
ABSTRACT
The tricarboxylic acid (TCA) cycle is a central route for generating cellular energy and precursors for biosynthetic pathways. Emerging evidences have shown that the aberrations of metabolic enzymes which affect the integrity of TCA cycle are implicated in various tumour pathological processes. Interestingly, several TCA enzymes exhibit the characteristics of RNA binding properties, and their long non-coding RNA (lncRNA) partners play critical regulatory roles in regulating the function of TCA cycle and tumour progression. In this review, we will discuss the functional roles of RNA binding proteins and their lncRNA partners in TCA cycle, with emphasis placed on the cancer progression. A further understanding of RNA binding proteins and their lncRNA partners in TCA cycle, as well as their molecular mechanisms in oncogenesis, will aid in developing novel layers of metabolic targets for cancer therapy in the near future.
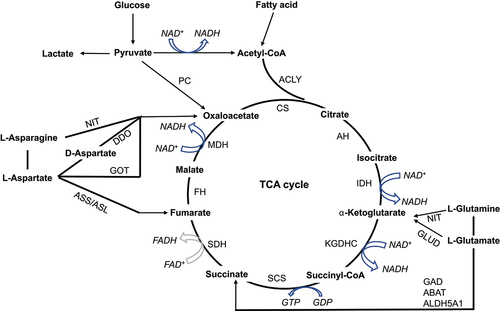
Abbreviations: CS: citrate synthase. AH: aconitase, including ACO1, and ACO2. IDH: isocitrate dehydrogenase, including IDH1, IDH2, and IDH3. KGDHC: α-ketoglutarate dehydrogenase complex, including OGDH, DLD, and DLST. SCS: succinyl-CoA synthase, including SUCLG1, SUCLG2, and SUCLA2. SDH: succinate dehydrogenase, including SDHA, SDHB, SDHC, and SDHD. FH: fumarate hydratase. MDH: malate dehydrogenase, including MDH1 and MDH2. PC: pyruvate carboxylase. ACLY: ATP Citrate Lyase. NIT: nitrilase. GAD: glutamate decarboxylase. ABAT: 4-aminobutyrate aminotransferase. ALDH5A1: aldehyde dehydrogenase 5 family member A1. ASS: argininosuccinate synthase. ASL: adenylosuccinate synthase. DDO: D-aspartate oxidase. GOT: glutamic-oxaloacetic transaminase. GLUD: glutamate dehydrogenase. HK: hexokinase. PK: pyruvate kinase. LDH: lactate dehydrogenase. PDK: pyruvate dehydrogenase kinase. PDH: pyruvate dehydrogenase complex. PHD: prolyl hydroxylase domain protein.
1. Introduction
Aberrant metabolism is a major hallmark of cancer [Citation1–3]. In the 1920s, Otto Warburg found that the glucose consumption of tumour tissues was remarkably higher than that of normal tissues [Citation4]. The increased glucose uptake and preferential utilization of glucose through aerobic glycolysis have been validated across diverse tumor types have been linked to an unfavourable prognosis [Citation5–7]. These early findings not only revealed the clinical relevance of aberrant tumour metabolism but also laid the groundwork for the application of tumour detection and imaging via positron emission tomography with radiolabeled glucose analogs [Citation8]. Recently, new insights are emerging into the involvement of TCA cycle in tumour metabolic reprogramming and cancer progression [Citation9]. It has been reported that tumour cells are capable of dissociating glycolysis from the TCA cycle, by utilizing alternative energy fuel sources (e.g. glutamine and fatty acids) in order to fulfill their elevated metabolic demands [Citation6,Citation10]. Importantly, extensive research has demonstrated that a variety of oncogenes and tumour suppressors play a vital role in regulating the uptake and breakdown of energy sources during the TCA cycle [Citation10–15]. The mutations and/or dysregulations of multiple TCA cycle enzymes, including fumarate hydratase (FH), isocitrate dehydrogenase (IDH), ketoglutarate dehydrogenase complex (KGDHC), and succinate dehydrogenase (SDH) etc., are commonly harboured in human cancers [Citation16–19]. Taken together, these studies have presented conclusive evidence to establish the significance of the TCA cycle in cancer metabolism and tumorigenesis.
Interestingly, several TCA cycle enzymes have the RNA binding capability, and are thus classified as RNA binding proteins (RBPs) [Citation14,Citation20]. RBPs have multiple roles in human cancer [Citation21]. The malleability of their RNA-binding domains, in combination with their structural flexibility, allows RBPs to govern the metabolism of a broad spectrum of transcripts. Meanwhile, they establish highly dynamic interactions with long non-coding RNAs (lncRNAs), as well as with coding RNAs and other proteins [Citation22]. LncRNAs constitute a large part of the human functional non-coding transcribed genome [Citation23]. It is well recognized that these large classes of RNA transcripts have been implicated in many pathological processes, especially in cancer metabolism and development [Citation12,Citation14,Citation24]. Cancer development has also been causally linked to disruptions in RBP-lncRNA network activity [Citation14,Citation25]. For instance, disrupting the interaction between lncRNA GAS5 and malate dehydrogenase 2 (MDH2) could affect FH – MDH2–citrate synthase (CS) complex formation and breast cancer tumorigenesis [Citation14]. Blocking lncRNA IDH1-AS1 dramatically reduced IDH1’s enzymatic activities and inhibited Hela cell proliferation [Citation25]. Hence, unravelling the complex web of interactions between RBPs and their associated lncRNAs in the TCA cycle may enhance our comprehension of tumour biology and reveal novel targets for cancer treatment. We will depict an overview of the roles played by RBPs and their lncRNA partners in TCA cycle and tumour progression.
2. Overview of TCA cycle in cancer
The typical hallmark of cancer is the uncontrolled growth and division of tumour cells, which requires a boost in both energy generation and macromolecular synthesis. In response to heightened metabolic demands, cancerous cells often undergo aerobic glycolysis, also referred to as the Warburg effect, as a means of generating energy, building blocks, and NADPH to support their survival and rapid proliferation while maintaining redox homoeostasis [Citation26]. Until recently, the role of the TCA cycle in cancer metabolism and tumorigenesis has been largely disregarded, probably due to the prevailing assumption that impaired mitochondrial activity is a ubiquitous hallmark of the Warburg effect and cancer progression. Based on 13C-labelled metabolomic analysis, recent research has challenged the notion that cancers have impaired mitochondrial activity, revealing that mitochondrial function is often fully intact in these diseases. More importantly, cancer cells are capable of decoupling glycolysis from the TCA cycle, by utilizing alternative energy sources (e.g. asparagine, glutamine and fatty acids) to satisfy their increased metabolic demands [Citation6,Citation10,Citation27,Citation28] (). Despite exhibiting a heightened demand for glucose, cancer cells differ from normal cells in how they metabolize glucose. While most cellular glucose in normal cells enters the TCA cycle as pyruvate, cancer cells often favour lactate production over acetyl-CoA generation even under normoxic condition [Citation29–32] ().
Figure 1. Biochemical reactions that drive the TCA cycle. By integrating the KEGG metabolic pathways (https://www.genome.jp/kegg/pathway.html#metabolism, hsa00010, hsa00020, hsa00071, and hsa00250), the biochemical reactions in TCA cycle were summarized. The TCA cycle is comprised of 8 steps, three of which are irreversible, including CS-driven the generation of citrate from oxaloacetate and acetyl-CoA; IDH-driven the conversion of isocitrate to α-KG; and KGDHC-driven the formation of succinyl-CoA from α-KG (hsa00020). Acetyl-CoA derived from pyruvate, which is the end product of glycolysis, is the typical input for the TCA cycle (hsa00010). In addition, outside sources, including the production of acetyl-CoA from β-oxidation of fatty acids (hsa00071), the production of fumarate, succinate, and α-ketoglutarate from protein catabolism (hsa00250), and the production of oxaloacetate from pyruvate (hsa00010), can also provide the intermediates in the TCA cycle.
Hypoxia is a hallmark of tumour microenvironment present in the majority of solid tumours [Citation33,Citation34]. Tumour cells have developed various adaptive mechanisms under hypoxic conditions to sustain their growth and outcompete normal cells. A crucial player in the hypoxia response of tumours is hypoxia-inducible factor alpha (HIFα) [Citation33]. Under low-oxygen conditions, HIFα proteins accumulate and migrate to the nucleus where they bind to the hypoxia response element (HRE) located in the 5’ regulatory regions of their downstream target genes, thus activating a diverse array of gene transcription events, such as the transcriptional regulation of glucose transporters (SLC2A1 and SLC2A3), glycolytic enzymes (e.g. hexokinase (HK), lactate dehydrogenase (LDH), and pyruvate kinase (PK)), and pyruvate dehydrogenase kinase (PDK) [Citation30] (). These enzymes, on the one hand, promote glycolysis and lactate production; on the other hand, suppress the activity of the pyruvate dehydrogenase complex (PDH), which is responsible for the conversion of pyruvate to acetyl-CoA. Hence, the activation of HIF coordinates a metabolic programme that enhances the breakdown of glucose via aerobic glycolysis, resulting in a diversion of glucose away from the TCA cycle (). As a means of compensating for the reduced glucose supply to the TCA cycle, tumour cells activated by HIF often augment their utilization of glutamine, which primarily feeds the TCA cycle through α-ketoglutarate (α-KG), thereby facilitating reductive carboxylation [Citation35,Citation36].
Figure 2. Interplay among TCA cycle, glycolysis, and tumorigenic signalling. Oncogenic mutations of IDH lead to the production of 2-HG. Deficiencies of SDH and FH in cancer cells elevate succinate and fumarate, respectively. Accumulation of 2-HG, succinate, and fumarate inhibits the degradation of HIF-1α, and results in HIF-1α’s nuclear translocation, which brings about the interaction of HIF-1α and the hypoxia response element (HRE) and thus promotes a plethora of tumour-related genes. The downstream genes of HIF-1α include glycolytic enzymes (e.g. HK, PK, LDH) and PDK, which promote the catabolism of glucose through aerobic glycolysis and shift glucose away from the TCA cycle. In addition, the accumulation of fumarate also stabilizes NRF2 through either altering cellular redox state via succination of GSH or a direct succination of its negative regulator KEAP1. Also, NRF2 can translocate to the nucleus and function as a transcriptional factor through interacting with the antioxidant response element (ARE) upon the promoter of multiple tumour-related genes.
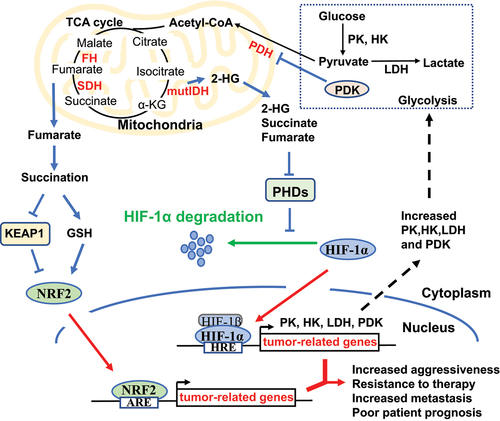
In addition to the HIF-regulated TCA cycle, mutations in the TCA cycle enzymes also affect the accumulation of HIF. For example, mutated IDH develops a neomorphic catalytic activity, enabling it to convert α-KG into the 2-hydroxyglutarate (2-HG) using NADPH as a cofactor [Citation37]. The accumulation of 2-HG inhibits the prolyl hydroxylase domain proteins (PHDs), which play critical roles in tumour metabolic adaptation, angiogenesis, proliferation, and metastasis via negatively regulating HIF-1α [Citation38]. However, pseudohypoxia, a state characterized by constitutive activation of HIF-1α under normoxic conditions, can manifest in certain types of cancer such as those with IDH mutations [Citation39]. Given the structural resemblance of succinate and fumarate to α-KG, their accumulation can trigger a pseudohypoxic reaction in tumours harbouring SDH and FH mutations [Citation40–42]. Thus, the establishment of a pseudohypoxic state driven by HIF, accompanied by tumour behavioural alterations, appears to be a common characteristic of various TCA cycle enzyme deficiencies and metabolic alterations in cancer (). Apart from affecting HIF-driven alterations, fumarate accumulation may also contribute to tumorigenesis by supporting the stabilization of the transcription factor nuclear factor erythroid 2-like 2 (NFE2L2, or NRF2), a key regulator of the cellular antioxidant defence system. Genetic targeting of NRF2 can govern a cellular detoxification and antioxidant defence programme and impair the K-Ras-induced lung and pancreatic tumorigenesis [Citation43,Citation44] (). Interestingly, inhibition of NRF2 can inactivate HIF-dependent lncRNA NLUCAT1, leading to higher reactive oxygen species (ROS), increased susceptibility to cisplatin-induced apoptosis, and lower proliferation rates of lung adenocarcinoma cells [Citation45]. In this case, the tumour metabolic lncRNA regulator NLUCAT1 is regulated by both NRF2 and HIFα. In fact, as master transcriptional regulators of energy metabolism in cancer, the activities of NRF2 and HIFα are closely correlated with the abundance of lncRNAs [Citation46–48]. Additionally, it has been widely demonstrated that lncRNAs also play critical roles in regulating metabolic processes, including TCA cycle [Citation14,Citation20,Citation25,Citation49–51].
3. Roles of lncRNAs in regulating TCA cycle
LncRNAs play regulatory roles at a plethora of cellular processes, most of which require the interaction with RBPs [Citation52]. RBPs are well known for their key roles in post-transcriptional events. During the structural and functional diversity, RBPs are essential for regulating RNA modification, localization, splicing, transport, translation, degradation, and stability [Citation21,Citation53]. Dysfunction of RBPs can lead to perturbations in RNA metabolism, resulting in wide alterations in the cell’s transcriptome and proteome. These changes can have significant implications for cell behaviour, including growth, proliferation, invasion, and apoptosis. The majority of RBPs that have been investigated in relation to cancer are considered canonical RBPs, which possess canonical RNA-binding domains (RBDs) such as RNA-recognition motifs (RRMs) [Citation54]. In addition to canonical RNA-binding proteins, many RBPs without canonical RBDs but endowed with RNA-binding activity and have well- defined cellular functions were also identified, including some TCA enzymes [Citation55].
TCA cycle occurs in the mitochondrial matrix and involves a sequence of biochemical reactions that are catalysed by several enzymes, including ATP Citrate Lyase (ACLY), CS, aconitase (AH), IDH, KGDHC, succinyl-CoA synthase (SCS), SDH, FH, and MDH (). An early example that highlighted the role of a metabolic enzyme as an RBP in post-transcriptional gene regulation was the discovery of ACO1, also referred to as iron regulatory protein 1 (IRP1). IRP1 could interact with mRNA to regulate the iron content in cells [Citation56]. Other enzymes of the TCA cycle, such as CS, MDH2, SUCLG1, and IDH, were consistently identified in RNA – protein interactome studies [Citation57,Citation58]. In addition to binding with exons, introns, and untranslated regions (UTR) of mRNA and regulating its processing, RBPs can also bind to lncRNAs and form a complex or interplay with each other to function in a variety of tumour-related biological processes. Current research indicates that TCA cycle enzymes interact with and are regulated by lncRNAs in different types of cancers, leading to specific metabolic changes that are closely related to tumorigenesis ( and ). In-depth understanding of the complex interplay between TCA cycle enzymes and lncRNAs will help provide a more comprehensive picture of how tumours undergo metabolic reprogramming.
Figure 3. Mechanisms of lncRNAs in regulating TCA cycle enzymes. TINCR binds to ACLY and protects it from ubiquitin degradation to maintain total cellular acetyl-CoA levels. LINC00477 interacts with ACO1 and suppresses the conversion ability from citrate to isocitrate by ACO1. IDH1-AS1 enhances IDH1 enzymatic activity through promoting its homodimerization, leading to increased production of α-KG. GAS5 inhibits FH – MDH2–CS complex formation by increasing SIRT3-mediated MDH2 deacetylation. AC020978 binds to MDH2 and protects it from ubiquitin degradation.
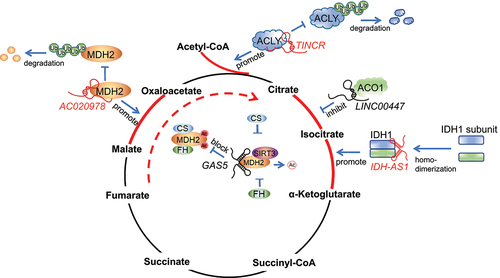
Table 1. The role of TCA cycle enzymes and their associated lncRNAs in tumour progression.
3.1. ACLY
ACLY is an enzyme located in the cytosol that facilitates the conversion of citrate and coenzyme A (CoA) to oxaloacetate (OAA) and acetyl-CoA in an Mg-ATP-dependent manner. This enzyme plays an essential role in cancer metabolism by potentially restricting cytosolic citrate levels, thereby regulating glycolysis and signalling pathways to promote cancer progression [Citation59,Citation60]. Its overexpression was correlated with poor prognosis and differentiation status of acute myeloid leukaemia and lung adenocarcinoma [Citation27,Citation61]. Human ACLY is a tetramer with a molecular weight of 480 kD and is composed of 1101 amino acids. It consists of a C-terminal citryl-CoA lyase (CCL) domain and an N-terminal citryl-CoA synthetase (CCS) module that is responsible for its catalytic activity [Citation62]. Zheng and co-workers found that the N-terminal of ACLY could interact with lncRNAs [Citation20]. By performing RNA pull-down assay followed by mass spectrometry (MS), Zheng et al. first identified the interaction between lncRNA TINCR and ACLY. Then, followed by the ACLY RNA immunoprecipitation (RIP) assay and the immunofluorescence (IF) of ACLY combined with TINCR fluorescence in situ hybridization (FISH) staining, Zheng et al. validated the endogenous colocalization and interaction of TINCR and ACLY. Next, Zheng et al. generated HA-tagged deletion mutants of ACLY to pinpoint the specific domains of the enzyme that interact with TINCR. Finally, RNA pull-down assays indicated that the N-terminal region of ACLY, which encompasses the ATP-binding, citrate-binding, and CoA-binding domains, was essential for the binding of the lncRNA TINCR. Mechanistically, TINCR binds to ACLY and protects it from ubiquitin degradation to maintain total cellular acetyl-CoA levels, which regulates a set of tumour-related events, including regulation of the PADI1–MAPK – MMP2/9 axis, histone H3K27 acetylation, and de novo lipid biosynthesis. Silencing TINCR inhibits nasopharyngeal carcinoma progression and cisplatin resistance [Citation20] (). In addition to TINCR, lncRNA FLJ22763 also regulates the mRNA and protein levels of ACLY. Ectopic expression of FLJ22763 suppresses the expression of ACLY, the biological malignant behaviour of gastric cancer cells and the xenograft tumour growth [Citation63]. However, it is unclear whether FLJ22763 can bind with ACLY, and whether the negative association between ACLY and FLJ22763 is due to their binding affinity.
3.2. ACO1
ACO1 is a protein located in the cytosol, which acts as a crucial enzyme in the TCA cycle and binds to mRNA to regulate intracellular iron concentrations [Citation56]. Under high iron levels, ACO1 interacts with a 4Fe-4S cluster and transforms into an active cytosolic aconitase enzyme [Citation64]. In contrast, when iron levels are insufficient, ACO1 binds to conserved stem-loop structures known as iron-responsive elements (IREs) in the UTRs of mRNAs responsible for iron metabolism. The impact of the IRE on regulation is determined by its position and context within the mRNA: ACO1-IRE located in the 5’ UTR of mRNA represses translation, while ACO1-IRE located in the 3’ end can indirectly activate translation via suppressing mRNA degradation [Citation64]. The dual roles of ACO1 in regulating TCA cycle and iron homoeostasis are closely related to tumorigenesis [Citation65,Citation66]. Interestingly, in addition to binding with mRNAs, ACO1 also interacts with a lncRNA variant of LINC00477, as predicted by RNA-binding Protein Database (RBPDB) and validated by RNA pull-down and RIP assays (). Gain-of-function and loss-of-function assays of the LINC00477 variant showed that it could suppress the functions of ACO1, including ferritin mRNA translation for cellular iron and glycol metabolism, conversion ability from citrate to isocitrate, and gastric cancer cell proliferation and migration [Citation49].
3.3. IDH1
IDH1 is a cytosolic enzyme that converts isocitrate to α-KG in TCA cycle [Citation67,Citation68]. The mutations in IDH1 are prevalent in various types of cancer [Citation69]. Mutant IDH1 reveals altered enzymatic activity leading to the synthesis of 2-HG, which acts as an oncometabolite that has pleotropic effects on tumorigenesis [Citation18]. The enzymatic activity of IDH1 is also regulated by lncRNA IDH1-AS1. By performing RIP and pull-down assays, Xiang et al. found that endogenous IDH1-AS1 was coprecipitated with IDH1 but not the mutant form of IDH1 (R132H) in Hela cells. To identify the WT IDH1 domain that physically interacts with IDH1-AS, Xiang et al. also used a domain mapping strategy of RIP assay, and finally revealed an intermediate part of IDH1 (103–286 aa), in which a helix structure formed by aa 271–286 was identified, which could bind with IDH1-AS1. Deletion of the residues 271–286 attenuated the interaction between IDH1 and IDH1-AS1, and inhibited the enzymatic activity of IDH1. This work eventually discovered that lncRNA IDH1-AS1 could bind and regulate IDH1 enzymatic activity by promoting its homodimerization, resulting in the elevated production of α-KG, downregulated expression of HIF-1a, and a decrease in glycolysis [Citation25] (). Besides, IDH1-AS1 has been found to regulate IDH1 in glioma. Wang et al. showed that IDH1-AS1 could regulate glioblastoma cell viability via IDH1. Although Wang et al. demonstrated the effect of IDH1-AS on IDH1 homodimerization and enzymatic activity in U251 cells, the interaction between IDH1-AS and IDH1 has not been validated in glioblastoma cells [Citation70].
3.4. MDH2
MDH2 is a mitochondrial enzyme essential for the conversion of malate to oxaloacetate [Citation71]. MDH2 has the potential to be a viable target for cancer treatment, owing to its effects on drug sensitivity and ATP production [Citation72]. For example, MDH2 is highly expressed in doxorubicin-resistant prostate cancer cells and uterine cancer cells. Overexpression of MDH2 enhances the P-glycoprotein’s ability to eliminate chemotherapeutic drugs, which can be a contributing factor to the shorter periods of relapse-free survival observed in patients following chemotherapy [Citation73]. MDH2 may also play a role in conferring resistance to docetaxel in prostate cancer cells, possibly through the JNK pathway [Citation74]. In recent years, the involvement of lncRNA in MDH2-directed regulations has been revealed. Xu et al. demonstrated that lncRNA AC020978 interacts with MDH2 and activates the AKT pathway by stabilizing MDH2, thus affecting the progression and metastasis of non-small cell lung cancer (NSCLC) [Citation50] (). The interaction between AC020978 and MDH2 was originally found by a biotin labelled AC020978 pull-down assay followed by MS analysis, which was further validated by MDH2 RIP assay and AC020978-FISH/MDH2-IF colocalization assay. Xu et al. investigated the binding sites of AC020978 and MDH2 by conducting a series of pull-down assays using truncated mutants of AC020978. The results identified the MDH2-specific binding sequence as being located within a 300-428nt long region in AC020978. Meanwhile, a set of truncated MDH2-based RIP assays were also performed. The findings demonstrated that the segment of MDH2 consisting of amino acids 121–260 was capable of binding with AC020978. In addition to AC020978, lncRNA GAS5 was also reported as an MDH2-interacted lncRNA, which was also originally identified by an RNA pull-down assay followed by MS analysis [Citation14]. After that, detection of RNA-protein interactions using cell lysates or recombinant MDH2 confirmed the direct interaction between GAS5 and MDH2. The MDH2-binding motif of GAS5 was also revealed (GAS5-Loop 2; 269–465 nucleotides) in this work. Functionally, GAS5 disrupts the metabolic enzyme tandem association of FH, MDH, and CS to modulate mitochondrial tricarboxylic acid flux via binding with MDH2 (). GAS5 expression shows a negative correlation with the levels of its associated mitochondrial metabolic enzymes, which improves the overall survival of patients with breast cancer [Citation14].
Protein interaction is a fundamental mechanism through which lncRNAs exert their biological functions. Thus, the identification of binding partners of lncRNAs is crucial for unravelling their biological functions and deciphering the underlying molecular mechanisms. In summary, the aforementioned lncRNA-binding TCA enzymes (ACLY, ACO1, IDH1, and MDH2) were all first identified based on RNA pull-down and MS analysis. As a matter of fact, more than a thousand novel RNA binding proteins lacking known RNA-binding domains have been characterized by MS analysis in large-scale studies [Citation75–86]. Several comprehensive databases documenting lncRNA-protein interactions have emerged in recent years, listing over 47 million entries of RNA interactions with DNAs, proteins, and other RNAs [Citation87–89]. In addition, a number of computational tools have been established to predict potential binding partners or even the binding sites [Citation90–92]. The advancements in technologies for assessing the interaction between lncRNA and RBPs have accelerated the progress in understanding the role of lncRNAs in the TCA cycle. Targeted analysis of TCA cycle enzymes associated with a specific lncRNA can be highly beneficial in uncovering the unknown functions of the lncRNAs under investigation. However, the biological functions of many novel lncRNA-TCA cycle enzyme partners discovered by large-scale studies or bioinformatics prediction remain elusive, and in-depth studies are warranted to gain a deeper understanding of the dynamics of these interactions and their roles in the development and progression of various tumours.
4. Alterations of TCA cycle enzymes are closely related to tumorigenesis
Numerous cancers harbour alterations that affect the integrity of TCA cycle have been found [Citation93–112]. For instance, the mutations of IDH1/2 have been commonly found in secondary glioblastoma and low-grade glioma, and relatively rare in angioimmunoblastic T-cell lymphomas, acute myeloid leukaemia (AML), prostate, colorectal, and thyroid cancer [Citation105,Citation113–116]. Patients with IDH1/2 mutations often have better outcomes than those carrying wild-type IDH genes [Citation18]. The mutations of SDH, which regulate the inactivation of protein function in tumour cells and thus affect angiogenesis, proliferation, and cell survival, have been widely identified in hereditary paraganglioma, pheochromocytoma, renal tumour, gastrointestinal stromal tumour, thyroid tumour, testicular seminoma, and neuroblastoma [Citation106,Citation117–123]. Another example is the mutations of FH, which often occur in bladder, breast, testicular, renal cancer, as well as cutaneous and uterine leiomyomas, can alter the morphological features of tumour cells, affect tumour cell migratory potential, and vary the ability to respond to oxidative stress and DNA damage [Citation107,Citation124–128].
In addition, the dysregulation or activity changes of TCA cycle enzymes (e.g. IDH1/2, CS, and KGDHC) have also been revealed in several studies. For example, it has been demonstrated that the overexpression of IDH1 and IDH2 in glioblastoma cells could inhibit the growth and migration of cancer cells [Citation129]. CS plays a pivotal role in the TCA cycle by catalysing a rate-limiting step. The overexpression or increased enzymatic activity of CS has been found in renal, ovarian, and pancreatic cancer, affecting the proliferation, migration, and invasion of tumour cells [Citation108,Citation130,Citation131]. KGDHC is also a TCA cycle rate-limiting enzyme, comprised of three components: dihydrolipoamide dehydrogenase (DLD), dihydrolipoamide S-succinyltransferase (DLST), and α-KG dehydrogenase (OGDH). The downregulation of OGDH, which regulates tumour cell cycle, migration, and viability, has been widely identified in colorectum, breast, oesophageal, lung, pancreatic, and cervical cancer [Citation109,Citation132–134]. Besides, lower expression of OGDHL, one of the rate-limiting components of OGDH complex, has also been observed in breast, cervical, lung, hepatocellular carcinoma, oesophageal, and pancreatic cancers, which is closely associated with advanced tumour stage [Citation110,Citation135]. DLST has been found to be upregulated in T-cell acute lymphoblastic leukaemia (T-ALL) and neuroblastoma. The elevated expression of DLST is an indicator of poor treatment response and a more aggressive disease course [Citation111,Citation136]. Inhibition of DLD has been found to decrease both ferrous iron accumulation and lipid peroxidation, resulting in the inhibition of ferroptosis in head and neck cancer [Citation137]. Unlike the well-explicit enzymes with certain oncogenic or suppressor roles, MDH2 has been shown to exhibit bifunctional roles. A previous study highlighted the downregulated expression of MDH2 in pheochromocytoma/paraganglioma caused by a germline mutation, and another study demonstrated the overexpression of MDH2 in clinical prostate cancer specimens [Citation74,Citation112]. The overexpression of MDH2 is considered an important factor for chemotherapy resistance in cancer. However, the in vitro experiments in prostate and doxorubicin-resistant uterine cancer cells also showed that MDH2 silencing could impair the viability of tumour cells [Citation74,Citation138].
Despite the prevalence of TCA cycle enzyme mutations and dysregulation in various malignancies, their actual roles in cancer development are still unclear, leaving many unanswered questions. Gaining a comprehensive understanding of the reasons behind the increased prevalence of specific mutations or dysregulation in certain cancers, compared to others, is crucial for researchers. This may be partly attributed the fact that different types of cancer have distinct underlying drivers and/or specific requirements on the path towards malignant transformation. Considering the expression specificity of lncRNAs in different types of cancers, lncRNAs may play a regulatory role in modulating the specificity of TCA cycle enzymes. Notably, cell death and inhibition of tumorigenesis may result from a significant level of mitochondrial dysfunction, whereas a moderate level of dysfunction could lead to an increase in mitochondrial ROS production and redox rebalancing, ultimately promoting tumour cell growth and invasiveness [Citation139]. Therefore, gaining a comprehensive comprehension of the functions of TCA cycle enzymes and their regulators in cancer is vital for devising efficacious therapeutic approaches to combat various forms of cancer in the future.
5. Targeting the TCA cycle enzymes and the associated lncRNAs
The TCA cycle plays a crucial role as the central hub for catabolic and anabolic reactions in tumour cells, and the dysregulation or mutation of enzymes involved in this pathway can significantly impact its modulation and functioning. Therefore, the TCA cycle enzymes and the regulators that cause alterations in these enzymes, such as lncRNAs, can serve as promising targets for the development of new cancer therapies [Citation140].
5.1. Targeting TCA cycle enzymes
In the case of IDH-mutated glioma cells, the administration of mutant IDH (mIDH) inhibitors resulted in suppressed growth and stimulated differentiation, whereas primary human AML cells with IDH mutation exhibited cell differentiation upon treatment [Citation141,Citation142]. These preclinical data revealing the targeted treatment of IDH mutations sheds light on advancing the clinical development of mIDH inhibitors. AG-221 was the first molecule to undergo clinical trials in late 2013. It is a selective, small molecule inhibitor of mutant IDH2 (). In 2014, AG-221 received orphan drug status for AML from the Food and Drug Administration (FDA), and later in 2019, the European Medicines Agency (EMA) also granted it a similar designation. Then, the mutant IDH1 and IDH2 inhibitor AG-881, which could penetrate the blood-brain barrier, entered the clinical trial (). Several studies have explored the potential of AG-881 in the treatment of glioblastoma and haematopoietic malignancy, leading up to a Phase III clinical trial that commenced in early 2020 to evaluate its efficacy in glioblastoma patients ().
Table 2. Selected clinical trials of small molecules targeting the TCA cycle enzymes for tumour.
Besides targeting mIDH, recent studies suggest that KGDHC represents a significant vulnerability in multiple cancers and holds potential as an effective therapeutic target. Anderson and co-workers utilized a MYC-driven model of T-ALL to demonstrate that the heterozygous inactivation of DLST led to a significant delay in tumour onset, without affecting the normal development of animals [Citation136]. Moreover, their study also revealed that the inactivation of DLST in T-ALL cells resulted in the disruption of the TCA cycle, deceleration of cell growth, and induction of apoptosis [Citation136]. In a recent study conducted by Ilic and colleagues, it was found that cancer cells with oncogenic PI3K mutations rely on all three components of KGDHC, particularly OGDH, to facilitate their proliferation [Citation143]. The results of the study provide support for the idea of targeting KGDHC as a potential approach for cancer therapy. Later, CPI-613, a multipurpose metabolic inhibitor (PDH and KGDHC) and α-lipoic acid (LA) derivative, was established. It is designed to target the dysregulated TCA cycle of tumour cells by inhibiting the enzymes PDH and KGDHC [Citation144,Citation145]. Clinical trials of CPI-613 were launched in the beginning of 2008, with a focus on advanced, metastatic, recurrent, or refractory diseases (). Thereafter, several Phase II and III trials were conducted to evaluate its safety and efficacy (). The FDA and EMA granted orphan drug status to CPI-613 for treating pancreatic cancer and AML [Citation146].
5.2. Targeting lncRNAs
LncRNAs exhibit tissue-, disease-, or developmental-specific expression patterns, which make them a promising candidate for use as a diagnostic biomarker and therapeutic target [Citation147]. The ability of lncRNA to exhibit specific expression patterns has been exploited to selectively target cancer cells, while sparing healthy cells, resulting in a potential strategy for the development of cancer therapies with fewer side effects. For instance, BC-819 (DTA-H19), a double-stranded DNA plasmid that contains the gene for diphtheria toxin-A under the influence of the H19 regulatory sequence, has been employed to target H19 overexpressing cancer cells. By intratumorally injecting BC-819, the expression of diphtheria toxin is induced, which effectively reduces the size of various tumour types [Citation148]. Aside from their potential for selectively eradicating cancer cells, the properties of lncRNAs, such as their expression patterns, functions, and structural characteristics, can also be investigated as potential tools for cancer diagnosis and treatment. For example, the expression of ACLY-binding lncRNA TINCR has been found to be associated with cancers, including prostate, gastric, hepatocellular carcinoma, oesophageal, lung, colon, breast, bladder, and oral squamous cell carcinoma [Citation149]. The diagnostic potential of TINCR was demonstrated in oesophageal squamous cell carcinoma and hepatocellular carcinoma, where TINCR levels were overexpressed at around 14-fold and 8.7-fold, respectively. Additionally, the significant correlations among TINCR dysregulation and clinicopathological characteristics, such as gender, age, histological grade, Ki-67 level, lymph node metastasis, tumour node metastasis stage, and tumour size in bladder and breast cancer were also validated [Citation150,Citation151]. However, the dysregulation patterns of TINCR can be varied across different cancer types. Due to the pleiotropic effects of TINCR dysregulation, efforts to target TINCR may lead to toxicity in both healthy cells and cancer cells. Thus, further investigations are needed to evaluate the potential of TINCR as an effective therapeutic target. Another example is MDH2-binding lncRNA GAS5. GAS5 is a lncRNA that exhibits diverse functions in the regulation of gene expression. Previous research has found that this lncRNA plays a role in the induction and suppression of cell apoptosis and tumorigenesis, respectively. Early investigations conducted in glioblastoma and NSCLC have suggested that the plasma levels of GAS5 may serve as a valuable diagnostic and prognostic biomarker for these diseases [Citation152]. Given the non-invasive nature of liquid biopsy compared to traditional biopsy methods, it is anticipated that the promising findings of these studies will be readily translated into clinical practice in the foreseeable future.
Although targeting lncRNAs has been the subject of extensive research over the past decade, most of the studies on lncRNA targeting are still in the preclinical stage [Citation153]. However, the expression of lncRNAs can be effectively repressed using small molecule inhibitors, antisense oligonucleotides, or RNA interference (RNAi) technology. Furthermore, gene therapy strategies can be employed to introduce beneficial tumour suppressor lncRNAs into cells for potential therapeutic benefit. In the future, lncRNAs are anticipated to expand the range of potential targets for RNAi and CRISPR-Cas genome editing technologies. In addition, to devise therapeutic approaches that target RBPs, researchers have screened RNA interference-based oligonucleotides and small molecule inhibitors that are designed to alter the levels of target RNAs and disrupt RBP-RNA interactions [Citation154]. The utilization of high-throughput techniques to identify RNA binding partners and the integration of multiple datasets can aid researchers in developing new prognostic biomarkers or therapeutic targets for various human cancers.
6. Conclusions and prospects
To date, as the repertoire of lncRNAs continues to expand, the process of comprehending their mechanisms of action is an ongoing endeavour, and we are still learning how to use them as weapons against human cancer. However, numerous pieces of evidence have demonstrated the power of lncRNAs in a variety of cancers, and the new advancement in computational algorithms, together with sophisticated massive sequencing and innovative techniques (e.g. CRISPR-Cas9), the prospect of discovering effective cures for cancer is within reach. And now, what has become apparent, even with the incomplete understanding of the molecular processes through which lncRNAs contribute to the specific tumour-related cellular activities, is that lncRNAs participate extensively in tumour-related regulatory networks, including cellular metabolism reprogramming.
Cellular metabolism reprogramming is a hallmark of cancer initiation and progression [Citation1–3]. Over the past decade, there has been an increasing recognition of metabolic phenotypes that distinguish cancer cells from adjacent normal tissues. When these phenotypes represent actionable therapeutic vulnerabilities, they hold significant potential for the development of novel cancer treatments. The TCA cycle is one of the crucial metabolic pathways that enable various cancers to utilize fatty acids, amino acids, and glucose to efficiently fulfill the cell’s bioenergetic, biosynthetic, and redox balance requirements [Citation140,Citation155]. In addition to investigating the function and regulation of TCA cycle components, there is also growing interest in developing therapies that target these components.
RBPs and their binding lncRNAs are recently identified as novel layers that cause direct drastic changes in TCA cycle enzymes and metabolites, and lead to distinct metabolic, genetic, and epigenetic alterations that are associated with the development and progression of tumours. Due to the functional versatility of lncRNAs, they played key roles in expanding the scope of RNA binding TCA cycle enzymes regulatory components in TCA cycle and tumour progression [Citation22,Citation156]. This represents a pivotal factor in the clinical development of metabolic inhibitors for oncology. Due to the compensatory pathways that cancer cells can employ to evade treatment and the context-dependent nature of their metabolic properties, developing effective therapies targeting cancer metabolism remains a significant challenge [Citation157–161]. Thus, a better understanding of the expression and function of the dysregulated pathways directed by TCA cycle enzymes and their binding lncRNAs in each cancer type may shed new light on the development of novel therapeutic strategy for cancer treatment and accelerating our race towards a healthier world.
In this review, we have provided an overview of the interplay between TCA cycle enzymes and their binding lncRNAs in tumorigenesis. The interactions between lncRNAs and TCA cycle enzymes have emerged as critical regulators in both tumour metabolism and tumorigenesis, and become the potential targets for anti-tumour treatment. However, we have limited our discussions to the interactions that have been experimentally validated. As a matter of fact, other TCA cycle enzymes, such as CS, SUCLG1, MDH3, etc., have also been identified as RNA binding proteins in the RNA interactome studies [Citation57,Citation58]. Nevertheless, the role of these TCA cycle enzymes in cancer, and the relevance of their RNA binding activities, are still elusive. Incorporating multiple RNA-binding, tumour dysregulation, and mutation data sources in the future will enhance our understanding of the relationships between TCA cycle enzymes and cancer.
Acknowledgments
We thank Dr. Rongkang Hu from Anhui Normal University for his constructive suggestions. We also thank the platforms and resources provided by the Outstanding Innovative Research Team for Molecular Enzymology and Detection in Anhui Provincial Universities, the Key Laboratory of Biomedicine in Gene Diseases and Health of Anhui Higher Education Institutes, and Anhui Provincial Key Laboratory of the Conservation and Exploitation of Biological Resources. The authors would also like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368. DOI:10.1126/science.aaw5473
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530.
- Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr. 2007;39:251–257.
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
- Som P, Atkins HL, Bandoypadhyay D, et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med. 1980;21:670–675.
- Papathanassiou D, Bruna-Muraille C, Jouannaud C, et al. Single-photon emission computed tomography combined with computed tomography (SPECT/CT) in bone diseases. Joint Bone Spine. 2009;76:474–480.
- Sajnani K, Islam F, Smith RA, et al. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie. 2017;135:164–172.
- Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta. 2012;1826:370–384.
- Eniafe J, Jiang S. The functional roles of TCA cycle metabolites in cancer. Oncogene. 2021;40:3351–3363.
- Hung CL, Wang LY, Yu YL, et al. A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci U S A. 2014;111:18697–18702.
- Park MK, Zhang L, Min KW, et al. (2021) NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab, 33, 2380–2397 e2389.
- Sang L, Ju HQ, Yang Z, et al. Mitochondrial long non-coding RNA GAS5 tunes TCA metabolism in response to nutrient stress. Nat Metab. 2021;3:90–106.
- Zhang X, Li Z, Xuan Z, et al. Novel role of miR-133a-3p in repressing gastric cancer growth and metastasis via blocking autophagy-mediated glutaminolysis. J Exp Clin Cancer Res. 2018;37:320.
- Eng C, Kiuru M, Fernandez MJ, et al. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202.
- Juang HH. Modulation of mitochondrial aconitase on the bioenergy of human prostate carcinoma cells. Mol Genet Metab. 2004;81:244–252.
- Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773.
- Tian C, Wen B, Bian M, et al. From a dimer to a monomer: construction of a chimeric monomeric isocitrate dehydrogenase. Protein Sci. 2021;30:2396–2407.
- Zheng ZQ, Li ZX, Guan JL, et al. Long noncoding RNA TINCR-Mediated regulation of acetyl-CoA metabolism promotes nasopharyngeal carcinoma progression and chemoresistance. Cancer Res. 2020;80:5174–5188.
- Qin H, Ni H, Liu Y, et al. RNA-binding proteins in tumor progression. J Hematol Oncol. 2020;13:90.
- Li L, Miao H, Chang Y, et al. Multidimensional crosstalk between RNA-binding proteins and noncoding RNAs in cancer biology. Semin Cancer Biol. 2021;75:84–96.
- Mattick JS, Amaral PP, Carninci P, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 2023. doi:10.1038/s41580-022-00566-8.
- Tan YT, Lin JF, Li T, et al. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun (Lond). 2021;41:109–120.
- Xiang S, Gu H, Jin L, et al. LncRNA IDH1-AS1 links the functions of c-Myc and HIF1alpha via IDH1 to regulate the Warburg effect. Proc Natl Acad Sci U S A. 2018;115:E1465–1474.
- Mullen AR, DeBerardinis RJ. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol Metab. 2012;23:552–559.
- Migita T, Narita T, Nomura K, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554.
- Corbet C, Feron O. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta Rev Cancer. 2017;1868:7–15.
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899.
- Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185.
- Gao C, Shen Y, Jin F, et al. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS ONE. 2016;11:e0154576.
- Kishton RJ, Barnes CE, Nichols AG, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016;23:649–662.
- Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer. 2016;2:758–770.
- Shen T, Xia W, Min S, et al. A pair of long intergenic non-coding RNA LINC00887 variants act antagonistically to control Carbonic Anhydrase IX transcription upon hypoxia in tongue squamous carcinoma progression. BMC Biol. 2021;19:192.
- Gameiro PA, Yang J, Metelo AM, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–385.
- Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121.
- Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:10.
- Cardaci S, Ciriolo MR. TCA cycle defects and cancer: when metabolism tunes redox state. Int J Cell Biol. 2012;2012:161837.
- Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–3658.
- Desideri E, Vegliante R, Ciriolo MR. Mitochondrial dysfunctions in cancer: genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer Lett. 2015;356:217–223.
- Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153.
- Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85.
- DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109.
- Saito R, Suzuki T, Hiramoto K, et al. Characterizations of three major cysteine sensors of keap1 in stress response. Mol Cell Biol. 2016;36:271–284.
- Moreno Leon L, Gautier M, Allan R, et al. The nuclear hypoxia-regulated NLUCAT1 long non-coding RNA contributes to an aggressive phenotype in lung adenocarcinoma through regulation of oxidative stress. Oncogene. 2019;38:7146–7165.
- Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5:2430–2435.
- Kacso TP, Zahu R, Tirpe A, et al. Reactive oxygen species and long non-coding RNAs, an unexpected crossroad in cancer cells. Int J Mol Sci. 2022;23:10133.
- Chen F, Xiao M, Feng J, et al. Different inhibition of Nrf2 by two keap1 isoforms alpha and beta to shape malignant behaviour of human hepatocellular carcinoma. Int J Mol Sci. 2022;23:10342.
- Zhao H, He Y, Li H, et al. The opposite role of alternatively spliced isoforms of LINC00477 in gastric cancer. Cancer Manag Res. 2019;11:4569–4576.
- Xu F, Hua Q, Zhang A, et al. LncRNA AC020978 facilitates non-small cell lung cancer progression by interacting with malate dehydrogenase 2 and activating the AKT pathway. Cancer Sci. 2021;112:4501–4514.
- Yang J, Liu F, Wang Y, et al. LncRNAs in tumor metabolic reprogramming and immune microenvironment remodeling. Cancer Lett. 2022;543:215798.
- Ferre F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform. 2016;17:106–116.
- Pereira B, Billaud M, Almeida R. RNA-Binding proteins in cancer: old players and new actors. Trends Cancer. 2017;3:506–528.
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490.
- Albihlal WS, Gerber AP. Unconventional RNA-binding proteins: an uncharted zone in RNA biology. FEBS Lett. 2018;592:2917–2931.
- Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406.
- Castello A, Hentze MW, Preiss T. Metabolic enzymes enjoying new partnerships as RNA-Binding proteins. Trends Endocrinol Metab. 2015;26:746–757.
- Ciesla J. Metabolic enzymes that bind RNA: yet another level of cellular regulatory network? Acta Biochim Pol. 2006;53:11–32.
- Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 2012;72:3709–3714.
- Icard P, Wu Z, Alifano M, et al. Gluconeogenesis of cancer cells is disrupted by citrate. Trends Cancer. 2019;5:265–266.
- Wang J, Ye W, Yan X, et al. Low expression of ACLY associates with favorable prognosis in acute myeloid leukemia. J Transl Med. 2019;17:149.
- Elshourbagy NA, Near JC, Kmetz PJ, et al. Cloning and expression of a human ATP-citrate lyase cDNA. Eur J Biochem. 1992;204:491–499.
- Zhang G, Wang Q, Lu J, et al. Long non-coding RNA FLJ22763 is involved in the progression and prognosis of gastric cancer. Gene. 2019;693:84–91.
- Volz K. The functional duality of iron regulatory protein 1. Curr Opin Struct Biol. 2008;18:106–111.
- Yuan C, Clish CB, Wu C, et al. Circulating metabolites and survival among patients with pancreatic cancer. J Natl Cancer Inst. 2016;108:djv409.
- Zhang T, Sun L, Hao Y, et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat Cancer. 2022;3:75–89.
- Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599–608.
- Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. 2021;18:645–661.
- Caravella JA, Lin J, Diebold RB, et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63:1612–1623.
- Wang J, Quan Y, Lv J, et al. LncRNA IDH1-AS1 suppresses cell proliferation and tumor growth in glioma. Biochem Cell Biol. 2020;98:556–564.
- Ait-El-Mkadem S, Dayem-Quere M, Gusic M, et al. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am J Hum Genet. 2017;100:151–159.
- Marquez J, Flores J, Kim AH, et al. Rescue of TCA cycle dysfunction for cancer therapy. J Clin Med. 2019;8:2161.
- Lo YW, Lin ST, Chang SJ, et al. Mitochondrial proteomics with siRNA knockdown to reveal ACAT1 and MDH2 in the development of doxorubicin-resistant uterine cancer. J Cell Mol Med. 2015;19:744–759.
- Liu Q, Harvey CT, Geng H, et al. Malate dehydrogenase 2 confers docetaxel resistance via regulations of JNK signaling and oxidative metabolism. Prostate. 2013;73:1028–1037.
- Bao X, Guo X, Yin M, et al. Capturing the interactome of newly transcribed RNA. Nat Methods. 2018;15:213–220.
- Castello A, Fischer B, Frese CK, et al. Comprehensive identification of RNA-Binding domains in human cells. Mol Cell. 2016;63:696–710.
- Conrad T, Albrecht AS, de Melo Costa VR, et al. Serial interactome capture of the human cell nucleus. Nat Commun. 2016;7:11212.
- Guo X, Tariq M, Lai Y, et al. Capture of the newly transcribed RNA interactome using click chemistry. Nat Protoc. 2021;16:5193–5219.
- He C, Sidoli S, Warneford-Thomson R, et al. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol Cell. 2016;64:416–430.
- Huang R, Han M, Meng L, et al. Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc Natl Acad Sci U S A. 2018;115:E3879–3887.
- Li X, Song J, Yi C. Genome-wide mapping of cellular protein-RNA interactions enabled by chemical crosslinking. Genomics Proteomics Bioinf. 2014;12:72–78.
- Queiroz RML, Smith T, Villanueva E, et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat Biotechnol. 2019;37:169–178.
- Trendel J, Schwarzl T, Horos R, et al. The human RNA-Binding proteome and its dynamics during translational arrest. Cell. 2019;176:391–403 e319.
- Yan S, Zhao D, Wang C, et al. Characterization of RNA-binding proteins in the cell nucleus and cytoplasm. Anal Chim Acta. 2021;1168:338609.
- Caudron-Herger M, Wassmer E, Nasa I, et al. Identification, quantification and bioinformatic analysis of RNA-dependent proteins by RNase treatment and density gradient ultracentrifugation using R-DeeP. Nat Protoc. 2020;15:1338–1370.
- Brannan KW, Jin W, Huelga SC, et al. SONAR discovers RNA-Binding proteins from analysis of large-scale protein-protein interactomes. Mol Cell. 2016;64:282–293.
- Kang J, Tang Q, He J, et al. Rnainter v4.0: rNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 2022;50:D326–332.
- Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–97.
- Teng X, Chen X, Xue H, et al. Npinter v4.0: an integrated database of ncRNA interactions. Nucleic Acids Res. 2020;48:D160–165.
- Zhang J, Chen Q, Liu B. iDRBP_MMC: identifying DNA-Binding proteins and RNA-binding proteins based on multi-label learning model and motif-based convolutional neural network. J Mol Biol. 2020;432:5860–5875.
- Suresh V, Liu L, Adjeroh D, et al. RPI-Pred: predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 2015;43:1370–1379.
- Livi CM, Klus P, Delli Ponti R, et al. catRAPID signature: identification of ribonucleoproteins and RNA-binding regions. Bioinformatics. 2016;32:773–775.
- Granchi C. ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018;157:1276–1291.
- Huang D, Wang Y, Thompson JW, et al. Cancer-cell-derived GABA promotes beta-catenin-mediated tumour growth and immunosuppression. Nat Cell Biol. 2022;24:230–241.
- Jansen MP, Sas L, Sieuwerts AM, et al. Decreased expression of ABAT and STC2 hallmarks ER-positive inflammatory breast cancer and endocrine therapy resistance in advanced disease. Mol Oncol. 2015;9:1218–1233.
- Jin L, Chun J, Pan C, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. 2018;69:87–99 e87.
- Kremer JC, Prudner BC, Lange SES, et al. Arginine deprivation inhibits the Warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep. 2017;18:991–1004.
- Lao-On U, Rojvirat P, Chansongkrow P, et al. C-Myc directly targets an over-expression of pyruvate carboxylase in highly invasive breast cancer. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165656.
- Qi L, Martin-Sandoval MS, Merchant S, et al. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell. 2021;28:1982–1999 e1988.
- Sawant Dessai A, Dominguez MP, Chen UI, et al. Transcriptional repression of SIRT3 potentiates mitochondrial aconitase activation to drive aggressive prostate cancer to the bone. Cancer Res. 2021;81:50–63.
- Tian X, Han Y, Yu L, et al. Decreased expression of ALDH5A1 predicts prognosis in patients with ovarian cancer. Cancer Biol Ther. 2017;18:245–251.
- You X, Tian J, Zhang H, et al. Loss of mitochondrial aconitase promotes colorectal cancer progression via SCD1-mediated lipid remodeling. Mol Metab. 2021;48:101203.
- Zheng B, Chai R, Yu X. Downregulation of NIT2 inhibits colon cancer cell proliferation and induces cell cycle arrest through the caspase-3 and PARP pathways. Int J Mol Med. 2015;35:1317–1322.
- Zou Z, Hu X, Luo T, et al. Naturally-occurring spinosyn a and its derivatives function as argininosuccinate synthase activator and tumor inhibitor. Nat Commun. 2021;12:2263.
- Ghiam AF, Cairns RA, Thoms J, et al. IDH mutation status in prostate cancer. Oncogene. 2012;31:3826.
- Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443.
- Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410.
- Lin CC, Cheng TL, Tsai WH, et al. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. 2012;2:785.
- Chang LC, Chiang SK, Chen SE, et al. Targeting 2-oxoglutarate dehydrogenase for cancer treatment. Am J Cancer Res. 2022;12:1436–1455.
- Dai W, Xu L, Yu X, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72:909–923.
- Anderson NM, Qin X, Finan JM, et al. Metabolic enzyme DLST promotes tumor aggression and reveals a vulnerability to OXPHOS inhibition in high-risk neuroblastoma. Cancer Res. 2021;81:4417–4430.
- Cascon A, Comino-Mendez I, Curras-Freixes M, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 2015;107. DOI:10.1093/jnci/djv053
- Kang MR, Kim MS, Oh JE, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–355.
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19:764–772.
- Yang M, Ternette N, Su H, et al. The succinated proteome of FH-mutant tumours. Metabolites. 2014;4:640–654.
- Yen KE, Bittinger MA, Su SM, et al. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29:6409–6417.
- Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54.
- Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–372.
- Baysal BE, Willett-Brozick JE, Lawrence EC, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39:178–183.
- Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020.
- Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142.
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270.
- Moog S, Lussey-Lepoutre C, Favier J. Epigenetic and metabolic reprogramming of SDH-deficient paragangliomas. Endocr Relat Cancer. 2020;27:R451–463.
- Buelow B, Cohen J, Nagymanyoki Z, et al. Immunohistochemistry for 2-Succinocysteine (2SC) and Fumarate Hydratase (FH) in cutaneous leiomyomas may aid in identification of patients with HLRCC (Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome). Am J Surg Pathol. 2016;40:982–988.
- Kancherla P, Daneshvar M, Sager RA, et al. Fumarate hydratase as a therapeutic target in renal cancer. Expert Opin Ther Targets. 2020;24:923–936.
- Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98:3387–3392.
- Carvajal-Carmona LG, Alam NA, Pollard PJ, et al. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab. 2006;91:3071–3075.
- Ylisaukko-Oja SK, Cybulski C, Lehtonen R, et al. Germline fumarate hydratase mutations in patients with ovarian mucinous cystadenoma. Eur J Hum Genet. 2006;14:880–883.
- Li S, Chou AP, Chen W, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 2013;15:57–68.
- Chen L, Liu T, Zhou J, et al. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE. 2014;9:e115708.
- Schlichtholz B, Turyn J, Goyke E, et al. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas. 2005;30:99–104.
- Fedorova MS, Kudryavtseva AV, Lakunina VA, et al. Downregulation of OGDHL expression is associated with promoter hypermethylation in colorectal cancer. Mol Biol (Mosk). 2015;49:678–688.
- Hoque MO, Kim MS, Ostrow KL, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661–2670.
- Ostrow KL, Park HL, Hoque MO, et al. Pharmacologic unmasking of epigenetically silenced genes in breast cancer. Clin Cancer Res. 2009;15:1184–1191.
- Snezhkina AV, Krasnov GS, Zaretsky AR, et al. Differential expression of alternatively spliced transcripts related to energy metabolism in colorectal cancer. BMC Genomics. 2016;17:1011.
- Anderson NM, Li D, Peng HL, et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia. 2016;30:1365–1374.
- Shin D, Lee J, You JH, et al. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020;30:101418.
- Ciccarone F, Vegliante R, Di Leo L, et al. The TCA cycle as a bridge between oncometabolism and DNA transactions in cancer. Semin Cancer Biol. 2017;47:50–56.
- Luo Y, Ma J, Lu W. The significance of mitochondrial dysfunction in cancer. Int J Mol Sci. 2020;21:5598.
- Anderson NM, Mucka P, Kern JG, et al. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9:216–237.
- Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630.
- Wang F, Travins J, DeLabarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626.
- Ilic N, Birsoy K, Aguirre AJ, et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci U S A. 2017;114:E3434–3443.
- Stuart SD, Schauble A, Gupta S, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2:4.
- Zachar Z, Marecek J, Maturo C, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl). 2011;89:1137–1148.
- Neitzel C, Demuth P, Wittmann S, et al. Targeting altered energy metabolism in colorectal cancer: oncogenic reprogramming, the central role of the TCA cycle and therapeutic opportunities. Cancers (Basel). 2020;12(7):12.
- Chandra Gupta S, Nandan Tripathi Y. Potential of long non-coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer. 2017;140:1955–1967.
- Mizrahi A, Czerniak A, Levy T, et al. Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J Transl Med. 2009;7:69.
- Sharma U, Barwal TS, Malhotra A, et al. Long non-coding RNA TINCR as potential biomarker and therapeutic target for cancer. Life Sci. 2020;257:118035.
- Tian T, Wang M, Lin S, et al. The impact of lncRNA dysregulation on clinicopathology and survival of breast cancer: a systematic review and meta-analysis. Mol Ther Nucleic Acids. 2018;12:359–369.
- Xia Y, Liu Z, Yu W, et al. The prognostic significance of long noncoding RNAs in bladder cancer: a meta-analysis. PLoS ONE. 2018;13:e0198602.
- Ghaforui-Fard S, Taheri M. Growth arrest specific transcript 5 in tumorigenesis process: an update on the expression pattern and genomic variants. Biomed Pharmacother. 2019;112:108723.
- Chen Y, Li Z, Chen X, et al. Long non-coding RNAs: from disease code to drug role. Acta Pharm Sin B. 2021;11:340–354.
- Hong S. RNA binding protein as an emerging therapeutic target for cancer prevention and treatment. J Cancer Prev. 2017;22:203–210.
- Cai Z, Li CF, Han F, et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol Cell. 2020;80:263–278 e267.
- Wang X, Song X, Glass CK, et al. The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harb Perspect Biol. 2011;3:a003756.
- Kishton RJ, Rathmell JC. Novel therapeutic targets of tumor metabolism. Cancer J. 2015;21:62–69.
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31.
- Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int J Biochem Cell Biol. 2015;59:167–181.
- Yuneva MO, Fan TW, Allen TD, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170.
- Zugazagoitia J, Guedes C, Ponce S, et al. Current challenges in cancer treatment. Clin Ther. 2016;38:1551–1566.