
ABSTRACT
Micro RNAs (miRNAs) are short, non-coding RNAs with significant potential as diagnostic and prognostic biomarkers. However, a lack of reproducibility across studies has hindered their introduction into clinical settings. Inconsistencies between studies include a lack of consensus on the miRNAs associated with a specific disease and the direction of regulation. These differences may reflect the heterogenous nature of pathologies with multiple phenotypes, such as amyotrophic lateral sclerosis (ALS). It is also possible that discrepancies are due to different sampling, processing, and analysis protocols across labs. Using miRNA extracted from L1CAM immunoaffinity purified extracellular vesicles (neural-enriched extracellular vesicles or NEE), we thrice replicated an 8-miRNA fingerprint diagnostic of ALS, which includes the miRNA species and direction of regulation. We aimed to determine if the extra purification steps required to generate NEE created a unique extracellular vesicle (EV) fraction that might contribute to the robustness and replicability of our assay. We compared three fractions from control human plasma: 1) total heterogenous EVs (T), 2) L1CAM/neural enriched EVs (NEE), and 3) the remaining total-minus-NEE fraction (T-N). Each fraction was characterized for size, total protein content, and protein markers, then total RNA was extracted, and qPCR was run on 20 miRNAs. We report that the miRNA expression within NEE was different enough compared to T and T-N to justify the extra steps required to generate this fraction. We conclude that L1CAM immunocapture generates a unique fraction of EVs that consistently and robustly replicates a miRNA fingerprint which differentiates ALS patients from controls.
1. Introduction
Micro RNAs (miRNAs) are short (~22 nucleotides) non-coding nucleic acid sequences that are post-transcriptional regulators of gene expression. miRNAs bind to complementary pairing sequences on messenger RNA (mRNA) and both inhibit translation and destabilize mRNA, thereby influencing downstream gene expression [Citation1]. In some cases, miRNA has also been found to up-regulate translation [Citation2]. miRNAs are present in the blood bound to proteins such as lipoproteins (where they are known as circulating miRNA), and packed into small extracellular vesicles (EVs) some of which could be termed exosomes. Exosomes are small (30–150 nm) lipid bilayer particles released by almost all tissue types and found in many biofluids including blood, urine, saliva, breast milk and cerebrospinal fluid (CSF) [Citation3]. Exosomes carry a cargo composed of proteins, lipids, DNA and RNA, including miRNA. Whilst initially thought to be in vitro artefacts or vesicles that contain discarded proteins and nucleic acids, it is now known that exosomes are actively involved in cell-to-cell communication, signalling, and contribute to the potentiation of pathology, including beta-amyloid 42, characteristic of some progressive neurodegenerative diseases [Citation4].
The differential expression of miRNAs in disease has been the subject of intense research since it was discovered they can be diagnostic and prognostic of disease, particularly for some cancers [Citation5]. Being nucleic acids, miRNAs are potentially labile and prone to degradation. We have found, however, that EVs provide protection for miRNAs from degradation during storage, extraction, and processing [Citation6].
The presence of miRNA containing EVs in multiple bodily fluids such as saliva, blood, urine, and breast milk make acquiring them a much less invasive process, especially when compared to specialized extraction of CSF. This accessibility of EVs makes them an attractive option for researchers seeking to understand pathological changes in the central nervous system, in particular neurodegenerative diseases such as ALS, Alzheimer’s Disease (AD), and Parkinson’s Disease (PD). EVs secreted by neurons in the central nervous system potentially provide a ‘window into the brain’ in living patients.
There is a potential for miRNAs to be used to assist neurologists in the diagnosis of progressive neurodegenerative diseases, such as ALS, where evidence of progression is typically required for accurate diagnosis. In the case of ALS, it often takes between 10 and 16 months for a patient to receive a diagnosis [Citation7]. This compounds stress and expense for the patient and their families, especially in cases when a misdiagnosis occurs [Citation7]. A long lag time before receiving an accurate diagnosis is particularly devastating for ALS patients who typically survive only 2–5 years after diagnosis [Citation8].
Using EVs extracted from blood plasma, we have now thrice replicated a miRNA fingerprint that differentiates ALS patients from control subjectsCitation6,Citation9]. The first of these experiments [Citation9] was conducted using L1CAM/CD171-enriched EVs (the fraction we term NEE) extracted from plasma of ALS patients obtained from a Phase IIa clinical trial sponsored by our discovery lab (ClinicalTrials.gov identifier: NCT03580616). In the first experiment, n = 10 patient samples and n = 10 control individuals were initially processed, then the experiment was replicated with a new cohort of patients to result in a final number of n = 20 patients and n = 20 controls. NEE isolation was conducted at the Brain Chemistry Labs and RNA extraction and qPCR quantification was conducted at Qiagen Genomic Services. In these first two studies, we identified eight miRNAs that significantly differentiated the ALS patients from controls.
For the third replication of these findings [Citation6], we sourced 50 ALS patient plasma samples from the Centers for Disease Control (CDC) National ALS Biorepository and compared them to plasma sourced from 50 control subjects. In our laboratory, we conducted the EV extraction and L1CAM/CD171 enrichment as before, with the total RNA extraction, cDNA synthesis, and qPCR also conducted in-house. With this larger sample size, statistical significance held for five of the previously identified eight miRNAs. Three miRNAs did not quite reach statistical significance; however, the direction of regulation for all eight miRNA was the same as in the original study.
Of note is that in the first two experiments [Citation9], we used blood samples as part of the clinical trial we sponsored in which ALS was diagnosed by a single physician and in which the conditions under which the plasma samples were collected, stored, and dispatched were strictly controlled. In the third experiment [Citation6], samples were sourced from the CDC National ALS Biorepository where ALS diagnosis and sample collection involved multiple health care professionals. In all cases, plasma samples from control individuals were purchased from the same contract research organization. Even though these three experiments were conducted in different laboratories using ALS patient samples sourced with different protocols by different investigators, we found that the miRNA fingerprint that distinguishes ALS and control blood samples was consistently and reliably reproduced, suggesting the suitability of this procedure for application as a diagnostic test.
We hypothesized that the extra steps undertaken to generate neural-enriched extracellular vesicles (NEE) from total extracellular vesicles (T) contributed to the reproducibility of this protocol by generating an EV fraction that was unique. We have now sought to systematically test this hypothesis.
We generated and compared three EV fractions from control plasma samples: 1) total heterogenous EV fraction generated using polyethylene glycol (PEG), termed ‘total’T; 2) L1CAM-enriched EVs which we have termed ‘neural-enriched extracellular vesicles’ (NEE), and 3) the remaining fraction when NEE is removed which we have termed, ‘total minus NEE’ (T-N).
We here report that L1CAM immunocapture generates an EV fraction with exosome-like properties that are unique and different to T and T-N. We conclude that L1CAM concentration of total EVs to produce NEE generates a fraction that contributes to the robustness and reproducibility of our miRNA fingerprint.
2. Materials and methods
2.1. Human plasma
Human plasma samples from 24 de-identified individuals (Blood Derived) K2 EDTA 10 ml (cat. no. IPLASK2E10ML) were obtained from Innovative Research Inc., (Novi, MI, USA). Plasma samples were stored at −80°C and freeze/thawed only once prior to EV extraction.
2.2. RNA extraction
RNA Tini Spin columns (cat. no. EZC107N) were from Enzymax LLC (Lexington, KY, USA). QIAzol™ Lysis Reagent (cat. no. 79306), Buffer RPE (concentrate 55 mL; cat. no. 1018013), and Buffer RWT (80 mL; cat. no. 1067933) were from Qiagen (Hilden, Germany). Chloroform 99%, stabilized, molecular biology grade (cat. no. 0219400225) was from MP Biomedicals (Santa Ana, CA, USA). Ethyl alcohol, Pure 200 proof for molecular biology (cat. no. E7023; Lot no. SHBJ8384), was from Sigma-Aldrich (St Louis, MO, USA).
2.3. Extracellular vesicle extraction and L1CAM immunocapture
EV extraction and L1CAM enrichment has been described previously [Citation9]. Briefly, 500 µL plasma samples were thawed at 4°C and each treated with 15 µL thrombin to remove fibrinogen. EVs were precipitated using polyethylene glycol (SBI ExoQuick, cat. no. EXOQ5TM–1, System Biosciences, Palo Alto, CA, USA). This fraction represents the total EV extraction. Enrichment of NEEs was accomplished by the addition of mouse anti-human CD171 (L1 cell adhesion molecule, L1CAM, neural adhesion protein) mono-clonal antibody (cat. no. eBIO5G3, 5G3, 13-1719-82, Biotin, eBioscienceTM Antibodies, Thermo Fisher Scientific, Waltham, MA, USA) followed by the addition of streptavidin-agarose resin (cat. no. 53116, Pierce Streptavidin Plus UltraLink Resin, Thermo Fisher Scientific) [Citation10]. Centrifugation of this mixture created the supernatant fraction which represents the total heterogeneous EV population minus the EVs with L1CAM neural surface proteins, a fraction which we designate as T-N. The pellet containing the NEEs was resuspended in 0.1 M glycine-HCl (pH 2.5) and the solution strongly vortexed and centrifuged to release the EVs from the streptavidin beads. The supernatant was recovered and neutralized with 1 M TRIS–HCl pH 8.0 to create the NEE fraction.
2.4. Nano Tracking Analysis (NTA) of three EV fractions.
A Zetaview® Particle Metrix instrument (Ammersee, Germany) was used to determine size and concentration of particles. The instrument was calibrated prior to each session using polystyrene beads (100 nm) and solvent blanks were verified following method parameters. Prior to scanning, EV fractions were mixed with an equal volume (Total = 4 µL; T-N = 5 µL; NEE = 10 µL) of CellMask Green (CMG: Thermo Fisher Scientific cat. no. C37608) diluted to 100 µg/ml in Dulbecco’s Phosphate Buffered Saline (DPBS) w/o calcium chloride and magnesium chloride, sterile-filtered, (Sigma-Aldrich cat. no. D8537) and incubated in the dark for one hour at room temperature on a shaker. Two exceptions to the volumes and ratio were tested during the optimization of conditions and included in the final analyses due to the precious nature of the samples and because the data were found to fall within the range of other replicate samples (1 NEE sample used 12 µL EV +12 µL CMG and 1 Total EV sample used 4 µL EV +1 µL CMG). Each sample was diluted 1/50 for NEE and 1/200 for T and T-N fractions and examined in both scatter mode (SM) and fluorescence mode (FL) using ZetaView software version 8.05.12 SP1, Camera 0.713 mum/px, and the following settings for collection and analysis: Max area = 1000; Min area = 10; Min Brightness = 30; Laser λ = 488 nm; Filter λ = 500 nm, for FL mode; Temp 25–27°C; size distribution 2 cycles in SM, 1 cycle in FL, with 11 positions measured; Shutter 100 (200 in two samples); tracelength = 15; and Frame Rate = 30. Multiple scans were taken for each sample and mode. For analyses, an inclusion criteria rubric was used to choose three scans for each mode which consisted of finding the first three scans with a minimum of 10 frame positions passing software elimination criteria (due to Grubbs analyses or minimum traces) with sensitivity settings at 55 for SM and 80 for FL. When insufficient scans met these criteria, inclusion criteria were expanded in order to create a replicate size of n = 3 (See Supplemental Data 1 for full details). These included one replicate where only nine frames passed the elimination criteria and three samples where only eight frames were included. In the majority of cases, more than 100 particles were traced, but in three cases scans with 69, 70 and 82 traced particles were included. Although a scanning method with sensitivity of 55 for SM and 80 for FL was initially attempted for all samples, in four samples these sensitivity parameters were insufficient to produce good scans and the sensitivity criteria were altered accordingly (See Supplemental Data 1).
2.5. Total protein.
Total protein was measured using Invitrogen™ Qubit™ Protein Assay 12.5 µg/mL to 5 mg/mL (cat. no. Q33211, Thermo Fisher Scientific) on a Qubit™ 3 fluorometer (cat. no. Q33216, Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions.
2.6. Neuro Exo-Check™ slot blot.
An antibody array, SBI Exo-Check™ Antibody Array (Neuro) Standard Kit (System Biosciences, cat. no. EXORAY500A–8) was used to assess the presence of EV-associated markers. Briefly, 100 µg of protein, as determined by the Qubit™ assay was loaded for each of the three EV fractions and the slot-blot developed according to the manufacturer’s instructions. Blots were developed using Clarity Max™ Western ECL Substrate (cat. no. 1705062, Bio-Rad Laboratories Hercules, CA, USA) and imaged on the BioRad ChemiDoc using the signal accumulation method (6 images, 300 seconds). Post-processing of the tif image, which consisted only of adjusting the levels, the white point, cropping the image, adding labels for each protein and exporting as a jpeg, was conducted in Adobe Photoshop version 23.4.2.
RNA extraction: Extraction of total RNA retaining short RNA species has been described previously and was conducted on three fractions of EVs (50 µL T, 100 µL T-N and 100 µL NEE) [Citation11]. RNA Tini Spin columns from Enzymax LLC were used in place of Qiagen columns and the extraction conducted according to the instructions described in Qiagen RNeasy Midi Kit Part 2: RNA isolation (a detailed protocol is available at www.qiagen.com/HB-2630 ‘exoRNeasy Midi/Maxi Handbook’ beginning page 21, step 6). For a detailed modified protocol see [Citation11].
cDNA synthesis: cDNA was synthesized using the miRCURY LNA RT Kit (Qiagen cat. no. 339340) according to the manufacturer’s instructions and as described previously [Citation11]. Modifications included the addition of 1 µL of a spike-in control mix containing UniSp6 and C. elegans cel-miR-39-3p to each cDNA reaction to monitor cDNA synthesis efficiency. We used 4 µL of total RNA rather than 2 µL, as recommended by the manufacturer, since we have previously determined this is more likely to return lower Cq values [Citation11]. Each 10 µL cDNA synthesis reaction was conducted in duplicate and pooled to create a total of 20 µL cDNA. Prior to storage at −20°C, cDNA was aliquoted into 3 µL aliquots to avoid future multiple freeze/thaw cycles.
2.7. Real-time quantitative PCR of miRNA.
qPCR for all quality control (QC), target and reference gene miRNAs was conducted using Qiagen miRCURY LNA miRNA SYBR PCR Assays (cat. no. 339306 and see Supplemental Data 5-T1 for GeneGlobe IDs for QC assays and Supplemental Data 5-T2 for targets and reference genes) and the miRCURY LNA™ SYBR Green PCR Kit (Qiagen cat. no. 339347) according to the manufacturer’s instructions and as described previously [Citation11]. qPCR was conducted on the Bio-Rad CFX Opus 384 in 384-well plates according to the reaction conditions described in Supplemental Data 5-T3 and using the sample maximization method [Citation12]. Duplicate no-template controls (NTCs) to check for contamination and melt curves to check for non-specific amplification were included for each primer pair. Six individual samples were blinded and run twice as technical replicates for internal control.
2.8. Relative quantitation using 2 –∆∆Cq.
A NormFinder analysis confirmed the stability of genes used for normalization (146a-5p, 29b-3p, 126-5p), as used in the prior experiments [Citation6,Citation9]. The geometric mean was used for relative quantitation/normalization and a suitability check was performed following Vandesompele [Citation13]. Since the standard deviation of the ratio V ¾ was less than 0.15, no additional reference genes were needed [Citation6,Citation9,Citation13]. Gene fold changes were calculated using 2–∆∆Cq, with ∆∆Cq calculated as the normalized sample Cq value minus the mean of the normalized control sample Cqs. Fold regulation was defined as equal to fold change when greater than one and as negative one divided by fold change when fold change was less than one.
2.9. Mass Spectometry (MS) protein confirmation.
SDS-PAGE: 20 µg of total protein from each fraction (T, T-N, and NEE) was separated by SDS-PAGE using BioRad Mini-PROTEAN TGX gels (4–15%, 10-well, 30 µL, cat. no. 4561083) and TRIS-glycine running buffer. The gel was run at 20 V constant current until the sample concentrated in the well, then the current was increased to 100 V until the dye front reached the bottom of the gel (approximately 45 mins). The gel was placed in a glass petri dish and washed twice with Milli-Q water for 5 minutes. The gel was silver stained using the Thermo Scientific Pierce™ Silver Stain kit for Mass Spectrometry (cat. no. 24600) according to the manufacturer’s instructions and imaged using a BioRad ChemiDoc. The protein bands were excised with a new sterile scalpel and cut into three segments, and each segment was placed into three separate 2 mL Protein LoBind tubes (cat. no. 022431081, Eppendorf, Hamburg, Germany). The gel pieces were destained using the Thermo Scientific Pierce™ Silver Stain kit for Mass Spectrometry (cat. no. 24600) according to the manufacturer’s instructions. The gel pieces were then digested overnight with trypsin using the Pierce In-Gel Tryptic Digestion Kit (cat. no. 89871, Thermo Scientific) according to the manufacturer’s instructions. The following day, the gel pieces were de-salted using ZipTip® C18 Pipette Tips (cat. no. ZTC18S960, MilliporeSigma, Burlington, MA, USA), then resuspended in 20 µL 0.2% formic acid and analysed on an orbital trap mass spectrometer.
Three gel fractions for each EV sample (T, T-N, and NEE) from a single individual were analysed using a Thermo Scientific Orbitrap Exploris 480 and a Thermo Easy nLC 1200 liquid chromatography system with Xcalibur software, a Thermo Nano Spray Flex source, and a Waters nano Ease M/Z HSS C18 T3 column (1.8 μM, 100 Å, 15 cm × 75 μm, part number 186008816). Three different methods were explored with varied gradients and MS resolution parameters (full method details available in Supplemental Data 2). All methods used Water, Optima™ LC/MS Grade, Fisher Chemical™ (Fisher Scientific cat. no. W6–1) containing 0.1% Pierce™ Formic Acid, LC-MS Grade (Thermo Fisher Scientific cat. no. 28905) as solvent A and 80% (v/v) acetonitrile in water (Acetonitrile, Optima™ LC/MS Grade, Fisher Chemical™ cat. no. A955–1 in Fisher Optima water) with the addition of 0.1% formic acid as solvent B. The flow rate was set to 300 nL/min and 1 μL sample was loaded. Full MS scans were 600–6000 m/z using a spray voltage of 1.4 kV, a capillary temperature of 250°C, and an S-lens RF set to 125% in positive mode. Data-dependent MS/MS (ddMS2) was employed with a default charge state of 2 and an isolation window of 2 m/z in fixed collision energy mode. Between samples and gel fractions, a 112 min wash method was run, and a water blank to check for carry-over before continuing with the next sample injection.
Raw data files from the orbital trap mass spectrometer were analysed using Thermo Proteome Discoverer 1.4.0.288 (Thermo Scientific). Three full scans of each gel fraction for each EV fraction (total of nine scans) were combined. Individual proteins were analysed against specific FASTA files obtained from the NIH National Library of Medicine, National Center for Biotechnology Information website (https://www.ncbi.nlm.nih.gov/ipg/AAB33925.2, see Supplemental Data 2 for full FASTA file information) as well as the entire human Swiss UniProt 9606. Maximum delta Cn was set to 0.05, precursor mass tolerance and fragment mass tolerance were set to 10 ppm and 0.02 Da respectively (See Supplemental Data 2 for full Proteome Discoverer method details). Blank gel pieces were cut from outside loaded lanes and analysed as described.
2.10. Statistical analyses.
Twenty miRNAs were examined from twenty-four individual plasma samples, each with three EV fractions (T, T-N, and NEE). We compared the median gene fold expression values for each of the twenty miRNAs in these twenty-four individual plasma samples to evaluate two alternative hypotheses:
Ho: the median gene fold expression of all three fractions (T, T-N, NEE) are the same; and
H1: the median gene fold expression of all three fractions (T, T-N, NEE) are not the same. Friedman’s non-parametric statistical test was chosen to evaluate these hypotheses.
3. Results
We characterized EVs from the three distinct isolated fractions: Total (T), Total-Neural (T-N), and Neural-enriched (NEE). These fractions differed in total protein, size, concentration, protein identification, and in miRNA cargo.
3.1. Total protein measure.
In accordance with the guidelines as described in minimal information for studies of extracellular vesicles 2018 (MISEV2018) [Citation3] we measured total protein amount for each EV fraction generated from four different patient samples (n = 4) (). Total protein was calculated from a standard curve generated simultaneously () using the Invitrogen™ Qubit™ Protein Assay on a QuBit™ 3 fluorimeter. As expected, and since NEE constitutes a small part of the total EV fraction, we report protein concentrations were lower for NEE when compared to T and T-N.
Table 1. Mean total protein concentrations of three EV fractions generated from four individual plasma samples. Total protein was measured using the Invitrogen™ Qubit™ Protein Assay on a Qubit™ 3 fluorimeter in accordance with the manufacturer’s instructions. A standard curve was generated simultaneously using 2 µl of manufacturer’s supplied standards in triplicate and RFUs were converted to µg/µl protein (). Both T and T-N fractions were diluted eight-fold with 2 µl analysed to fit on the standard curve. NEE was read neat with 2 µl of sample measured ().
3.2. Nano tracking analysis (NTA) of three EV fractions.
Nano tracking analysis was conducted using the Zetaview® Particle Metrix instrument for all three fractions of EVs using both fluorescence and scatter modes (, Supplemental Data 1). Since the samples were frozen and diluted in DPBS prior to analysis, we expected some degree of clumping in all fractions. The size distribution of particles within these fractions generally falls within the range of exosomes which are characterized as being 30–150 nm [Citation14]; however, aggregates were also detected. The T-N fraction consistently had the smallest particles and the highest concentration of particles. The fluorescence mode marks intact lipid membranes and identified the greatest variability among EV fractions.
Table 2. Nano-tracking analyses (NTA) of extracellular vesicle (EV) fractions: Total, total minus neural enriched, and neural enriched EV (NEE). Concentration was adjusted to reflect equivalent calculations based on particles/ml of original 500 μl plasma sample (see supplemental data 1 for raw NTA data). Data represents mean of the EV fractions from five plasma samples ± standard deviation. Size is measured in nm, while concentration is measure in (µg/µl).
3.3. Neuro Exo-Check™ screening of three EV fractions.
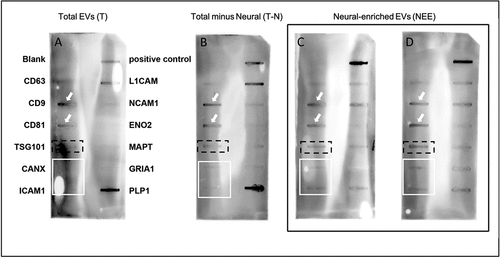
In order to characterize the EVs, we screened the three fractions for proteins using the Exo-Check™ Exosome Antibody Array (Neuro) slot-blots. We report the presence of the tetraspanins CD9, and CD81 in all three fractions (, see white arrows) where Panel A is T, Panel B is T-N, and Panels C and D are NEE. Another tetraspanin, CD63, commonly used for EV characterization [Citation3] was barely visible in any of the membranes. The ESCRT-I subunit, TSG101, which is a cytosolic protein commonly recovered in EV preparations, was present in both NEE fractions (, Panels C and D) with a barely detectable presence in T (, Panel A) and T-N (, Panel B) (dashed black lines). Calnexin (CANX) and ICAM1 were not visible in T or T-N, but slightly visible in NEE, as indicated by the white rectangles. L1CAM, the protein captured during purification/concentration, was present in all three fractions but was abundant in the T-N fraction. The neuronal markers NCAM1, ENO2, MAPT, and PLP1 were present in the NEE fraction, with variation in density compared to the T-N, and T.
Figure 1. Neuro Exo-Check™ of all three EV fractions demonstrated the presence of proteins consistent with exosomes. Panel A, Total (T); Panel B, Total minus neural (T-N): Panels C and D, neural-enriched extracellular vesicles (NEE). Panel A, B and C represent fractions prepared from the same plasma sample; Panel D is NEE prepared from a different sample. Each “slot” represents a different protein or control as indicated: blank (negative control for background); CD63, LAMP-3, tetraspanin 30; CD9, tetraspanin 29; CD81, tetraspanin 28; TSG101, VPS23, tumor susceptibility gene 101; CANX, calnexin; ICAM1, CD54, intracellular adhesion molecule 1; PC, positive control for HRP detection; L1CAM, CD171, L1 transmembrane protein; NCAM1, CD56, neural cell adhesion molecule; ENO2, enolase 2; MAPT, total tau protein; GRIA1, glutamate receptor 1; PLP1, proteolipid protein 1. 100 µg of protein (as determined using the Qubit™ assay) was loaded on each membrane and slot-blot developed according to the manufacturer’s instructions. Order of protein “slots” as indicated in Panel A is the same for all blots.
3.4. Mass spectrometry protein data
Data from orbital trap mass spectrometry identified proteins consistent with EVs and components of non-EV co-isolated structures (Supplemental Data 3) as identified in Théry et al. [Citation3]. The tetraspanin CD63 was found in NEE fractions while CD81 was found in the T-N. No tetraspanins were found in either the blank or the total fraction. HLA-A and Flotillin1 were found in all EV fractions. Albumin and apolipoproteins were apparent in all EV fractions and albumin was noted in the blank piece of gel suggesting some diffusion of proteins in, or on, the gel itself. The integrins, ADAM10, LAMP1, LAMP2 were not found in the NEE, but were present in both the T and T-N fractions.
The presence or absence of the various targeted proteins varied among fractions. The results generally support the slot blot study in that EV markers are present, but differences in specific protein identification were noted and require further investigation.
3.5. Real-time qPCR quality control
Prior to running qPCR of 20 target miRNAs, we conducted quality control (QC) to ensure all samples were suitable for downstream analysis.
QC for RNA extraction spike-ins, UniSp2, 4, and 5. We conducted qPCR of spike-ins where a 1 µL mix of UniSp2, 4, and 5 was added to each 700 µL Qiazol lysis reagent and extraction conducted as normal. qPCR of the spike-ins was conducted to check for RNA extraction efficiency across all samples and the raw Cqs compared. QC of sample 70 revealed inefficient RNA extraction [where mean raw Cq for UniSp2 for samples S61–72 (excluding S70) was 16.02 and for S70 was 17.28, mean raw Cq for UniSp4 for samples S61–72 (excluding S70) was 23.79 and for S70 was 31.58, and mean raw Cq for UniSp5 for S70 was not detected]. As a result, another aliquot was obtained, and the RNA extraction was repeated. The repeated sample (S73) had values that fell within an acceptable range for QC raw Cqs. Results for all samples for RNA extraction spike-ins QC, including raw Cqs, amplification curves and melt curves can be found in Figures S1-S63A in Supplemental Data 5.
QC for cDNA synthesis efficiency spike-ins, UniSp6, and cel-miR-39-3p. A mixture of 1 µL of cDNA synthesis control spike-ins containing UniSp6 and cel-miR-39-3p was added to each cDNA synthesis reaction to check for reverse transcription efficiency. qPCR was run on all samples including S73 which was a repeat of S70 (see above) and all samples returned acceptable raw Cqs. The raw Cqs, no-template controls, amplification and melt curve results for all samples can be found in Figures S1-S63A in Supplemental Data 5. All samples passed the spike-in QC for RNA extraction and cDNA synthesis.
QC for RNA sample signal. Since traditional methods for RNA quantitation (virtual gel electrophoresis such as Bioanalyzer) are not optimized for short RNA species such as miRNA, we measured the miRNA signal in each sample using qPCR of six miRNA which we have previously used to determine sample signal: hsa-miR-142-3p, hsa-miR-451a, hsa-miR-23a-3p, hsa-miR-30c-5p, hsa-miR-103a-3p, and hsa-miR19-5p [Citation15]. All samples passed QC for sufficient miRNA to proceed with downstream qPCR (see Figures S1-S63A in Supplemental Data 5).
Haemolysis check. During collection and processing of blood samples, erythrocytes are prone to lysis, releasing miRNA that has the potential to create artifactually high signals in downstream qPCR. To determine if samples had undergone haemolysis, we calculated ΔCq (hsa-miR-23a – hsa-miR-451a) [Citation16]. Any samples that returned a value of 7 or greater may have undergone haemolysis and were carefully considered before use in downstream analysis. We report four samples returned ∆Cq greater than seven (S34–36 and S38) but we determined these values were insufficient to eliminate them from downstream qPCR analysis (see Supplemental Data 5-T4).
Technical Replicates. Cq scores calculated for 18 miRNAs from six samples, each blinded and run twice as technical replicates, were compared (Supplemental Data 4). Differences between replicates were generally small and with few exceptions. Low concentrations of miRNA, represented by high Cq values, had larger variations between replicates suggesting sampling error due to low copy numbers in the initial sample.
3.6. qPCR differential expression of miRNAs in three EV fractions
To determine if the three EV fractions, T, T-N, and NEE were sufficiently different from each other, we ran qPCR targeting 20 miRNAs (). In eight of the 20 miRNAs, the null hypothesis was rejected suggesting that PEG EV precipitation along with L1CAM immunoprecipitation steps did change the gene fold expression values in individual plasma samples. Three of the eight miRNA found to be differentially expressed between ALS patients and controls [Citation9] were found to be differentially expressed between the different EV fractions.
Table 3. Twenty miRNA were examined from 24 individual plasma samples, each with three fractions: Total (T), Total minus Neural (T-N), and Neural Enriched EV (NEE). Gene fold expression values were compared with two hypotheses: Ho: the median gene fold expression of all three fractions (T, T-N, NEE) were the same; and H1: the median gene fold expression of all three fractions (T, T-N, NEE) were different. We evaluated these alternative hypotheses using a non-parametric Friedman’s test with the critical value of p ≤ 0.05. * represents 8 miRNA identified as potential ALS biomarker in [Citation9]; ^ represents the 5 miRNA confirmed in a broad sample of ALS patients [Citation6]. NS = not significant.
4. Discussion
miRNA biomarker data sometimes suffers from a lack of reproducibility [Citation17–20]. This has the potential to hinder the introduction of diagnostic and prognostic miRNA assays into clinical settings. We have now replicated a miRNA ALS biomarker fingerprint thrice using EVs isolated from human plasma and immunocaptured with the adhesion molecule, L1CAM, to generate a fraction we term NEE. We hypothesized that the extra steps required to generate this fraction from the total heterogenous EVs contributes to the reproducibility of our assay, but this hypothesis needed rigorous testing. We here report that the NEE fraction used in the miRNA ALS biomarker has different profiles as represented by size, protein content, protein markers, and miRNA cargo, indicating that L1CAM immunocapture generated a population of EVs with exosome-like properties that was consistent with a reproducible diagnostic miRNA fingerprint.
Of the twenty miRNAs we measured using qPCR, a total of eight were significantly different across the three EV fractions (). This included three of the original miRNAs which we previously described as differentiating ALS patient samples from controls [Citation6,Citation9]. However, not all miRNA differed in this study between the three fractions suggesting that it may be possible to produce an ALS biomarker using rigorous laboratory methods that require fewer processing steps.
In order to characterize the three fractions of EVs, we isolated from control plasma, we measured total protein, size, protein identification, and qPCR of miRNA with an eye towards fulfilling the criteria suggested in the most recent minimal information for studies of extracellular vesicles [Citation3]. The guidelines recommend that general characterization of EVs should encompass at least three positive markers of EVs, including at least one transmembrane/lipid bound protein or a cytosolic protein, and at least one negative protein marker (simply meaning a protein not expected to be present in EVs/endosomes).
Using the Neuro Exo-Check™ Antibody arrays, all three EV fractions were found to be positive for the tetraspanins and general markers of EVs, CD9 and CD81 (). We also report the presence of a cytosolic protein recovered in EV preparations, TSG101, but note that TSG101 was not detected in the preliminary mass spectrometry analysis from a single donor sample. Slot blots that were run using three EV fractions extracted from the same plasma sample clearly show an enrichment of the neural-markers, NCAM1, MAPT, and PLP1 in two different NEE fractions (), however, using mass spectrometry, PLP1 was not found in any fraction and NCAM1, and MAPT were only identified in the total EV fraction. We also note that the slot-blot data come from a commercial SBI kit that may have its own inherent complications. It is possible that the antibodies used in these kits have insufficient selectivity. In addition, the positive controls did not always work despite adequate protein loading and the appearance of positive signals in other lanes. At this point, the combined data from these two experiments suggest that the EV samples contain exosome markers; however, a more thorough analysis and protein identification is warranted.
L1CAM was originally identified on primary cortical neurons [Citation21–24], and later shown to be specifically found on EVs collected from mature cortical neurons within the 4–6 density gradient fraction [Citation25]. However, the use of L1CAM as a marker for neural-derived EVs has been questioned because although it is highly expressed in the brain, it is also expressed in other tissues including the tibial nerve, and the gastrointestinal tract (https://gtexportal.org/home/gene/L1CAM). A recent paper by Norman et al. [Citation26] reported L1CAM was not associated with EVs in plasma or CSF but was predominantly present in a soluble form [Citation26]. Cleavage of the extracellular domain of L1CAM by the metalloproteinases ADAM10 and ADAM17 results in the release of a 200 kDa soluble ectodomain leaving behind a 32 kDa transmembrane stub that is retained in the plasma membrane of EVs [Citation27]. The L1CAM antibody used in this study and by other researchers (5G3) is raised against the L1 ectodomain meaning it can bind to this section of L1CAM whether it is in the transmembrane form or cleaved. Thus, the suitability of L1CAM as a marker for neural-enriched EVs is under question and alternative markers are being sought. As such, we do not classify our EV preparations as neural-derived, but rather we use the term ‘neural-enriched EVs’ since they do display some neuronal markers, but their precise biogenetic origin has not been established. Through mass spectrometry, we identified L1CAM in all exosome fractions, and its presence in the blank suggests some degree of leaching within the gel membrane itself.
Similarly, although the isolation procedures used to create the ALS miRNA fingerprint contains small particles bearing exosome markers, it could contain circulating miRNA possibly bound to apolipoproteins, and miRNA from microvesicles, apoptotic bodies, and aggregates. We note that the results presented herein are from control subjects without known neurological illness. Data from ALS patients may have an increase in neural exosome release and apoptotic bodies as a result of the disease process and this may also influence the ability to differentiate ALS from controls using these methods. These results suggest that L1CAM immunocapture increases the purity of the EV sample and thereby adds to the robustness of this assay rendering the method of blood collection, plasma extraction and storage prior to EV extraction less critical. Although the EV yield is reduced with this method, quantification of miRNA from limited amounts of starting material is possible due to optimized total RNA extraction and cDNA synthesis methods to increase the sensitivity of our assay [Citation11].
Further analyses of the predictive value of these miRNAs compared to ALS-like diseases is a necessary next step. In addition, further analyses of whether a subset of these miRNAs could be used to diagnose ALS using total EVs should also be considered.
Highlights
Extracellular vesicles isolated from human plasma were purified using L1CAM.
Fractionation of extracellular vesicles through immunoprecipitation increases replicability of miRNA signatures.
The resultant isolated fractions have unique profiles including size, protein, and miRNA.
Robust methods have led to the development of a blood diagnostic test for ALS which consistently replicates a miRNA fingerprint.
Supplemental Material
Download Zip (11.8 MB)Disclosure statement
The not-for-profit research institute Brain Chemistry Labs has applied for a patent on the use of this biomarker.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2198805.
Additional information
Funding
References
- Fabian MR, Sundermeier TR, Sonenberg N. Understanding how miRnas post-transcriptionally regulate gene expression. Prog Mol Subcell Biol. 2010;50:1–20.
- Vasudevan S, Tong Y, Steitz JA. Cell cycle control of microRNA-mediated translation regulation. Cell Cycle Georget Tex. 2008;7:1545–1549.
- Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. DOI:10.1080/20013078.2018.1535750
- Sardar Sinha M, Ansell-Schultz A, Civitelli L, et al. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol (Berl). 2018;136(1):41–56. DOI:10.1007/s00401-018-1868-1
- Wang M, Yu F, Ding H, et al. Emerging function and clinical values of exosomal MicroRNAs in cancer. Mol Ther Nucleic Acids. 2019;16:791–804.
- Banack SA, Dunlop RA, Stommel EW, et al. miRNA extracted from extracellular vesicles is a robust biomarker of amyotrophic lateral sclerosis. J Neurol Sci. 2022;442:120396.
- Richards D, Morren JA, Pioro EP. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J Neurol Sci. 2020;417:117054.
- Mehta P, Antao V, Kaye W, et al. Prevalence of amyotrophic lateral sclerosis - United States, 2010-2011. MMWR. 2014;Suppl 63(7):1–14.
- Banack SA, Dunlop RA, Cox PA. An miRNA fingerprint using neural-enriched extracellular vesicles from blood plasma: towards a biomarker for amyotrophic lateral sclerosis/motor neuron disease. Open Biol. 2020;10(6). DOI:10.1098/rsob.200116
- Mustapic M, Eitan E, Werner JK Jr, et al. Plasma Extracellular Vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017;11:278.
- Dunlop RA, Banack SA, Cox PA. A comparison of the efficiency of RNA extraction from extracellular vesicles using the Qiagen RNeasy MinElute versus Enzymax LLC RNA Tini Spin columns and qPCR of miRNA. Biol Methods Protoc. 2021;6(1):1–9.
- Hellemans J, Mortier G, De Paepe A, et al. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8(2):R19. DOI:10.1186/gb-2007-8-2-r19
- Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. DOI:10.1186/gb-2002-3-7-research0034
- Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88(1):487–514.
- Matias-Garcia PR, Wilson R, Mussack V, et al. Impact of long-term storage and freeze-thawing on eight circulating microRnas in plasma samples. PLoS ONE. 2020;15:e0227648.
- Blondal T, Jensby Nielsen S, Baker A, et al. Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods. 2013;59:S1–6.
- Basso D, Padoan A, Laufer T, et al. Relevance of pre-analytical blood management on the emerging cardiovascular protein biomarkers TWEAK and HMGB1 and on miRNA serum and plasma profiling. Clin Biochem. 2017;50(4–5):186–193. DOI:10.1016/j.clinbiochem.2016.11.005
- Chen Y, Gelfond JA, McManus LM, et al. Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genomics. 2009;10:407.
- Godoy PM, Barczak AJ, DeHoff P, et al. Comparison of reproducibility, accuracy, sensitivity, and specificity of miRNA quantification platforms. Cell Rep. 2019;29(12):4212–4222.e5. DOI:10.1016/j.celrep.2019.11.078
- Witwer KW, Halushka MK. Toward the promise of microRnas – Enhancing reproducibility and rigor in microRNA research. RNA Biol. 2016;13(11):1103–1116.
- Faissner A, Kruse J, Nieke J, et al. Expression of neural cell adhesion molecule L1 during development, in neurological mutants and in the peripheral nervous system. Brain Res. 1984;15(1):69–82.
- Fauré J, Lachenal G, Court M, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006;31(4):642–648. DOI:10.1016/j.mcn.2005.12.003
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10(1):19–26.
- Rathjen FG, Schachner M. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. Embo J. 1984;3(1):1–10.
- Lachenal G, Pernet-Gallay K, Chivet M, et al. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci. 2011;46(2):409–418. DOI:10.1016/j.mcn.2010.11.004
- Norman M, Ter-Ovanesyan D, Trieu W, et al. L1CAM is not associated with extracellular vesicles in human cerebrospinal fluid or plasma. Nat Methods. 2021;18(6):631–634. DOI:10.1038/s41592-021-01174-8
- Kiefel H, Bondong S, Hazin J, et al. L1cam. Cell Adhes Migr. 2012;6(4):374–384. DOI:10.4161/cam.20832