupts oncogenic transformation and Warburg effect by direct modulation of multiple genes of tumor-promoting pathways. Cancer Res. 2019;79(8):1844–1856.
- Sikand K, Slaibi JE, Singh R, et al. Shukla GC. miR 488* inhibits androgen receptor expression in prostate carcinoma cells. Int J Cancer. 2011;129(4):810–819.
- Fletcher CE, Sulpice E, Combe S, et al. Androgen receptor-modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene. 2019;38(28):5700–5724.
- Ostling P, Leivonen SK, Aakula A, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71(5):1956–1967.
- Yang Y, Liu KY, Liu Q, et al. Androgen receptor-related non-coding RNAs in prostate cancer. Front Cell Dev Biol. 2021;9:660853.
- Kishore S, Luber S, Zavolan M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief Funct Genomics. 2010;9(5–6):391–404.
- Wurth L. Versatility of RNA-binding proteins in cancer. Comp Funct Genomics. 2012;2012:178525.
- Pereira B, Billaud M, Almeida R. RNA-binding proteins in cancer: old players and new actors. Trends Cancer. 2017;3(7):506–528.
- Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed). 2012;17(1):189–205.
- Wang H, Zeng F, Liu Q, et al. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr D Biol Crystallogr. 2013;69(3):373–380.
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58(2):266–277.
- Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65(20):3168–3181.
- Ripin N, Boudet J, Duszczyk MM, et al. Molecular basis for AU-rich element recognition and dimerization by the HuR C-terminal RRM. Proc Natl Acad Sci U S A. 2019;116(8):2935–2944.
- Wang J, Guo Y, Chu H, et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14(5):10015–10041.
- Wu M, Tong CWS, Yan W, et al. The RNA binding protein HuR: a promising drug target for anticancer therapy. Curr Cancer Drug Targets. 2019;19(5):382–399.
- Lopez de Silanes I, Fan J, Yang X, et al. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene. 2003;22(46):7146–7154.
- Jakstaite A, Maziukiene A, Silkuniene G, et al. HuR mediated post-transcriptional regulation as a new potential adjuvant therapeutic target in chemotherapy for pancreatic cancer. World J Gastroenterol. 2015;21(46):13004–13019.
- Hwang CK, Wagley Y, Law PY, et al. Phosphorylation of poly(rC) binding protein 1 (PCBP1) contributes to stabilization of mu opioid receptor (MOR) mRNA via interaction with AU-rich element RNA-binding protein 1 (AUF1) and poly A binding protein (PABP). Gene. 2017;598:113–130.
- Du Z, Fenn S, Tjhen R, et al. Structure of a construct of a human poly(C)-binding protein containing the first and second KH domains reveals insights into its regulatory mechanisms. J Biol Chem. 2008;283(42):28757–28766.
- Yeap BB, Voon DC, Vivian JP, et al. Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3’-untranslated region of the androgen receptor messenger RNA. J Biol Chem. 2002;277(30):27183–27192.
- Mitsunari K, Miyata Y, Asai A, et al. Human antigen R is positively associated with malignant aggressiveness via upregulation of cell proliferation, migration, and vascular endothelial growth factors and cyclooxygenase-2 in prostate cancer. Transl Res. 2016;175:116–128.
- Niesporek S, Kristiansen G, Thoma A, et al. Expression of the ELAV-like protein HuR in human prostate carcinoma is an indicator of disease relapse and linked to COX-2 expression. Int J Oncol. 2008;32(2):341–347.
- Barbisan F, Mazzucchelli R, Santinelli A, et al. Overexpression of ELAV-like protein HuR is associated with increased COX-2 expression in atrophy, high-grade prostatic intraepithelial neoplasia, and incidental prostate cancer in cystoprostatectomies. Eur Urol. 2009;56(1):105–112.
- Epis MR, Barker A, Giles KM, et al. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem. 2011;286(48):41442–41454.
- Sutherland JM, McLaughlin EA, Hime GR, et al. The musashi family of RNA binding proteins: master regulators of multiple stem cell populations. Adv Exp Med Biol. 2013;786:233–245.
- Sakakibara S, Nakamura Y, Satoh H, et al. RNA-binding protein musashi2: developmentally regulated expression in neural precursor cells and subpopulations of neurons in mammalian CNS. J Neurosci. 2001;21(20):8091–8107.
- Kharas MG, Lengner CJ, Al-Shahrour F, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16(8):903–908.
- Wuebben EL, Mallanna SK, Cox JL, et al. Musashi2 is required for the self-renewal and pluripotency of embryonic stem cells. PLoS One. 2012;7(4):e34827.
- Hope KJ, Cellot S, Ting SB, et al. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell. 2010;7(1):101–113.
- Ito T, Kwon HY, Zimdahl B, et al. Regulation of myeloid leukaemia by the cell-fate determinant musashi. Nature. 2010;466(7307):765–768.
- Byers RJ, Currie T, Tholouli E, et al. MSI2 protein expression predicts unfavorable outcome in acute myeloid leukemia. Blood. 2011;118(10):2857–2867.
- Emadi-Baygi M, Nikpour P, Mohammad-Hashem F, et al. MSI2 expression is decreased in grade II of gastric carcinoma. Pathol Res Pract. 2013;209(11):689–691.
- Fox RG, Lytle NK, Jaquish DV, et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534(7607):407–411.
- He L, Zhou X, Qu C, et al. Musashi2 predicts poor prognosis and invasion in hepatocellular carcinoma by driving epithelial-mesenchymal transition. J Cell Mol Med. 2014;18(1):49–58.
- Zhao J, Zhang Y, Liu XS, et al. RNA-binding protein musashi2 stabilizing androgen receptor drives prostate cancer progression. Cancer Sci. 2020;111(2):369–382.
- Culig Z, Hobisch A, Cronauer MV, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54(20):5474–5478.
- Jenster G. Ligand-independent activation of the androgen receptor in prostate cancer by growth factors and cytokines. J Pathol. 2000;191(3):227–228.
- Craft N, Shostak Y, Carey M, et al. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5(3):280–285.
- Signoretti S, Montironi R, Manola J, et al. Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92(23):1918–1925.
- Zhang Y, Fondell JD, Wang Q, et al. Repression of androgen receptor mediated transcription by the ErbB-3 binding protein, Ebp1. Oncogene. 2002;21(36):5609–5618.
- Morote J, de Torres I, Caceres C, et al. Prognostic value of immunohistochemical expression of the c-erbB-2 oncoprotein in metastasic prostate cancer. Int J Cancer. 1999;84(4):421–425.
- Mellinghoff IK, Vivanco I, Kwon A, et al. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6(5):517–527.
- Heery DM, Kalkhoven E, Hoare S, et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733–736.
- Zhang Y, Wang XW, Jelovac D, et al. The ErbB3-binding protein Ebp1 suppresses androgen receptor-mediated gene transcription and tumorigenesis of prostate cancer cells. Proc Natl Acad Sci U S A. 2005;102(28):9890–9895.
- Zhou H, Mazan-Mamczarz K, Martindale JL, et al. Post-transcriptional regulation of androgen receptor mRNA by an ErbB3 binding protein 1 in prostate cancer. Nucleic Acids Res. 2010;38(11):3619–3631.
- Yeap BB, Wilce JA, Leedman PJ. The androgen receptor mRNA. Bioessays. 2004;26(6):672–682.
- Tut TG, Ghadessy FJ, Trifiro MA, et al. Long polyglutamine tracts in the androgen receptor are associated with reduced trans-activation, impaired sperm production, and male infertility. J Clin Endocrinol Metab. 1997;82(11):3777–3782.
- Hsing AW, Gao YT, Wu G, et al. Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk: a population-based case-control study in China. Cancer Res. 2000;60(18):5111–5116.
- Sasaki M, Kaneuchi M, Sakuragi N, et al. The polyglycine and polyglutamine repeats in the androgen receptor gene in Japanese and Caucasian populations. Biochem Biophys Res Commun. 2003;312(4):1244–1247.
- Bluemn EG, Coleman IM, Lucas JM, et al. Androgen receptor pathway-independent prostate cancer Is sustained through FGF signaling. Cancer Cell. 2017;32(4):474–89 e6.
- Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front Genet. 2014;5:423.
- Cordin O, Banroques J, Tanner NK, et al. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37.
- Shih JW, Wang WT, Tsai TY, et al. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem J. 2012;441(1):119–129.
- Vellky JE, Ricke EA, Huang W, et al. Expression and Localization of DDX3 in prostate cancer progression and metastasis. Am J Pathol. 2019;189(6):1256–1267.
- Vellky JE, McSweeney ST, Ricke EA, et al. RNA-binding protein DDX3 mediates posttranscriptional regulation of androgen receptor: a mechanism of castration resistance. Proc Natl Acad Sci U S A. 2020;117(45):28092–28101.
- Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18(5):R183–96.
- Hornberg E, Ylitalo EB, Crnalic S, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6(4):e19059.
- Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–2730.
- Lu C, Brown LC, Antonarakis ES, et al. Androgen receptor variant-driven prostate cancer II: advances in laboratory investigations. Prostate Cancer Prostatic Dis. 2020;23(3):381–397.
- Sharp A, Coleman I, Yuan W, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129(1):192–208.
- Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–2313.
- Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–1038.
- Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287(23):19736–19749.
- Liu LL, Xie N, Sun S, et al. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33(24):3140–3150.
- Nadiminty N, Tummala R, Liu C, et al. NF-kappaB2/p52:c-Myc:hnRNPA1 pathway regulates expression of androgen receptor splice variants and enzalutamide sensitivity in prostate cancer. Mol Cancer Ther. 2015;14(8):1884–1895.
- Stockley J, Markert E, Zhou Y, et al. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci Rep. 2015;5(1):13426.
- Takayama KI. Splicing factors have an essential role in prostate cancer progression and androgen receptor signaling. Biomolecules. 2019;9(4):131.