
ABSTRACT
CRISPR systems elicit interference when a foreign nucleic acid is detected by its ability to base-pair to crRNA. Understanding what degree of complementarity between a foreign nucleic acid and crRNA is required for interference is a central question in the study of CRISPR systems. A clear description of which target–crRNA mismatches abrogate interference in type III, Cas10-containing, CRISPR systems has proved elusive due to the complexity of the system which utilizes three distinct interference activities. We characterized the effect of target–crRNA mismatches on in vitro cyclic oligoadenylate (cOA) synthesis and in vivo in an interference assay that depends on cOA synthesis. We found that sequence context affected whether a mismatched target was recognized by crRNA both in vitro and in vivo. We also investigated how the position of a mismatch within the target–crRNA duplex affected recognition by crRNA. Our data provide support for the hypothesis that a Cas10-activating region exists in the crRNA–target duplex, that the Cas10-proximal region of the duplex is the most critical in regulating cOA synthesis. Understanding the rules governing target recognition by type III CRISPR systems is critical: as one of the most prevalent CRISPR systems in nature, it plays an important role in the survival of many genera of bacteria. Recently, type III systems were re-purposed as a sensitive and accurate molecular diagnostic tool. Understanding the rules of target recognition in this system will be critical as it is engineered for biotechnology purposes.
Introduction
CRISPR-Cas systems provide prokaryotes an adaptive immune defence against bacteriophage and mobile genetic elements by utilizing a crRNA to detect foreign genes and Cas proteins to elicit an interference response against them[Citation1], [Citation2]. CRISPR-Cas systems can be organized into two classes and six types. In Class 1 systems, a crRNA in the presence of a multi-protein complex is used for interference, while in Class 2 systems, a single polypeptide bound to a crRNA carries out interference [Citation3]. The subdivision of CRISPR systems into six types is based upon the presence of a signature Cas effector in each type, a protein which is structurally and evolutionarily unique to the type. The opportunistic pathogen, Staphylococcus epidermidis strain RP62A, possesses a CRISPR system categorized as type III-A due to the presence of the Cas10 protein, the signature of type III systems and the Csm2 protein which groups the system with III-A members. Type III systems are notable for their elaborate and multi-faceted interference activities.
The S. epidermidis CRISPR locus encodes a multi-protein Cas effector complex made up of the proteins Cas10, Csm2, Csm3, Csm4 and Csm5 bound to a crRNA originating from one of the three spacer genes of the locus () [Citation4]. The Cas10-Csm complex surveils the cell for foreign RNA able to base-pair to its crRNA and upon its detection activates three interference activities: a single-stranded DNA cleavage activity in the HD domain of Cas10, a cyclic oligoadenylate synthesis (cOA) activity originating from the Palm2 domain of Cas10 and cleavage of the bound foreign RNA via an active site in the Csm3 protein [Citation2]. The cOAs, 3-mer to 6-mer oligomers of adenosine monophosphate whose tail and head are bonded to form a cyclic structure, act as second messenger molecules binding to the Csm6 RNase and activating it to indiscriminately cleave cellular RNA driving the cell into dormancy [Citation5–8]. The relative importance of the three interference activities in Cas10 systems has been debated and may be context dependent [Citation9–13].
Figure 1. Interference mediated by the S. epidermidis Cas10-Csm complex. (A) The type III-A CRISPR-Cas locus in S. epidermidis contains a repeat-spacer region with three spacers (coloured) and four repeats (grey) and nine CRISPR-associated genes. The repeat-spacer is transcribed and processed during crRNA biogenesis into mature crRNA of primarily 37 and 43 nucleotides. The crRNA combine with five Cas proteins to form the Cas10-Csm effector complex which carries out interference against foreign genetic elements. An active site in the Csm3 protein cuts bound target RNA (black triangles) and Csm6, once stimulated by cyclic oligoadenylates (cOAs) indiscriminately cuts RNA inducing cell dormancy. Cas10 also possesses a ssDNA cutting activity that is not pictured. (B) The 8 nucleotides on the 5’ end of crRNA are referred to as the 5’ tag or the proto-spacer flanking sequence (PFS) and are not involved in base-pairing to cognate targets. The first nucleotide following the 5’ tag can engage in base-pairing to a cognate target and is numbered +1. Each six nucleotides of crRNA behaves as a structural unit with five nucleotides base-pairing and the sixth unpaired. This pattern repeats throughout the crRNA–target duplex and each six-nucleotide segment can be assigned a number 1–5.
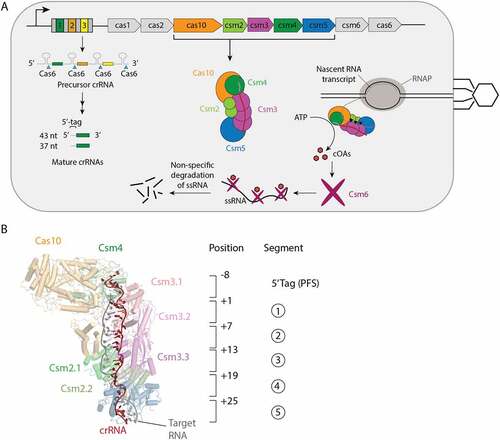
When a cognate target base-pairs to crRNA, by convention the base pair involving the first crRNA nucleotide after the 5’ tag region, is denoted as the +1 pair with subsequent pairs counting up from this position towards the 3’ end of crRNA (). Structurally, each six nucleotides of the crRNA–target duplex form a discrete unit: five nts engage in base-pairs with A-form geometry and the sixth nt is flipped out of the stack and unpaired. The pattern of five-paired nts and an unpaired sixth nt continues throughout the crRNA–target duplex and therefore each six nt unit can be termed a segment.
Since the crucial first step in interference in all CRISPR systems is the recognition of a foreign gene by its base-pairing with cRNA, understanding the extent of base-pairing necessary for interference is a central question for all systems. For Cas9 and Cas12 systems, which are used extensively in genome editing, the question of target specificity takes on great significance because off-target gene cuts are deleterious to the precision of genome editing and therefore have been investigated extensively [Citation14–18]. The effect of mismatches on interference by type III systems has been tested in microbiological assays that capture the effect in the complex context of an attack on the cell [Citation19–21]. These assays, however, do not indicate to what degree each of the distinct interference activities is affected by mismatches since their individual contributions to interference are difficult to disentangle. Using in vitro biochemistry methods, the effect of mismatches on the DNA cleavage activity of Cas10 and the Csm3/Cmr4-mediated target RNA cutting activity has each been measured [Citation22–25]. However, previous studies on the effect of mismatches on cOA synthesis were limited in scope not thoroughly addressing the effect of mismatch position within the crRNA–target duplex and the potential effect of sequence context [Citation7,Citation25–27]. To address these shortcomings, we investigated the effect of single mismatches across thirty nucleotides of the crRNA–target duplex in two sequence contexts. In vitro assays provided evidence for a Cas10-activating region, a location in the duplex with heightened sensitivity to mismatches; however, these mismatches minimally affect the affinity of target binding to cRNA [Citation25]. We performed a cell-based transformation efficiency assay that depends on cOA synthesis. This assay allowed us to compare the effects of mismatches on cOA synthesis in vitro with their effect on cOA-driven interference in cells. The cell-based assay did not display the strong positional effects seen in vitro but confirmed the finding that sequence context is an important variable affecting how Cas10-Csm responds to mismatches.
Results
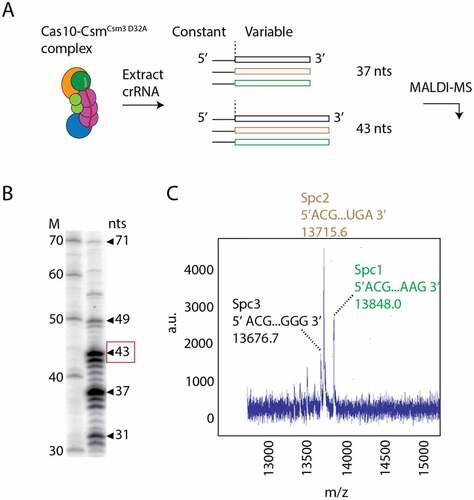
Previously, we described a method for purification of the Cas10-Csm effector complex from S. epidermidis cells that is active in cOA synthesis [Citation7]. The method utilizes the pcrispr plasmid wherein the S. epidermidis CRISPR-Cas10 locus has been relocated from genomic DNA and csm2 has been fused to a hexahistidine tag. We used immobilized metal affinity chromatography (IMAC) followed by ultracentrifugation to separate, from other cellular components, the ribonucleoprotein complex which is expected to contain a mixture of the three crRNA species. To confirm that purified Cas10-Csm complexes contain the expected crRNAs, we extracted cRNAs from the Cas10-Csm complex with phenol–chlorofom and analysed them by urea-PAGE and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) (). shows a distribution of crRNA lengths consistent with previous analysis of S. epidermidis Cas10-Csm with 37 nts and 43 nts as the most abundant lengths. MALDI-MS of the extracted cRNAs allows precise measurement of molecular mass and the unique nucleotide sequences of each crRNA should produce a unique m/z signature (). For example, in the family of peaks corresponding to the 43-nt crRNAs we observed three peaks by MALDI-MS each with an m/z value within 0.3 to 2.5 m/z units of the value predicted by the gene sequence of each spacer (Table S1). These data confirmed that the Cas10-Csm we isolated possessed crRNAs targeting the nickase (Nes) mRNA (targeted by spc1) and the Cn20 mRNA (targeted by spc2) which we intended to use as model targets for interference.
Figure 2. Identification of crRNAs bound to Cas10-Csm by mass spectrometry. (A) Cas10-CsmCsm3 D32A, a variant of the complex that is without Csm3 catalysed target RNase activity, co-purifies from S. epidermidis cells bound to crRNAs derived from three spacers with lengths of predominantly 37 and 43 nucleotides. The 5’ tag region of crRNA has a constant sequence derived from the repeat regions that flank spacers in the genome. Spacer regions in the CRISPR-Cas loci encode for unique crRNAs of variable sequence. (B) 32P-labelled, extracted crRNAs were visualized on a urea-PAGE gel revealing a distribution of intermediate and mature species with the 37 and 43 nts being most prominent. The 43-mer crRNA band, for which mass spectrometry data is shown, is highlighted with a red box. (C) MALDI mass spectrometry performed on the mixture of crRNAs identifies distinct peaks associated with the unique sequence of each RNA.
Mismatches proximal to Cas10 can reduce cOA synthesis
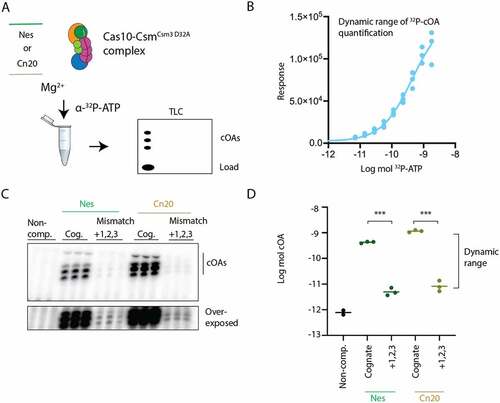
Next, we sought to develop a quantitative assay for cOA synthesis by S. epidermidis Cas10-Csm that could conveniently analyse the results of dozens of reactions in a few hours. To this end, we adapted a published protocol for separation of cOA products from the α-[28]P-ATP reactant by thin layer chromatography () [Citation28]. To gauge the dynamic range of the assay, we performed titrations of α-32PATP in triplicate, spotted the solutions on the TLC plates and integrated the phosphorimaging results (). Interpolation of the dose–response curve demonstrated that the technique had a dynamic range of approximately two orders of magnitude. We had previously shown that when Cas10-Csm was bound to a Nes target with mismatches in segment 1, which was recently termed the Cas10-activating region, dramatically lower amounts of cOA were produced [Citation7,Citation25]. Therefore, we reasoned that three consecutive mismatches in segment 1 would inhibit in vitro cOA synthesis by Cas10-Csm (). The Csm3 protein of Cas10-Csm contains a divalent metal dependent active site that cleaves bound target RNA thereby inhibiting cOA synthesis [Citation7]. Since this activity would likely be affected by the mismatches and make it difficult to resolve the effect of mismatches solely on activating Cas10 for cOA synthesis, we performed our assays with complex in which the Csm3 protein possesses the site-directed mutant D32A (Cas10-CsmCsm3 D32A). We assayed the effect of a triple mismatch in segment 1 in two sequence contexts, feeding Cas10-Csm a synthetic RNA mimicking the Nes transcript with three mismatches or a synthetic RNA mimicking the Cn20 transcript with three mismatches. In both cases, only, an extremely small amount of cOAs were produced an amount at the very low end of the dynamic range of our assay. We conclude that a triple mismatch in segment 1 has at least a two orders of magnitude effect on cOA synthesis in vitro.
Figure 3. A triple mismatch in segment 1 of the crRNA–target duplex inhibits cOA synthesis. (A) cOA synthesis was assayed by incubating either cognate target–RNAs or RNAs containing mismatches at positions +1, +2 and +3 with Cas10-Csm complex and α-32P-ATP. Radiolabeled cOA products were separated on a TLC plate and visualized by phosphorimaging. (B) A serial dilution of known amounts of 32P-ATP was applied to a TLC plate and quantitated to establish the dynamic range of the assay. (C) An image of cOA synthesis reactions, performed in triplicate, in the presence of cognate (Cog.) target–RNA or triple mismatch target–RNA. Data are shown for target RNAs complementary to the spacer-1 crRNA (Nes) and the spacer-2 crRNA (Cn20). The lower image shown is the same TLC plate overexposed. (D) Reactions shown in (C) were quantified using ImageJ and interpolated using the curve in (B) to moles of product generated. ***P <0.001.
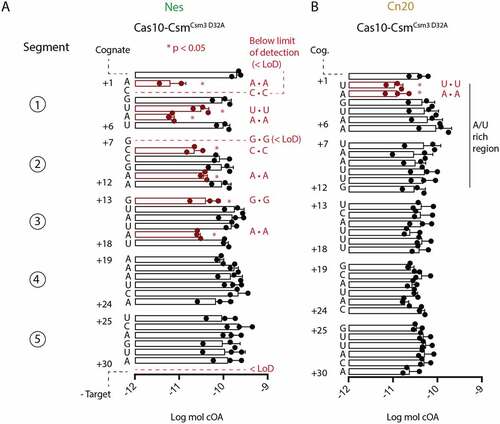
We next sought to characterize across the length of the crRNA–target duplex how more subtle, single mismatches may affect cOA synthesis. A library of RNAs mimicking the Nes and Cn20 transcripts was produced by in vitro transcription from synthetic DNA duplexes coding for each transcript (Fig. S3). We systematically introduced a single mismatch into each position from +1 to +30 of both the Nes and Cn20 transcripts by changing each transcript nucleotide to its Watson–Crick base-pair partner and fed these targets to Cas10-CsmCsm3 D32A to initiate cOA synthesis (). The products of three technical replicates were separated by TLC and quantitated (Figs. S4, S5). For the Nes transcript, we identified nine positions for which there was a statistically significant reduction in cOA synthesis compared to a cognate target (). These positions, all occurring in segments 1–3, are biased towards the 5’ end of the crRNA–target duplex, in and near the region termed the Cas10-activating region [Citation25]. Two Nes mismatches produced cOA levels equivalent to our no-target negative control, amounts below our level of detection, the +2 and +7 mismatched positions (). We characterized the effect of single mismatches across the length of the Cn20 transcript and observed a different pattern. In the Cn20 sequence context, two mismatches at positions +1 and +2 had a statistically significant reduction in cOA levels, but the amount of the reduction was about fivefold, less than the dramatic decreases seen in some Nes mismatches (see Nes positions +1, +2, +5 and +7) (). However, in keeping with the pattern observed for Nes mismatches, the only Cn20 mismatches which reduced cOA synthesis were in the Cas10-activating region. The segmented 6 nt structure of the crRNA–target duplex described previously dictates that each sixth nt is not base-paired. Therefore, introduction of a mismatch into position +6, +12, +18 or +24 is not expected to have an effect on cOA synthesis and indeed no statistically significant differences between these mismatched positions were observed in either the Nes or Cn20 context, a powerful internal control in our experiment. An additional control experiment was performed: for mismatch positions in the Cas10-activating region with large effects on cOA synthesis, we repeated the cOA synthesis experiment with synthetic target RNAs to ensure that it was not heterogeneity in the IVT RNAs producing the synthesis defect (Fig. S6). Using the TLC-based assay, we observe varying absolute amounts of cOA produced from experiment to experiment and between Nes targets versus Cn20 targets. This can be observed in the log moles cOA in and S6. The cause of this difference is unclear but appears to be a systematic difference and therefore does not affect our main goal to identify large differences in the effect of mismatches on cOA synthesis within a particular target RNA. Our studies affirm that the Cas10-activating region of the crRNA–target duplex plays a special role in stimulating cOA synthesis but demonstrate that sequence context also matters: the effect of a single mismatch on cOA synthesis is determined by position within the duplex and some contribution from local sequence context. We speculate that local GC content is the relevant parameter.
Figure 4. Single mismatches in the crRNA–target duplex inhibit cOA synthesis in a position- dependent and sequence-dependent manner. (A) Triplicate cOA synthesis reactions were performed with Nes target RNAs containing a single mismatch at position +1 to position +30 in the presence of α-32P-ATP and the cOA products were resolved by TLC and quantitated by phosphorimaging. Targets which produce cOA levels significantly different than cognate target RNA are shown in red. Mismatches at +2 or +7 or reactions conducted without any target were below the level of detection (<LoD). Mismatches significantly different than cognate have the identity of the mismatch given (for example A • A indicates two adenosines mismatched with each other). (B) Triplicate cOA synthesis reactions with Cn20 target RNAs containing single mismatches from position +1 to position +30. As above, targets which produce cOA levels significantly different than cognate target RNA are shown in red. *P < 0.05.
The binding affinity of mismatched targets is minimally affected
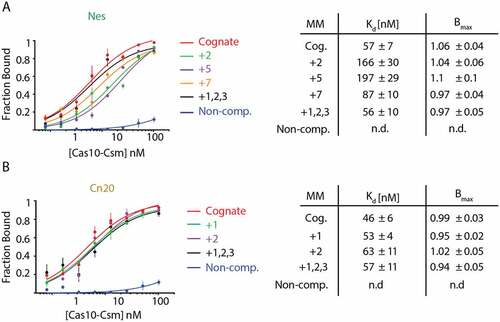
Two potential mechanisms could explain the decrease in cOA synthesis caused by mismatches at some positions within the crRNA–target duplex: either the mismatch fails to induce a conformational change in Cas10-Csm necessary for cOA synthesis or the mismatch causes a dramatic decrease in binding affinity of the target for Cas10-Csm. To test the latter mechanism, we performed an affinity binding assay comparing the mismatches to cognate target RNA (). We used synthetic RNAs mimicking mismatched transcripts of interest, those with the largest cOA synthesis defects. Some mismatches in the Nes transcript displayed a modest increase in the Kd for binding to Cas10-Csm; however, the approximately threefold differences observed for +2 and +5 mismatched transcripts would have a modest effect on the amount of target bound in the cOA synthesis assay since the target was present at 400 nM, enough to still drive binding of most transcript to the effector complex. The Nes +7 and +1,2,3 (triple) mismatched transcripts did not show a meaningful change in Kd and neither did any of the Cn20 mismatched transcripts. This was expected since when Cas10-Csm is bound to a 37 nt cRNA, five segments of crRNA–target can bind, and in this context, a loss of one to three base-pairs is unlikely to dramatically alter the Kd of target for the crRNA bound Cas10-Csm. Since binding affinity is not dramatically changed due to the presence of mismatches, we conclude that Watson–Crick base-pairing between crRNA and its target in the Cas10-activating region is sensed by Cas10-Csm as a critical pre-condition for cOA synthesis, albeit in a manner dependent on sequence context.
Figure 5. Single and triple mismatch target–RNAs show only modest changes in binding affinity towards Cas10-CsmCsm3 D32A compared to cognate targets. (A) 32P-labelled Nes targets were incubated with increasing amounts of Cas10-Csm and bound target was isolated with a nitrocellulose and Hybond double membrane assay. Fraction bound was found by quantitating the amount of 32P-target on the nitrocellulose membrane (Cas10-Csm bound) versus the total amount of target (nitrocellulose plus Hybond) by phosphorimaging. (B) An affinity binding assay was also performed with 32P-labelled Cn20 targets.
Mismatching segments are required in cells for the loss of cOA-mediated interference
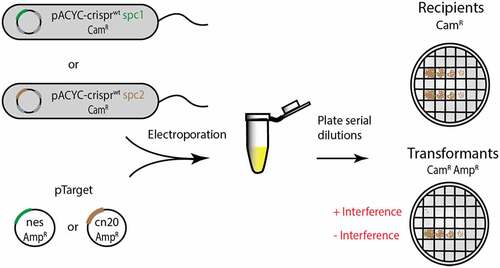
In vivo assays of the effect of crRNA–target mismatches on interference are critical towards understanding how Cas10-Csm protects prokaryotes from bacteriophage and mobile genetic elements and also indicating the conditions under which interference can be bypassed, for example by a phage that is genetically divergent from an ancestor targeted by CRISPR. However, interpretation of in vivo interference assays can be challenging because context dictates how much each of Cas10-Csm’s three interference activities contributes to the outcome of the experiment [Citation10–12]. To reduce the number of variables contributing to interference, we re-purposed an E. coli-based transformation efficiency assay developed by Terns and co-workers [Citation29,Citation30]. In this assay, E. coli-bearing S. epidermidis CRISPR-Cas10 on a plasmid with a chloramphenicol marker is challenged by transformation with a plasmid encoding a target of crRNA on a plasmid with an ampicillin marker. In the assay, quantitation of colony-forming units (CFUs) on a Cam plate indicates the number of recipient cells and CFUs on a Cam/Amp plate indicate the number successfully transformed by the target plasmid. Terns and co-workers have shown interference in the assay is only dependent on the cOA-mediated RNase activity of Csm6 which is known to broadly target most RNAs of the cell [Citation30]. We modified the published assay by introducing mismatches into the nes target gene and we constructed a pair of new reagents to probe interference in an alternate sequence context: a new pACYC-crispr containing strain harbouring the spc2 gene and a pTarget containing the cn20 gene ().
Figure 6. Schematic of the cOA-mediated interference assay. E. coli cells harbouring pACYC-crispr spc1 or pACYC-crispr spc2 are electroporated with the corresponding pTarget and transformation efficiency (colony-forming units per mL) is scored as a measure of interference. Cells transformed with pTarget have a chloramphenicol-resistant (CamR) and ampicillin-resistant (AmpR) phenotype. The pACYC-crispr plasmids are labelled as wt because each encodes a catalytically active Csm3 rather than the D32A target RNase-dead variant used for in vitro assays.
We wondered to what degree crRNA–target mismatches would affect cOA-mediated interference with the goal of making comparisons between interference in vivo and our in vitro cOA synthesis assays. As before, we were also interested in how sequence context may impact the effect of mismatches on interference. We began by introducing a triple mismatch into the segment 1 coding region of the nes gene in pTarget. Whereas, a triple mismatch in vitro reduced cOA synthesis levels so much that they were below the level of accurate quantitation, the triple mismatch at positions +1 to +3 had no effect on interference (). We then introduced successively mismatching segments into the nes gene of pTarget. A fully mismatched nes segment 1 also had no effect on interference but if both segments 1 and 2 were mismatched interference receded to background levels. The introduction of mismatches simultaneously into segments 1 to 3, 1 to 4 and 1 to 5 also resulted in no detectable interference (). We performed the same experiment with pTarget now encoding a fragment of the cn20 gene and cells carrying S. epidermidis CRISPR-Cas10 engineered with a spacer targeting this bacteriophage gene. As observed before, a triple mismatch at positions +1 to +3 in segment 1 had no effect on interference (). However, if the cn20 gene was altered so that the entirety of segment 1 was mismatched, interference was dramatically reduced (). The successive introduction of mismatches in segments 1 to 2, 1 to 3, 1 to 4 and 1 to 5 caused a loss of interference in all cases. The experiments with the cn20 target, however, revealed a critical distinction between the nes and cn20 sequence contexts with segment 1 being critical to interference in the cn20 but not nes sequence context.
Figure 7. The effect of crRNA–target mismatches on cOA-mediated interference in cells. (A) A schematic of the base-pairing between crRNA and target RNAs encoded in the assay. Mismatching segments were successively introduced into the target gene beginning with segment 1. The effect of mismatched segments on interference was measured by the transformation efficiency of mismatched versions of pTarget harbouring a fragment of the nes gene. Recipient colony-forming units (CFUs) indicate the number of cells available for transformation and the transformants CFUs indicate pTarget uptake. (B) The same assay was performed but now pACYC-crisprwt carried the spc2 gene which encodes a crRNA targeting the transcript of the cn20 gene, a fragment of which is encoded on pTarget cn20. (C) A schematic of pTarget constructs used in a second interference assay is shown that introduced successive mismatches beginning with segment 5. The assay was performed with pTarget encoding nes and (D) pTarget encoding cn20.
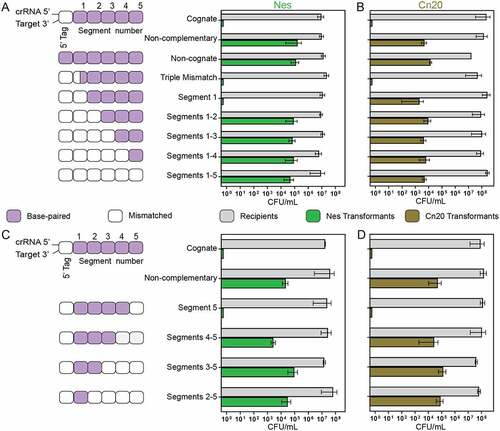
We next performed an experiment designed to test whether, in the cOA-mediated interference assay, Cas10-proximal segments 1 and 2 played a more critical role in interference than segments 3 to 5. That is, interference was lost when segments 1 and 2 were mismatched in nes or only segment 1 in cn20 and we wondered how many segments must be mismatched to inhibit interference if we began successively mismatching segments starting with segment 5 (). The loss of base-pairing in segment 5 had no effect on interference in either the nes or cn20 sequence contexts; however, the loss of base-pairing in additional segments reduced interference to background levels (). Taken together, the two experiments utilizing a nes target indicate that the loss of two base-paired segments causes a loss of interference and it does not matter whether the target is mismatched at segments 1 and 2 or 4 and 5. The two experiments in the cn20 sequence context produced a slightly different result: these experiments indicate that base-pairing in the Cas10-proximal region (including segment 1) has a greater effect on interference than base-pairing in the Cas10-distal region (including segment 5). Loss of base-pairing in segment 1 alone caused a loss of interference in the cn20 target but when the segments were successively removed from the Cas10-distal region, a loss of base-pairing in segments 4 and 5 were required to abrogate interference.
Discussion
The effect of mismatches between crRNA and its target on interference by type III systems has been previously investigated but drawing wide-ranging conclusions from these experiments was not possible due to various limitations such as only one target (sequence context) was investigated, mismatches were only examined over a portion of the crRNA–target duplex, or mismatching was investigated in vivo but not with a complementary in vitro assay. By assaying the effects of mismatches on a specific interference activity, cOA-synthesis, across the length of the crRNA–target duplex, in two sequence contexts by complementary approaches, we believe we have filled an important gap in the field.
Evidence for a Cas10-activating region in positions +1 to +7 of crRNA
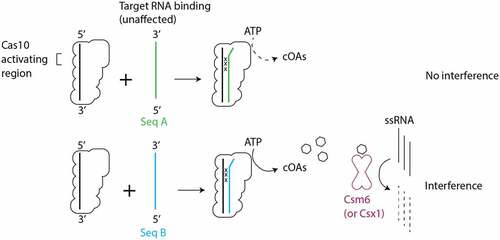
It has recently been argued that type III CRISPR systems contain two functionally important regions of crRNA, a Cas10-activating region in the 5’ region of crRNA and a seed sequence in the 3’ region. The Cas10-activating region is located at positions +1 to +7, wherein activation of cOA synthesis is highly sensitive to mismatches [Citation7,Citation25,Citation27]. The in vitro cOA synthesis assays we performed revealed that single mismatches between crRNA and target capable of substantially reducing cOA synthesis were located in +1 to +7 strongly supporting the existence of a Cas10-activating region. Additionally, our affinity binding assays provide a mechanistic clue as to how mismatches in the Cas10-activating region regulate cOA synthesis. The fact that we do not see substantial loss of binding affinity between targets mismatched in segment 1 and crRNA, suggests that correct base-pairing in segment 1 drives a conformational change in Cas10 required for cOA synthesis rather than being critical for tight binding of the target (). Our cell-based interference assays also provide support for the Cas10-activating region’s critical role in interference, albeit modest support. When nes was used as the target, similar sensitivity was seen in both the 5’ and 3’ end of the crRNA; mismatches in two contiguous segments on the 5’ end of crRNA (segments 1 and 2) and on the 3’ end of crRNA (segments 4 and 5) were sufficient to eliminate interference.
Figure 8. Intrinsic features of the sequence of target RNAs determine the effect of mismatches on cOA-mediated interference. Mismatches between crRNA and target RNA in the Cas10-activating region did not lead to substantial defects in target binding. However, cOA synthesis is sensitive to crRNA–target mismatches in the Cas10-activating region in a manner that depends on intrinsic features of the target sequence. Seq A and Seq B indicate two hypothetical target RNA sequences which differ in their ability to stimulate cOA synthesis. The ‘x’ symbol indicates a crRNA–target RNA mismatch.
However, when cn20 was used as the target mismatches in segment 1 were sufficient for loss of interference, while on the 3’ end of the crRNA loss of pairing in both segments 4 and 5 were required for loss of interference consistent with a special role for positions +1 to +7 in activating cOA synthesis by Cas10. A caveat must be attached to our in vivo interference results that they are performed with recombinantly expressed CRISPR-Cas10 which may affect the amount of cOA synthesized. For example, it may be the case that overexpression causes an abundance of cOA to be made underestimating the effect mismatches have on interference.
RNA-based regulatory systems typically identify their nucleic acid binding partners, among the complex milieu of the cell, by utilizing seed sequences, short nucleotide segments involved in the initial search for complementary partners [Citation31]. For example, Cas9 binds its crRNA so that nucleotides 11–20, which are adjacent to its PAM (protospacer-adjacent-motif) sequence are pre-organized into an A-form helix and displayed towards solvent to facilitate initial binding to its DNA target [][Citation32]. Evidence has been presented for and against the existence of a seed sequence on the 3’ end of cRNA in type III systems [Citation21,Citation24,Citation25,Citation33]. Our cell-based interference experiments did not produce evidence for a 3’ a seed sequence since we did not see heightened sensitivity to mismatches at the 3’ end of crRNA and, in the case where cn20 was used as the target, observed less sensitivity to mismatches at the 3’ end of crRNA. Pyenson et al. performed a transformation efficiency assay utilizing S. epidermidis CRISPR-Cas10, as we did, however the assay was different in several key aspects [Citation21]. These investigators data agree with ours as they also observed the 5’ end of crRNA was more sensitive to mismatches than the 3’ end. The varied findings on the importance of the 3’ region of cRNA in type III systems can be rationalized several ways. It has been shown that target concentration can affect how sensitive type III systems are to mismatches. In a transformation efficiency assay, You et al. showed that pairing from positions +1 to +23 was required for interference when target levels were high but pairing from +1 to +26 was required when target levels were low indicating a greater sensitivity to mismatches at the 3’ end of crRNA when target levels are low. Therefore, varying levels of target among the assays could explain some divergent results. Additionally, it has been argued that the prominent role of pairing at the 3’ end of crRNA is masked by the existence of multiple oligomeric states of type III complexes, a phenomenon that is hard to control for in vivo [Citation25].
Sequence context affects the sensitivity to mismatches in type III systems
Our work highlights an underappreciated aspect of type III CRISPR systems: conclusions are often drawn from mechanistic experiments performed with a single target sequence. Our results highlight that in vitro and cell-based assays of interference can return different results depending on the target sequence used. Stated differently: intrinsic aspects of the target sequence can affect its ability to stimulate interference (Compare Seq A to Seq B in ). Prior to the discovery that cOAs are a critical driver of type III interference, Maniv et al. presented data from assays in staphylococcal cells which hinted that intrinsic aspects of a sequence could affect its sensitivity to mismatches [Citation20]. The investigators used the nes and cn20 genes as model targets, as we did, and found that mismatches at position +2 to +4 of nes caused a loss of interference but mismatches at +2 to +4 of cn20 did not. Surprisingly, the phenomenon depended on gene orientation, whether the gene was on the leading versus lagging strand, which the investigators believed was connected to the fact that the gene was being replicated by rolling circle replication. We have shown intrinsic sequence effects on interference in two assays that differ markedly from those in Maniv et al.: a plasmid transformation efficiency assay that depends on theta replication and an in vitro cOA synthesis assay. Taken together, these data argue that there are several layers of properties that effect how a target sequence will respond to mismatches. Gene orientation is one, but our in vitro results suggest that some target sequences may be less able to induce the conformational changes in Cas10 required for activation as efficiently as others when mismatches occur.
In conclusion, we present data relevant to the ongoing debate over the location and mechanism of specific regions of crRNA that affect interference in type III systems and our data argue for the existence of a Cas10-activating region at positions +1 to +7. Additionally, we present data which argue that the propensity of mismatches within the Cas10-activating region to block interference is an intrinsic feature of the sequence that may vary widely. Resolving the debate over how specific regions of crRNA affect interference is critical since it impacts our understanding of how natural selection drives both mutations in mobile genetic elements to evade type III CRISPR systems and may affect and reinforce the selection of particular spacers by prokaryotes which provide robustness to immunity. Understanding the significance and impact of mismatches in type III systems is also relevant to new biotechnologies being developed with these CRISPR systems [Citation25,Citation27,Citation34,Citation35].
Materials and methods
Purification of Cas10-Csm
Cas10-Csm expression and immobilized metal affinity chromatography (IMAC) were performed as previously described [Citation36]. Briefly, overnight cultures of S. epidermidis LM1680, which carry the plasmid pcrispr/csm3D32A but lack a genomic crispr locus, were used to inoculate 1 L of Brain Heart Infusion media containing appropriate antibiotics. Growth was continued at 37°C until OD600 ~ 2.0 was reached and cells were harvested by centrifugation. Lysis was achieved by sonication and the addition of lysostaphin to 28 μg/mL. Following centrifugation, lysate was subjected to IMAC which used wash buffer containing 100 mM NaH2PO4 pH 8.0, 600 mM NaCl and 40 mM imidazole and elution buffer with the same components except 250 mM imidazole was present. Cas10-Csm-enriched fractions were pooled and layered onto a 5–20% (w/v) sucrose gradient composed of 50 mM Tris HCl pH 8.0, 150 mM NaCl and 5% (v/v) glycerol. Ultracentrifugation was performed for 41 h at 118,000 x g on a Beckman SW-32TI rotor. Fractions containing intact Cas10-Csm complex were identified by A280 measurements and appeared near the bottom of the gradient consistent with a molecular weight of ~300 kDa. Sample purity was analysed by SDS-PAGE and purified Cas10-Csm was flash-frozen and stored at −80°C.
Purification and mass spectrometry analysis of crRNAs
CrRNAs were extracted from purified Cas10-CsmCsm3D32A with 1:1 (v/v) ratio of phenol–chloroform–isoamyl alcohol (25:24:1) twice, followed by one extraction with 1 vol. chloroform. For PAGE analysis, crRNAs were 32P-labelled by incubation with γ-32P-ATP (3000 Ci/mmol) and T4 Polynucleotide kinase at 37°C for 1 h. Extracted crRNAs were mixed 1:1 with formamide dye (5 mM EDTA pH 8.0, 95% formamide v/v) and heated at 70°C for 2 min before loading to a 12% acrylamide, 8 M urea gel. Invitrogen Decade markers were added to the gel to infer crRNA sizes. Electrophoresis was carried out at 50 W for approximately 90 min. Phosphorimaging was performed with storage phosphor screens and a Typhoon FLA 7000 both from GE Healthcare.
For mass spectrometry analysis, crRNAs were desalted with C18 ziptips according to the manufacturer’s instructions. One microlitre of matrix (9 parts of 50 mg/mL 3-hydroxypicolinic acid in 50% acetonitrile/water and 1 part 50 mg/mL ammonium citrate in water) was spotted on a matrix-assisted laser desorption ionization (MALDI) target and was dried by a stream of nitrogen gas. One microlitre of desalted crRNAs was then layered on the dried matrix and the mixture was subjected to MALDI mass spectrometry with a Bruker RapifleX MALDI TOF/TOF.
cOA synthesis
All reactions were carried out in 50 mM Tris HCl pH 8.0, 150 mM NH4Cl, 5% (v/v) glycerol, 500 μM ATP, 30 nM α-32P-ATP 3000 Ci/mmol and 10 mM Mg2+ (TNG buffer) at 37°C. All reactions were performed with 100 nM Cas10-CsmCsm3D32A and 400 nM target RNA as indicated in TNG buffer for 1-h incubation. cOA reaction products were separated by thin-layer chromatography using previously described methods [Citation28]. Briefly, a chamber containing TLC running buffer composed of 0.2 M ammonium bicarbonate pH 9.3, 70% ethanol and 30% water was pre-warmed to 35°C to saturate the chamber with buffer vapour. Samples were spotted on a silica gel TLC plate 2 cm from the bottom of the plate and were air-dried. The plate was then placed in the chamber for 2 h until the solvent front was approximately 5 cm from the top. The plate was removed from the chamber, dried with a gentle air stream and cOAs were visualized by phosphorimaging.
Target RNA mismatch library
The target RNA library containing mismatches that were used in cOA synthesis assays is given in supporting information. RNAs are either derived from the sequence of the nickase (nes) gene of the pG0400 conjugative plasmid that can be targeted by spc1 crRNA of pcrispr [Citation4,Citation37] or derived from the sequence of the cn20 gene of the staphylococcal phage CNPH82 that can be targeted by spc2 crRNA. DNA templates (Table S2) used to synthesize the library by in vitro transcription were ordered from Eurofins genomics. In vitro transcription was performed using the Promega RiboMax kit. Clean-up of the in vitro transcription products was performed by DNase I digestion, phenol–chloroform extraction and ethanol precipitation according to the manufacturer’s instructions except phenol–chloroform–isoamyl alcohol 25:24:1 was used for extractions. Synthetic RNAs used in were made by Horizon Discovery. The quality of the RNAs was checked by a 12% acrylamide urea-PAGE run at 300 V for 30 min, stained with SYBR green II (Lonza) and imaged on a Typhoon FLA 7000 imager.
Affinity binding assays
Varying concentrations of Cas10-CsmCsm3 D32A (8–1024 nM) were incubated with 4 nM 32P-radiolabeled ssRNA at 0°C for 30 min. The binding buffer contained 50 mM Tris HCl, pH 8.0, 10 mM Mg2+, 5% (v/v) glycerol and 0.5 mM EDTA. Protein-RNA complexes were separated and quantified by a double-membrane binding assay [Citation38]. A sandwich of Amersham Protran 0.45 μm nitrocellulose and Amersham Hybond N+ membranes were soaked in binding buffer and were assembled in a BioRad BioDot vacuum filtration device. Reactions were applied to the membrane sandwich, filtered and the membranes were then washed with 100 μL cold buffer. The membrane sandwich was disassembled, dried and then subjected to phosphorimaging. The fraction bound in each titration series was calculated after integration of the membrane arrays in GE ImageQuant software by dividing the nitrocellulose counts by the sum of the counts from both membranes.
Interference assays in E. coli
A pACYC vector containing a chloramphenicol resistance marker and encoding S. epidermidis crispr-cas10 for expression in E. coli was the gift of Dr Michael Terns of the University of Georgia. The crispr-cas10 locus is modified to facilitate expression in E. coli including the inclusion of T7 promoters, codon optimization of the open reading frames and the inclusion of only a single spacer gene, spc1 which targets the Nes transcript [Citation29]. We refer to this plasmid as pACYC-crisprwt spc1 (). This was modified for experiments targeting the Cn20 transcript: a GeneArt string (ThermoFisher Scientific) encoding spc2, was ligated into the plasmid utilizing the NcoI and PspXI sites to create pACYC-crisprwt spc2 (). This construct was verified by Sanger sequencing with a set of primers that covered the entire plasmid.
A pTrc vector containing an ampicillin marker and a segment of the nes gene which we refer to as pTarget-nes was also the gift of Dr Michael Terns of the University of Georgia () [Citation29]. This plasmid was modified by systematically altering segments of the nes gene to introduce mismatches between its transcript and crRNA. Mismatches at positions +1, +2 and +3 and +1 to +6 were introduced by QuikChange. Additional nes mismatches, segments 1 and 2 or segments 1 to 3, etc., were introduced by Gibson assembly using PCR-linearized pTrc-nes and GeneStrands (Eurofins Genomics) [Citation39]. For experiments in which interference was initiated by the cn20 gene, the nes gene of pTrc-nes was swapped for the cn20 gene by Gibson assembly using PCR-linearized vector and a GeneStrand encoding a fragment of cn20 (). PTrc-cn20 variants encoding mismatches between the Cn20 transcript and spc2-derived crRNA were constructed by QuikChange (+1, +2 and +3 mismatch and the +1 to +6 mismatch) or by Gibson assembly (segment 1 and 2 or segments 1 to 3 etc.) with GeneStrands. All constructs were verified by Sanger sequencing.
Interference in E. coli cells was scored by a transformation efficiency assay. Electrocompetent BL21(DE3) cells harbouring pACYC-crisprwt plasmids were transformed by electroporation with 40 ng of pTrc-nes or pTrc-cn20. The transformed cells were then suspended in 1 mL of SOC medium and incubated at 37°C, shaking at 250 rpm for 1 h. Ten-fold serial dilutions were then performed in LB medium and the resulting suspensions were plated on LB-agar plates containing either chloramphenicol (17 µg/mL) or chloramphenicol (17 µg/mL) and ampicillin (50 µg/mL). The plates were incubated overnight at 37°C then colony-forming units per mL (CFU/mL) were quantitated for three replicate transformations of each target plasmid.
Data availability
PAGE, TLC and membrane (affinity binding) images are available at: https://doi.org/10.5281/zenodo.7041988.
Supplemental Material
Download MS Word (3.5 MB)Acknowledgments
We thank Dr Asma Hatoum-Aslan for the kind gift of S. epidermidis LM1680 carrying pcrispr/csm3D32A. We also thank Dr Michael Terns for kind gift of pACYC-crispr and pTrc-target.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2022.2150812
Additional information
Funding
References
- Wright AV, Nunez JK, Doudna JA. Biology and applications of crispr systems: harnessing nature’s toolbox for genome engineering. Cell. 2016;164(1–2):29–44.
- Nussenzweig PM, Marraffini LA. Molecular mechanisms of CRISPR-Cas immunity in bacteria. Annu Rev Genet. 2020;54(1):93–120.
- Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18(2):67–83.
- Hatoum-Aslan A, Samai P, Maniv I, et al. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J Biol Chem. 2013;288(39):27888–27897.
- Kazlauskiene M, Kostiuk G, Venclovas Č, et al. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017;357(6351):605–609.
- Niewoehner O, Garcia-Doval C, Rostol JT, et al. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017;548(7669):543–548.
- Nasef M, Muffly MC, Beckman AB, et al. Regulation of cyclic oligoadenylate synthesis by the Staphylococcus epidermidis Cas10-Csm complex. RNA. 2019;25(8):948–962.
- Athukoralage JS, White MF. Cyclic oligoadenylate signalling and regulation by ring nucleases during type III CRISPR defence. RNA. 2021.
- Deng L, Garrett RA, Shah SA, et al. A novel interference mechanism by a type IIIB CRISPR-Cmr module in sulfolobus. Mol Microbiol. 2013;87(5):1088–1099.
- Hatoum-Aslan A, Maniv I, Samai P, et al. Genetic characterization of antiplasmid immunity through a type III-A CRISPR-Cas system. J Bacteriol. 2014;196(2):310–317.
- Jiang W, Samai P, Marraffini LA. Degradation of Phage transcripts by CRISPR-Associated RNases enables type III CRISPR-Cas Immunity. Cell. 2016;164(4):710–721.
- Rostol JT, Marraffini LA. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR–Cas immunity. Nat Microbiol. 2019;4(4):656–662.
- Liu TY, Liu -J-J, Aditham AJ, et al. Target preference of Type III-A CRISPR-Cas complexes at the transcription bubble. Nat Commun. 2019;10(1):3001.
- Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832.
- Pattanayak V, Lin S, Guilinger JP, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843.
- Kim D, Kim J, Hur JK, et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34(8):863–868.
- Kleinstiver BP, Tsai SQ, Prew MS, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34(8):869–874.
- Murugan K, Seetharam AS, Severin AJ, et al. CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects. J Biol Chem. 2020;295(17):5538–5553.
- Manica A, Zebec Z, Steinkellner J, et al. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon sulfolobus solfataricus. Nucleic Acids Res. 2013;41(22):10509–10517.
- Maniv I, Jiang W, Bikard D, et al. Impact of different target sequences on type III CRISPR-Cas immunity. J Bacteriol. 2016;198(6):941–950.
- Pyenson NC, Gayvert K, Varble A, et al. Broad targeting specificity during bacterial type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe. 2017;22(3):343–53 e3.
- Johnson K, Learn BA, Estrella MA, et al. Target sequence requirements of a type III-B CRISPR-Cas immune system. J Biol Chem. 2019;294(26):10290–10299.
- Staals RJ, Zhu Y, Taylor DW, et al. RNA targeting by the type III-A CRISPR-Cas csm complex of thermus thermophilus. Mol Cell. 2014;56(4):518–530.
- Pan S, Li Q, Deng L, et al. A seed motif for target RNA capture enables efficient immune defence by a type III-B CRISPR-Cas system. RNA Biol. 2019;16(9):1166–1178.
- Steens JA, Zhu Y, Taylor DW, et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat Commun. 2021;12(1):5033.
- Rouillon C, Athukoralage JS, Graham S, et al. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. Elife. 2018;7. DOI:10.7554/eLife.36734
- Gruschow S, Adamson CS, White MF. Specificity and sensitivity of an RNA targeting type III CRISPR complex coupled with a NucC endonuclease effector. Nucleic Acids Res. 2021;49(22):13122–13134.
- Rouillon C, Athukoralage JS, Graham S, et al. Investigation of the cyclic oligoadenylate signaling pathway of type III CRISPR systems. Methods Enzymol. 2019;616:191–218.
- Ichikawa HT, Cooper JC, Lo L, et al. Programmable type III-A CRISPR-Cas DNA targeting modules. PLoS One. 2017;12(4):e0176221.
- Foster K, Kalter J, Woodside W, et al. The ribonuclease activity of Csm6 is required for anti-plasmid immunity by Type III-A CRISPR-Cas systems. RNA Biol. 2019;16(4):449–460.
- Gorski SA, Vogel J, Doudna JA. RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol. 2017;18(4):10509–10517.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna J A, Charpentier E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–21. 10.1126/science.1225829
- Mogila I, Kazlauskiene M, Valinskyte S, et al. Genetic dissection of the type III-A CRISPR-Cas system csm complex reveals roles of individual subunits. Cell Rep. 2019;26(10):2753–654.
- Santiago-Frangos A, Hall LN, Nemudraia A, et al. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Reports Medicine. 2021;2:100319.
- Woodside WT, Vantsev N, Catchpole RJ, et al. Type III-A CRISPR systems as a versatile gene knockdown technology. RNA. 2022;28:1074–1088.
- Chou-Zheng L, Hatoum-Aslan A. Expression and purification of the cas10-Csm complex from staphylococci. Biol Protoc. 2017;7(11). DOI:10.21769/BioProtoc.2353
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322(5909):1843–1845.
- Wong I, Lohman TM. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc Natl Acad Sci U S A. 1993;90(12):5428–5432.
- Gibson DG, Young L, Chuang R-Y, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345.