
ABSTRACT
Adiponectin, an adipocyte-specific secretory protein encoded by the ADIPOQ gene has a causal role in insulin resistance. Anti-diabetic drugs increase plasma adiponectin by a poorly understood, post-transcriptional mechanism enhancing insulin sensitivity. Deletion analysis of a reporter bearing the mouse Adipoq mRNA 5’-leader identified an inhibitory cis-regulatory sequence. The 5’-leader harbours two potential upstream open reading frames (uORFs) overlapping the principal downstream ORF. Mutation of the uORF ATGs increased reporter translation ~3-fold, indicative of a functional uORF. uORFs are common in mammalian mRNAs; however, only a select group resist translational repression by the integrated stress response (ISR). Thapsigargin (TG), which induces endoplasmic reticulum (ER) stress and the ISR, enhanced expression of a reporter bearing the Adipoq 5’-leader; polysome profiling verified translation-stimulation. TG-stimulated translation was absent in cells defective in Ser51 phosphorylation of eukaryotic initiation factor 2α (eIF2α), required for the ISR. To determine its role in expression and function of endogenous adiponectin, the upstream uORF was disrupted by CRISPR-Cas9-mediated mutagenesis of differentiated mouse 3T3-L1 adipocytes. uORF disruption in adipocytes increased adiponectin expression, triacylglycerol accumulation, and glucose uptake, and inhibited paracrine muscle and liver cell expression of gluconeogenic enzymes, establishing an important role of the uORF in adiponectin-mediated responses to stress.
Introduction
Translation-initiation generally occurs when the 48S translation initiation complex, containing the small, 40S ribosome subunit, recognizes the cap structure at the 5’ terminus of mRNA, and scans the leader sequence until it encounters the first AUG codon [Citation1]. About half of human and mouse mRNAs contain ATGs upstream of the principal AUG, i.e. in the 5’leader, that if initiated, determines an upstream open reading frame (uORF) [Citation2]. uORFs are generally conserved between species, suggesting important functional activities [Citation3,Citation4]. Ribosome profiling studies have indicated that non-canonical initiation codons, e.g. CUG, TUG, and GUG, also can direct premature initiation [Citation5–7]. A major function of uORFs appears to be inhibition of translation of the major ORF by masking the canonical AUG, for example, by overlap of the uORF with the canonical AUG or by early ribosome termination in non-overlapping cases [Citation2,Citation3,Citation8,Citation9]. Importantly, certain environmental stresses inhibit global mRNA translation, but simultaneously stimulate translation of specific uORF-containing transcripts [Citation10]. The integrated stress response (ISR) is an elaborate regulatory system that ‘funnels’ multiple stress types into a single response dependent on phosphorylation of eIF2α, and consequent global inhibition of translation-initiation. Despite the prevalence of uORFs, only a limited set of uORF-containing mRNAs are resistant to global translation shut-down during the ISR [Citation11,Citation12]. uORF dysregulation is implicated in multiple patho-physiological conditions including defective liver regeneration, increased oxidative metabolism, protection of kidney injury, and impaired osteoclast differentiation [Citation13]. Despite the limited number of preferentially translated targets during the ISR, the targets identified to date are almost without exception hubs of critical pathways regulating gene transcription, cell viability, nutrient transport, DNA damage response, and tRNA charging during translation [Citation14]. Identification of new uORF-containing, ISR-resistant genes certainly increases the understanding of the stress-mediated regulation of those genes, but more can also provide new connectors linking these critical hubs.
Adiponectin is an adipocyte-specific cytokine, i.e. adipokine, that regulates glucose and lipid metabolism, sensitizes insulin signalling, stimulates mitochondria biogenesis, and suppresses inflammation, among other critical homoeostatic functions [Citation15–17]. Low plasma levels of adiponectin are associated with obesity, insulin-resistance, and type 2 diabetes. Adiponectin influences energy metabolism by inducing AMP-activated protein kinase (AMPK)-mediated transcription, following adiponectin receptor 1 (AdipoR1) binding in skeletal muscle and liver [Citation18]. AMPK activation increases glucose uptake, and favours ATP production via activation of catabolic pathways, while inhibiting anabolic reactions. Also, adiponectin enhances insulin-sensitivity by activation of peroxisome proliferator-activated receptor (PPAR)α, which induces long-chain fatty acid uptake, transport, and oxidation, while inhibiting fatty acid synthesis. Together, these responses provide fuel for biochemical activities in target tissues, for example, muscle contraction, and reduce lipid accumulation in adipose tissue. Although AdipoR1 is highly expressed in human adipocytes, limited information is available on autocrine or juxtacrine influence of adiponectin [Citation19,Citation20].
Plasma levels of adiponectin are 3 to 6 orders of magnitude higher than most hormones and cytokines, e.g. leptin, insulin, and interleukins [Citation21]. Despite the exceptionally high level, plasma adiponectin is stringently regulated in response to nutritional status. In mice, serum and cerebrospinal fluid adiponectin levels increase during fasting and are restored after re-feeding [Citation22]. Likewise, in rats and teleost fish, long-term food restriction increases circulating adiponectin [Citation23,Citation24]. Studies of individuals with eating disorders, e.g. anorexia nervosa, show increased serum adiponectin [Citation25], and an opposite response is observed in women with binge eating disorder [Citation26]. The mechanisms regulating plasma adiponectin level are not well understood. Insulin-sensitizing, anti-diabetic drugs, e.g. thiazolidinediones (TZDs), increase plasma levels of adiponectin and other adipokines [Citation27–29]. TZDs increase adipose tissue ADIPOQ mRNA in some studies [Citation30,Citation31], but others suggest the moderate increase in mRNA cannot account for the observed increase in plasma adiponectin [Citation32,Citation33]. Similarly, PPARγ agonists increase plasma adiponectin levels without altering in ADIPOQ mRNA [Citation27,Citation34,Citation35]. The PPAR agonist pioglitazone increases ADIPOQ mRNA translation by about 2- to 3-fold, by an as-yet undefined mechanism [Citation36]. Post-transcriptional regulation of ADIPOQ mRNA by elements in the 3’-untranslated region (UTR) has been reported, but little is known about the potential regulatory function of the 5’-UTR [Citation37–39].
Here, we describe translational control of murine Adipoq mRNA by a 5’-UTR uORF responsive to the eIF2α-mediated ISR. The uORF contains a pair of in-frame ATGs and is out-of-frame with and overlapping the principal adiponectin ORF. The uORF profoundly inhibits adiponectin expression as shown by relief by start site mutations in a reporter of in vitro translation and by a knock-in mutation in 3T3-L1 adipocytes. Most importantly, an ISR agonist induced translation of a uORF-containing reporter and endogenous mouse 3T3-L1 adipocyte Adipoq mRNA by ~2-fold. These results indicate Adipoq is a new member of the small family of uORF-containing genes resistant to the ISR, and consistent with the protective activity of adiponectin against oxidative and endoplasmic reticulum (ER) stress [Citation40,Citation41].
Results
Regulatory elements in Adipoq mRNA
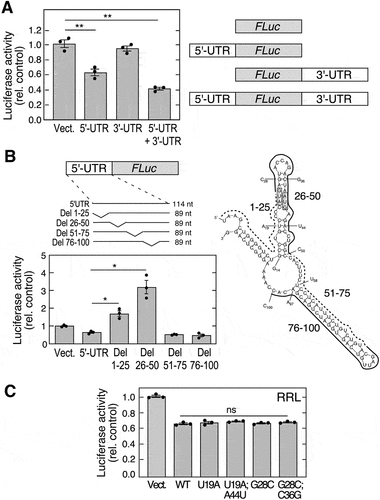
Translational control mechanisms generally depend on sequence or structural elements in the 5’- or 3’-UTR [Citation42,Citation43]. The critical regulatory region in mouse Adipoq mRNA (GenBank accession no. NM_009605) was investigated using a reporter system. Differentiated 3T3-L1 adipocytes were transfected with polyadenylated firefly luciferase (FLuc) cDNA bearing an upstream 5’-UTR, or downstream 3’-UTR, or both, and co-transfected with renilla luciferase (Rluc) as transfection efficiency control. Significant inhibition of expression was observed with the construct containing the 114-nt 5”-UTR, but not with the 3’-UTR (). The presence of both 5’- and 3’-UTRs showed a trend towards further suppression of translation, suggesting a potential role for mRNA circularization in translational control [Citation42,Citation44]. To further investigate the specific cis-acting RNA element, constructs were generated containing sequential 25-nt deletions from the 5’-UTR. Deletion of nts 1–25 and 26–30 both increased luciferase expression indicating that the inhibitory element likely spans the 1–50 nt region (, left). To provide a clue to the function of the 1–50 nt region, the 5’-UTR was subjected to computational RNA folding analysis using mfold [Citation45]. The computed Adipoq 5’-UTR forms two hairpin stem-loops spanning regions 14–50 nt and 58–97 nt (, right). Based on the predicted RNA secondary structure, we used a mutagenesis approach to determine the structural requirement for the stem-loop spanning the 14–50 nt region. In vitro translation by rabbit reticulocyte lysate (RRL) was determined using reporters generated by in vitro transcription. An ~40% inhibition was observed with the construct containing the full-length, 114-nt 5’-UTR (). The inhibition was comparable to that observed in transfected adipocytes suggesting that adipocyte-specific trans-acting factors are not required. Secondary structures calculated by RNAfold [Citation46] predict that mutation of U19A and G28C disrupt the stem-loop, and the compensatory mutations, U19A;A44U and G28C;C36G, restore the stem-loop structure (Fig. S1). None of the mutations reversed the inhibition of translation by the Adipoq 5’-UTR (), suggesting that secondary structure does not have a major role in translation-inhibition of adiponectin, and that linear sequence-dependent mechanisms might be critical.
Figure 1. Mouse Adipoq 5’-UTR harbours a translational inhibitor element. (A) CMV-driven plasmids containing polyadenylated FLuc vector with 5’-UTR, 3’-UTR, or 5’− and 3’-UTRs (right) of AdipoQ were transfected into 3T3-L1 adipocytes. FLuc activity was normalized to expression of co-transfected RLuc (left; mean ± SEM, n = 3, **P < 0.01). (B) plasmids containing 25-nt deletions of 5’-UTR (left-top) were transfected into adipocytes. FLuc activity was normalized by expression of co-transfected RLuc (left-bottom; mean ± SEM, n = 3, *P < 0.05). Secondary structure of Adipoq 5’-UTR was determined by mfold (right). Deletions (solid and dashed curves), mutations (individual labels), and start codons (shaded boxes) are highlighted. (C) plasmids containing mutations that disrupt or restore secondary structure of Adipoq 5’-UTR were used to in vitro synthesize RNA. in vitro translation was done using RRL, and luciferase activity normalized to vector control (mean ± SEM, n = 3: ns, not significant).
Adipoq mRNA is translationally repressed by an uORF in the 5’-leader
The mouse Adipoq mRNA spans 3 exons. The 5’-UTR includes the 106-nt exon 1 and the 5’-most 8-nt of exon 2; the Adipoq coding sequence (CDS) consists of the remainder of exon 2 and part of exon 3 (). Inspection of the cis-acting regulatory region of Adipoq mRNA reveals two upstream AUG sites at 5’-leader nt positions 24 and 39 which are in-frame with each other, but out-of-frame with Adipoq CDS, which initiates at nt position 115 in exon 2. The upstream 5’-leader AUG and the canonical adiponectin AUG are predicted to have moderately strong Kozak consensus sequences (Kozak Similarity Score equals 0.61 and 0.67, respectively; range = 0 to 1 where 1 is optimal), but the downstream 5’-leader AUG is not predicted to be an initiation site [Citation48]. The resulting ORF terminates with a UGA at nt position 171 in exon 2, and if translated would encode a 49- (or possibly 44-) amino acid peptide. Because the uORF initiates upstream of the Adipoq AUG, and terminates within the CDS, it potentially inhibits Adipoq mRNA translation as described for multiple uORFs [Citation10]. We generated a reporter construct in which a CMV promoter drives the Renilla luciferase (RLuc) CDS downstream of the Adipoq wild-type (WT) 5’-leader, and a GUG mutant construct in which both 5’-leader AUGs are mutated to GUG (A24G, A39G) (, top). In all constructs, RLuc CDS is preceded by an identical Kozak sequence surrounding the ATG corresponding to the Adipoq major AUG (AUGApN). Because RLuc is out-of-frame with the ATG sites in the 5’-leader, translation will be terminated by a stop codon in the RLuc CDS. Mutation of both AUG sites to GUG increased RLuc expression ~3.2-fold, consistent with relief of uORF-mediated inhibition (, bottom). To directly investigate translation driven by the Adipoq uORF, a fusion construct was generated in which the AUG start site preceding RLuc was deleted, as well as the G nt immediately downstream to bring the 5’-leader and the RLuc CDS into the same reading frame (ΔAUGG; the uORF is in reading frame 3 whereas RLuc CDS is in reading frame 1) (, schematics). Translation initiating at an uORF start codon in the ΔAUGG construct should generate RLuc with a 44- or 49-amino acid N-terminus extension. Indeed, RLuc immunoblot (, middle) showed RLuc translated from this fusion construct exhibited a greater molecular weight than RLuc translated from the canonical start codon, confirming uORF start site recognition by ribosomes; equal loading is shown by Ponceau stain (, bottom).
Figure 2. Mouse adiponectin expression is inhibited by uORF element in the 5’-leader. (A) adiponectin is encoded on 3 exons. 5’-UTR (leader) sequence in exons 1 and 2 is shown above, sequence of exon 1 with the uORF ATGs (bold) are shown below. (B) an RLuc reporter with 5’-leader bearing A-to-G mutations in both uORF sites was generated; the vertical shift in the schematic indicates the altered reading frame (top). The mutated and control constructs were subjected to in vitro translation in a RRL, and luciferase activity determined (bottom; mean ± SEM, n = 3, **P < 0.01). (C) reporter fusion constructs were engineered with (AUGApN) without (ΔAUGG) the AUG of adiponectin (ApN), and with a single following ‘G’ to restore RLuc ORF in the same reading frame as the uOrfs. A triplet in RLuc that acts as a stop codon in the uORF reading frame is indicated (*) (schematic at top). RLuc immunoblot following translation by RRL (middle) and Ponceau stain (bottom). (D) effect of Adipoq uORF peptide on translation of luciferase mRNA in RRL (mean ± SEM, n = 3, ns, not significant). (E) effect of translation of uORF region as a fusion protein of reporter (ΔATGG) has a stimulatory influence on translation (mean ± SEM, n = 3, *P < 0.01). (F) published ribosome profiling data [Citation47] (available on-line at GSE89108 | GEO | 2017/08/04) show elongating ribosomes on the AdipoQ transcript from 0-day (top) and 5-day (middle) differentiating primary mouse adipocyte cultures, and mouse adipose tissue (bottom). The data was visualized using UCSC genome browser.
![Figure 2. Mouse adiponectin expression is inhibited by uORF element in the 5’-leader. (A) adiponectin is encoded on 3 exons. 5’-UTR (leader) sequence in exons 1 and 2 is shown above, sequence of exon 1 with the uORF ATGs (bold) are shown below. (B) an RLuc reporter with 5’-leader bearing A-to-G mutations in both uORF sites was generated; the vertical shift in the schematic indicates the altered reading frame (top). The mutated and control constructs were subjected to in vitro translation in a RRL, and luciferase activity determined (bottom; mean ± SEM, n = 3, **P < 0.01). (C) reporter fusion constructs were engineered with (AUGApN) without (ΔAUGG) the AUG of adiponectin (ApN), and with a single following ‘G’ to restore RLuc ORF in the same reading frame as the uOrfs. A triplet in RLuc that acts as a stop codon in the uORF reading frame is indicated (*) (schematic at top). RLuc immunoblot following translation by RRL (middle) and Ponceau stain (bottom). (D) effect of Adipoq uORF peptide on translation of luciferase mRNA in RRL (mean ± SEM, n = 3, ns, not significant). (E) effect of translation of uORF region as a fusion protein of reporter (ΔATGG) has a stimulatory influence on translation (mean ± SEM, n = 3, *P < 0.01). (F) published ribosome profiling data [Citation47] (available on-line at GSE89108 | GEO | 2017/08/04) show elongating ribosomes on the AdipoQ transcript from 0-day (top) and 5-day (middle) differentiating primary mouse adipocyte cultures, and mouse adipose tissue (bottom). The data was visualized using UCSC genome browser.](/cms/asset/1a08666f-563a-409e-8a78-c89e67c1a63c/krnb_a_2256094_f0002_b.gif)
The role of the uORF coding sequence in translation-inhibition was investigated. Translation-inhibition by the Adipoq 5’-leader could be due to (i) uORF-mediated generation of a short inhibitory peptide, (ii) ribosomal stalling during elongation due to codon usage bias or RNA secondary structure in the uORF, or (iii) bypass of the canonical Adipoq ATG start site. Pre-incubation of translation complexes with a 49-aa uORF peptide generated by in vitro translation did not inhibit control reporter translation in an RRL (). Similarly, translation of the 5’-leader as a fusion with the reporter is stimulatory, eliminating stalling due to rare codons as an inhibitory mechanism (). Reduced RLuc expression in the non-fused, out-of-frame construct is likely due to non-productive translation at the uORF ATGs that bypasses the RLuc start site, and terminates in the RLuc CDS. In a previous report, mouse primary white adipocyte tissue cultures were subjected to ribosome profiling, and ribosome occupancy of the Adipoq 5’-leader was determined [Citation47]. The non-adipocyte, stromal vascular fraction from 2–3-week C57BL/6 mice did not show significant ribosome occupancy; however, in vitro differentiation of pre-adipocytes to adipocytes markedly increased ribosome occupancy after 5 d (, top 2 panels). Ribosome occupancy in the 5’-UTR was exceptionally high in white adipose tissue isolated from 6-week mice (, bottom). These results are consistent with ribosomal engagement with an upstream start codon in the mouse Adipoq mRNA, indicating physiological utilization of uORFs overlapping the Adipoq CDS.
Adipoq mRNA translation resists repression by the integrated stress response
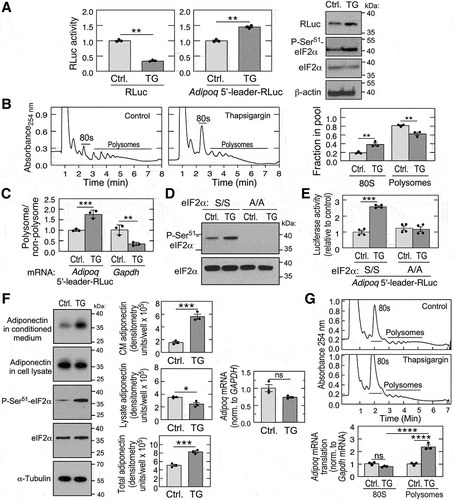
Phosphorylation of eukaryotic initiation factor-2α (eIF2α) at Ser51 exhibits a critical role in translation-initiation, integrating nutritional status, growth factors, and other stress signals, thereby acting as a checkpoint for translation [Citation49]. The resulting inhibition of global protein synthesis, termed the integrated stress response (ISR), reduces cellular energy expenditure, and facilitates a switch to pathways that direct cell resistance and recovery from stress. The switch is driven in part by a small family of uORF-containing transcripts that are not inhibited, or even stimulated, by the stress. Four kinases are responsible for eIF2α phosphorylation and the ISR, namely, haem-regulated inhibitor (HRI) kinase, general control non-derepressible 2 kinase (GCN2), protein kinase RNA-activated (PKR), and protein kinase R-like endoplasmic reticulum kinase (PERK) [Citation50]. Although about half of mouse and human transcripts contain uORFs [Citation2], their presence is not sufficient to determine response to P-eIF2α [Citation14]. To explore the potential participation of Adipoq mRNA and its uORF in the ISR, HEK293T cells were transfected with RLuc reporters, and then treated with thapsigargin, an inducer of PERK and the ISR. As expected for a repressor of global protein synthesis, thapsigargin decreased activity of a leader-less control RLuc reporter by about 75% (, left). In contrast, thapsigargin increased activity of the leader-containing RLuc reporter by about 40% compared to untreated control cells (, centre). Increased RLuc expression and eIF2α Ser51 phosphorylation by thapsigargin was shown by immunoblot (, right). Reduced global translation by thapsigargin was established by polysome profiling of HEK293T cell lysates which showed reduced RNA in polysome fractions, and a concomitant increase in monosomes (). The specific influence of thapsigargin on translation of Adipoq 5’leader-RLuc and Gapdh mRNAs was determined by qRT-PCR analysis of combined polysome profile fractions. Thapsigargin induced a ~75% increase in the polysome-to-non-polysome ratio of Adipoq 5’-leader-RLuc transcript; in contrast, the polysome-to-non-polysome ratio of GAPDH mRNA was reduced by ~65% (). To evaluate the role of eIF2α in uORF-regulated translation, we took advantage of phospho-null MEFs which introduced a Ser51-to-Ala (A/A) mutation in the eIF2α locus [Citation51]. As expected, thapsigargin induced phosphorylation of eIF2α at Ser51 in WT (S/S) MEFs, but P-Ser51-eIF2α was not detected in A/A mutant MEFs (). Thapsigargin increased RLuc activity from Adipoq 5’leader-RLuc by ~160% in WT MEFs, but no induction was observed in A/A cells (). These results indicate that the Adipoq 5’-leader directs preferential translation of adiponectin in response to stress-mediated eIF2α phosphorylation. The effect of stress on the expression of endogenous adiponectin was investigated in differentiated mouse 3T3-L1 adipocytic cells. The cells were treated with thapsigargin for 3 h, and adiponectin in the conditioned medium and cell lysate determined by immunoblot. A marked increase in adiponectin in the conditioned medium was observed, coupled with a minor decrease in the cell lysate (, left). Combining the amount in the lysate and conditioned medium revealed an ~60% increase in adiponectin expression (, centre), despite the minor thapsigargin-mediated reduction in Adipoq mRNA (, right). The thapsigargin-mediated increase in total adiponectin was shown to be primarily due to enhanced translation of Adipoq mRNA as shown by polysome profiling ().
Figure 3. Adiponectin is a positive responder to eIf2α phosphorylation and the ISR. (A) HEK293T cells were transfected with control (left) or 5’-leader-containing (centre) reporter, and treated with thapsigargin (TG) for 6 h, and luciferase activity determined (mean ± SEM, n = 3, **P < 0.01). Immunoblot analysis of 5’-leader-containing reporter (right). (B) polysome fractionation of control or TG-treated HEK293T cells; 80S and polysome peaks are indicated (left 2 panels). Relative rates of total protein synthesis were determined by quantification of areas under the curves for 80S and polysome peaks using NIH imageJ (right; mean ± SEM, n = 3, *P < 0.01). (C) Adipoq-5’-leader-RLuc and gapdh mRnas in polysomal and non-polysomal fractions were determined by RT-qPCR using TaqMan probes (mean ± SEM, n = 3, **P < 0.001, **P < 0.01). (D) immunoblot analysis of P-Ser51-eIF2α in wild-type (S/S) and phospho-deficient MEFs (A/A) treated with TG (left). (E) RLuc activity of S/S and A/A MEFs transfected with 5’-leader construct and treated with TG (mean ± SEM, n = 4, **P < 0.001). (F) differentiated 3T3-L1 adipocytes were treated with TG for 3 h, and adiponectin in the cell lysate and conditioned medium (CM), as well as eIf2a and P-Ser51-eIF2α, determined by immunoblot (left). Adiponectin in each fraction was quantitated by densitometry and corrected for aliquot size (centre). Adipoq mRNA was determined by RT-qPCR (right). (mean ± SEM, n = 3, **P < 0.05, ***P < 0.001, ns = non-significant). (G) translation rate of Adipoq mRNA was investigated by polysome fractionation of lysates from differentiated 3T3-L1 adipocytes subjected to control condition or treated with TG (top 2 panels). Adipoq and gapdh mRnas in polysomal and 80S monosomal fractions were determined by RT-qPCR using TaqMan probes (bottom; mean ± SEM, n = 3, **P < 0.0001).
Deletion of the Adipoq uORF increases adipocyte adiponectin expression and modulates downstream autocrine and paracrine responses
To investigate uORF-mediated depletion of endogenous adiponectin expression by adipocytes, and the physiological significance of such depletion, we generated a uORF-defective 3T3-L1 pre-adipocytic cell line using CRISPR-Cas9n-directed mutagenesis (). Guide RNA targeting the Adipoq 5’-leader deleted 11 nt (nt 22–32) of the 5’- leader, thereby eliminating the upstream ATG (, S2). A downstream nick in the 5’-leader removed 4 nt (nt 56–59) placing the 5’-leader downstream ATG in the same reading frame as the major ATG, without an intervening stop codon (Fig. S2). As expected, significant translation-initiation at the downstream 5’-leader ATG does not take place since the expected fusion protein containing a 43-amino acid N-terminus extension is not observed by immunoblot in differentiated 3T3-L1ΔuORF cells (, top), despite clear resolution of a fusion protein of nearly the same size (). This observation is consistent with the Kozak sequence-based prediction that the downstream ATG is not an initiation site [Citation48]. Importantly, adiponectin expression in the mutated cells was ~3-fold higher compared to untargeted 3T3-L1 cells consistent with uORF disruption, facilitating functional analysis of differential expression (, bottom). 3T3-L1 cells transduced with Adipoq lentivirus, or subjected to exogenous adiponectin, exhibit autocrine responses, including rapid differentiation, and increased triacylglycerol accumulation and glucose uptake [Citation20]. We thus investigated if increased expression of adiponectin mediated by uORF disruption was sufficient to alter autocrine responses in 3T3-L1 cells. The 3T3-L1ΔuORF cells exhibited a marked increase in neutral lipid accumulation as shown by Oil Red O stain (). Likewise, a 3-fold higher amount of triacylglycerol (TAG) in 3T3-L1ΔORF cells was shown by enzymatic assay following solvent extraction (). Basal and insulin-stimulated glucose uptake were substantially higher in differentiated 3T3-L1ΔORF adipocytes compared to untargeted 3T3-L1 control cells ().
Figure 4. Deletion of AdipoQ uORF increases adiponectin levels and insulin sensitivity. (A) schematic of region targeted by guide RNA. (B) Sanger sequencing of the 3T3-L1ΔORF exon 1 region of Adipoq. (C) immunoblot analysis of conditioned media from differentiated 3T3-L1 and 3T3-L1ΔORF adipocytes (left). Densitometric quantitation of adiponectin in conditioned media (right; mean ± SEM, n = 3, **P < 0.05). (D) Oil Red O staining of differentiated 3T3-L1 and 3T3-L1ΔORF adipocytes. (E) TAG accumulation in 3T3-L1 and 3T3-L1ΔORF adipocytes was determined after differentiation using enzymatic method (mean ± SEM, n = 3, *P < 0.05). (F) glucose uptake in untreated and insulin-treated 3T3-L1 and 3T3-L1ΔORF adipocytes (mean ± SEM, n = 3, **P < 0.05, **P < 0.01). (G) medium conditioned by 3T3-L1 and 3T3-L1ΔORF adipocytes for 24 h was incubated with C2C12 cells for 24 h, and G6PC1 and PCK1 mRNA determined by qRT-PCR (2nd and 3rd panels, mean ± SEM, n = 6 to 8, **P < 0.01). Similar experiment was repeated in Huh7 cells (4th and 5th panels, mean ± SEM, n = 3 to 5, **P < 0.01, ***P < 0.05).
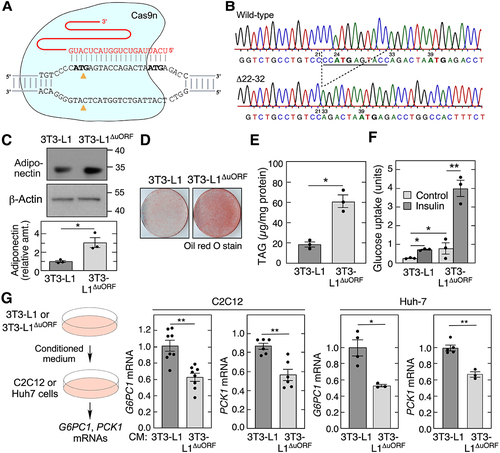
Adiponectin contributes to insulin sensitivity by repression of gluconeogenesis, primarily in liver and muscle cells [Citation15,Citation16]. The influence of Adipoq uORF on adiponectin paracrine function was investigated by its influence on expression of rate-limiting enzymes of gluconeogenesis, i.e. G6PC1 mRNA (encodes glucose-6-phosphatase) and PCK1 mRNA (encodes phosphoenolpyruvate carboxykinase) [Citation52]. C2C12 cells, a murine myoblast cell line, and Huh cells, a human hepatocyte-derived carcinoma cell line, were incubated for 24 h with medium conditioned for 24 h by differentiated 3T3-L1 or 3T3-L1ΔORF cells (, left). uORF disruption markedly reduced C2C12 cell expression of G6PC1 and PCK1 mRNA as determined by qRT-PCR, consistent with adiponectin-mediated inhibition of gluconeogenesis (, 2nd and 3rd panels). A similar effect on gluconeogenic enzymes was observed in hepatocyte-derived Huh-7 cells exposed to the conditioned media (, 4th and 5th panels). Together, these results indicate the Adipoq uORF is a potent negative regulator of basal and agonist-stimulated, adiponectin-dependent autocrine and paracrine metabolic functions.
Discussion
Gene expression is regulated at transcriptional and post-transcriptional steps, including translation. Compared to transcription, translational control permits a more rapid regulatory response to environmental stimuli. Translation-initiation is the principal rate-limiting step that determines protein synthesis rate and is subject to multiple control mechanisms, often involving upstream or downstream untranslated regions. Here, we show that basal adiponectin protein expression is repressed by a uORF in the 5’-leader of mouse Adipoq mRNA. However, expression is stimulated by thapsigargin, an inducer of the ISR and global inhibitor of protein synthesis. About half of human and mouse mRNAs contain one or more uORFs [Citation2], but a much more limited group has been shown experimentally to be resistant to repression by the ISR. Ribosome profiling revealed translation of 10 mRNAs was resistant to stress induced by sodium arsenite [Citation12]. In six transcripts, translation was increased; in the remaining four, translation was reduced to a much lesser extent than the global reduction. Importantly, nearly all stress-resistant mRNAs possessed one or more translated uORFs that suppressed translation of the major ORF under non-stress conditions [Citation12]. A second study evaluated the global translation response following induction of the unfolded protein response by tunicamycin, a toxin that induces ER stress by inhibiting N-linked glycosylation of ER-resident proteins. This study showed substantially altered translation in 78 mRNAs following ER stress in HEK293T cells [Citation11]. Thus, discovery and experimental validation of a new uORF-mediated, ISR-regulated transcript represents a significant contribution despite the near-omnipresence of uORFs.
The Adipoq uORF consists of a pair of ATGs spaced 15 nts apart in exon 1 and just 24 nt downstream of the transcript 5 end. The uORF terminates in a TGA stop codon at nt position 171 in exon 2, potentially encoding peptides of length 44 and 49 amino acids. Importantly, the uORF overlaps and is out-of-frame with the major Adipoq ORF. The overlap of the uORF with the major ORF is similar to other reported examples of uORF-mediated repression, e.g. ATF4 and EPRS1 [Citation53,Citation54]. Multiple experiments provide evidence for substantial uORF-mediated repression of adiponectin expression. In vitro translation of a reporter bearing the Adipoq 5’-leader was increased about 3-fold when the ATG start codons were eliminated by mutation to GTG. The result suggests that adipocyte-specific factors were not required for the observed suppression. Likewise, a 3-fold increase in endogenous adiponectin expression was observed in differentiated 3T3-L1 adipocytes in which the upstream ATG in the 5’-leader was deleted by CRISPR-Cas9-directed mutagenesis. The latter result is particularly important since adipocytes are the only cells that express Adipoq mRNA and protein. The result suggests the upstream ATG in the 5’-leader is essential for gene repression since the mutagenesis did not delete the downstream ATG. The elevated expression of adiponectin in the absence of the uORF was shown to be functional in autocrine and paracrine assays. uORF deletion increased TAG accumulation and glucose uptake in differentiated adipocytes. Likewise, conditioned medium from uORF-defective adipocytes inhibited the expression of gluconeogenic enzymes in muscle- and liver-derived cells.
Expression of a reporter driven by the Adipoq 5’-leader was enhanced by thapsigargin. Polysome profiling showed the stimulation by thapsigargin was at the level of translation. Moreover, the requirement for eIF2α revealed that Adipoq is a new uORF-mediated target of the ISR. Several mechanisms of initiation site selection directed by eIF2α-P accumulation in the ISR have been proposed. According to one mechanism, the high-level eIF2α-P delays formation of the eIF2 ternary complex, facilitating bypass of the inhibitory uORFs to promote initiation at the major translation-initiation site, as reported for ATF4 and GCN4 production [Citation14]. According to a second proposed mechanism, eIF2α-P destabilizes the interaction between the scanning ribosomal subunit and the mRNA at the −3 position of the start codon Kozak sequence where a purine (A/G) is essential for optimal initiation [Citation55]. Importantly, both ATG initiation sites in the uORF have a non-purine ‘C’ in the −3 position, whereas the purine ‘A’ occupies that position in the initiation site of the major ORF in mouse Adipoq. Possibly, eIF2α-P has a greater inhibitory effect on initiation at the weak Kozak sequences in the uORF compared to the major ORF, thereby enhancing leaky scanning. Consistent with this mechanism, weak Kozak sequences in uORFs often indicate susceptibility to stimulation by eIF2α-P [Citation14].
The widespread prevalence of uORFs in mammalian genes suggests they must confer a significant advantage to well-being of the host. However, the specific advantages conferred remain unclear. Certainly, expression of a uORF-encoded peptide that is coordinated with expression of the principal protein has potential functionality. In fact, nascent peptides encoded by uORFs can stall ribosomes by a metabolite-regulated mechanism [Citation56]. However, a multi-species genomic and proteomic analysis led to the conclusion that only a small fraction of uORFs (<1%) encode peptides maintained during evolution by natural selection [Citation57]. This conclusion is consistent with the result shown here that the uORF-directed peptide of Adipoq is not responsible for diminished synthesis of adiponectin. There is substantial evidence for peptide-independent mechanisms of uORF-mediated regulation of translation. As mentioned above, a major mechanism involves altered expression following stress-dependent accumulation of P-eIF2α in the ISR. An advantage of this mechanism is the increased expression of a post-transcriptional family of proteins that specifically respond to the stress, despite the overall inhibition of protein synthesis to conserve energy expenditure. A second advantage of the uORF-ISR pathway is the speed of response. Response speed is a well-known advantage of translational control mechanisms compared to transcriptional control, as the former eliminates a major pathway step, i.e. transcript formation. In a mechanism analogous to relieving a brake in an automobile, the relief of uORF-mediated inhibition of translation is extremely rapid as it is dependent on a single catalytic reaction, i.e. phosphorylation of eIF2α, that rapidly overcomes the genetically programmed brake. Moreover, translational control mechanisms uniquely permit condition-dependent, extra-nuclear localization of the response, whereas transcriptional responses are confined to the nucleus.
Activation of the ISR by cell-extrinsic factors, for example, hypoxia, amino acid or glucose deprivation, and viral infections, triggers a multi-level protective response. The rapid phase of the response is mediated by kinase-activated phosphorylation of eIF1α and relief of uORF-mediated inhibition of translation, followed by a slower phase dependent on ATF4-mediated transcriptional induction of gene expression [Citation58]. Several activities of adiponectin are reliant on rapid response, and thus are candidates for uORF-mediated translational activation. During amino acid starvation, preferential translation of Adipoq mRNA would suppress gluconeogenesis in the target organs, liver and muscle, by inhibiting G6Pase and PEPCK, thereby increasing the cellular pool of amino acids. Moreover, as a paracrine signalling agent that stimulates fatty acid β-oxidation in skeletal muscle, a rapid response to exercise is advantageous.
Beyond its important role in lipid and glucose homoeostasis, adiponectin influences multiple pathological conditions and disorders, including carcinogenesis. Elevated adiponectin levels are associated with lower risk for multiple cancers including ovarian, breast, and prostate [Citation59,Citation60]. Decreased risk is consistent with the robust inhibition of in vitro cancer cell proliferation by adiponectin, and its stimulation of apoptosis [Citation60]. The specific role of ISR-mediated induction of adiponectin in tumorigenesis has not been explored. In view of our finding that the ISR increases adiponectin expression, one might predict that the ISR would likewise decrease cancer risk. In contrast, recent investigations reveal the ISR is pro-tumorigenic [Citation59,Citation60] and has been proposed as a therapeutic option for KRAS-driven lung cancer [Citation61]. The apparent conflict is almost certainly due to the complexity of the ISR, particularly, the selective increase in expression of a few proteins, but inhibition of synthesis of the vast majority. Adipocytes themselves secrete a host of pro- and anti-inflammatory cytokines [Citation62], but the effect of the ISR on individual adipokines, as well as the net effect, is not known. Moreover, stress-induced expression of adiponectin is likely to be transient, whereas tumorigenesis is chronic. Lastly, although the tumour cells are negatively affected by the ISR, there is no evidence that nearby or distal adipocytes are similarly influenced during tumorigenesis.
In summary, Adipoq is subject to transcriptional activation by transcription factors involved in adipogenesis, and is suppressed by transcriptional repressors and pro-inflammatory agents [Citation63–65]. Here, we report an additional layer of rapid control of adiponectin expression at the level of translation. Post-transcriptional regulation of adiponectin is likely to be critical for rapid adaptation of serum levels and tissue responses during dynamic metabolic demands initiated by stress.
Materials and methods
Cells, reagents, and antibodies
Mycoplasma-tested mouse pre-adipocytes (3T3-L1, ATCC-CL-173) were cultured under standard conditions in high-glucose DMEM (GIBCO, Life Technologies, Carlsbad, CA) supplemented with 10% foetal bovine serum, penicillin/streptomycin, 4 mM L-glutamine without sodium pyruvate. HEK293F (ThermoFisher, Waltham, MA) and Huh7 (JCRB Cell Bank, JCRB0403) cells were cultured in high-glucose DMEM supplemented with 10% foetal bovine serum and 4 mM L-glutamine. For differentiation of 3T3-L1 cells into adipocytes, cells at 75% confluence were cultured in medium containing DMEM and 10% FBS supplemented with 1× solutions of insulin, dexamethasone, and 3-isobutyl-1-methylxanthine (Cayman, Ann Arbor, MI) for 72 h, as described [Citation66,Citation67]. The medium was replaced with 10% FBS and DMEM containing insulin, and maintained for a wk with three medium changes. Adipocytes were maintained in DMEM with 10% calf serum and 1× antibiotic/antimycotic for at least 3 d before use. Rabbit reticulocyte lysate and luciferase reagents were purchased from Promega (Madison, WI). Modified or unmodified RNA and DNA oligonucleotides were obtained from Integrated DNA Technologies (IDT; Coralville, IA). Antibodies against FLAG peptide (GenScript; Piscataway, NJ), AMPKα, P-Thr172 AMPK (Cell Signaling, Danvers, MA), Rluc, GAPDH, α-tubulin, and β-actin were from Proteintech (Rosemont, IL), and against adiponectin from (R&D systems, Minneapolis, MN). Mouse embryo fibroblasts (MEFs) bearing a non-phosphorylatable Ser51Ala mutation in EIF2α were generated by homologous recombination [Citation51].
cDNA cloning, constructs, and peptide expression
All Adipoq constructs were cloned by double-stranded gene fragment synthesis (IDT, Coralville, IA) and assembled using NEBuilder® HiFi DNA Assembly (NEB, Ipswich, MA). Mutations in the 5’-leader were constructed using Gblock gene fragments and verified by Sanger sequencing. The 49-aa uORF peptide was generated by an in vitro coupled transcription-translation reaction (500 ng vector/reaction) using TNT T7-coupled rabbit reticulocyte lysate (Promega). A similar reaction using the empty vector served as a control.
Real-time PCR analysis
Total cellular RNA was isolated with Trizol or RNeasy mini kit (Qiagen, Valencia, CA), and subjected to reverse transcription and real-time PCR using AgPath-ID One-Step RT-PCR reagent (Ambion). TaqMan probes specific for Adipoq, Pparg, Cebp1a, Srebp1c, luciferase, G6PC1, PCK1, β-actin, and GAPDH (Applied Biosystems) were used. For qPCR analysis of polysome fractions, total RNA was isolated from combined fractions by extraction with Trizol LS reagent, and purified by RNeasy minikit (Qiagen). RNA was quantitated and used for real-time PCR analysis.
Immunoblot analysis
Cell lysates were denatured and resolved on 4–20% gradient SDS-PAGE. After transfer, the blots were probed with specific primary antibody followed by HRP-conjugated secondary antibody, and developed using ECL or ECL plus reagent (GE Healthcare).
Luciferase assays
Plasmids containing luciferase were transfected into HEK293 cells in 24-well plates using Lipofectamine 2000 (ThermoFisher). Fluc plasmid (100 ng/well) was co-transfected as transfection efficiency control. After 24 h, cells were lysed and RLuc and FLuc activity measured using Renilla-Glo luciferase assay system and luciferase assay reagent (Promega), respectively, in a Victor3 1420 Multilabel Plate Counter (Perkin-Elmer). The same constructs were subjected to in vitro transcription and translation using T7 RNA polymerase (Takara Bio USA, San Jose, CA) and rabbit reticulocyte lysate (Promega). RNAs were generated as described [Citation68–70]. For TG experiments, cells were treated 4 h after transfection for 6 h, and lysed using passive lysis buffer (Promega).
Polysome profiling
Ribosome-rich, translationally-active and ribosome-poor, inactive mRNA pools were isolated by sucrose gradient fractionation. Cycloheximide (100 µg/mL) was added to 107 HEK293F cells or differentiated 3T3-L1 adipocytes for 20 min, and cells collected by low-speed centrifugation and washed twice with cycloheximide-containing, ice-cold phosphate-buffered saline (PBS). Cell pellets were suspended in 350 µL of lysis buffer (10 mM Tris pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% Triton X-100, 0.5% deoxycholate, 2 mM DTT, 100 µg/mL cycloheximide, and RNAse inhibitor), and incubated for 5 min on ice. The lysates were centrifuged at 15,000 × g for 10 min and supernatants collected. RNase inhibitor (5 µL, 40 U/µL) and cycloheximide (100 µg/mL) were added to 50 mL each of freshly prepared 10% and 50% sucrose gradient solutions (in 20 mM HEPES, pH 7.4, 100 mM KCl, 5 mM MgCl2, and 2 mM DTT) just before use. Lysates were loaded onto the sucrose gradient and centrifuged at 36,000 rpm for 2 h, and ~0.75 mL fractions collected. Fractions containing light ribonucleoproteins, 40S, 60S, and 80S ribosomes formed the translationally inactive pool, and heavy polysome fractions formed the translationally active pool.
Generation of AdipoqΔuORF 3T3-L1 pre-adipocyte cell line
Adipoq (GenBank accession no. NM_009605) was mutated in 3T3-L1 cells by CRISPR-Cas9n genome editing [Citation71]. sgRNAs targeting Adipoq uORF were designed by web-based tool (https://www.benchling.com/), and cloned into lentiCRISPR v2 vector (Addgene) with paired D10A Cas9n nickases, a Cas9 version that improves efficiency and reduces off-target effects [Citation72]. The AdipoqΔuORF cell line was generated following lentiviral transduction of lentiCRISPR v2-Cas9 D10A-AdipoqΔuORF sgRNA in 3T3-L1 cells. AdipoqΔuORF single-cell clones were isolated and expanded following fluorescence-activated cell sorting (FACS). Genomic DNA was extracted from 26 isolated clones using Adipoq uORF-specific primers. The PCR product was gel-purified with gel extraction kit (ThermoFisher), and Sanger sequenced using the following primers designed to give a unique product with length 408 bp:
Primers used for gene editing are as follows:
AdipoqΔuORF Nick-5F: CACCGTCATTAGTCTGGTACTCATG
AdipoqΔuORF Nick-5 R: AAACCATGAGTACCAGACTAATGAC
AdipoqΔuORF Nick-3F: CACCGTCTGTCTGTACGATTGTCAG
AdipoqΔuORF Nick-3 R: AAACCTGACAATCGTACAGACAGAC
Oil red O staining
Differentiated 3T3-L1 adipocytes were washed with 1 ml of PBS (Corning, Corning, NY) before fixation for 15 min with 4% paraformaldehyde in PBS at room temperature. Fixed cells were washed three times with 1 ml of PBS and once with 60% isopropanol, and thoroughly air-dried. Cells were stained with 60% Oil Red O (Sigma) in double-distilled H2O for 10 min at room temperature. Excess stain was removed with four washes with deionized water.
Adipocyte triacylglycerol determination
Briefly, lipids from 5 × 106 cells were extracted in chloroform/methanol (2:1, v/v) at room temperature overnight. The extract was quantitatively separated from protein, dried under N2, and re-dissolved in chloroform/methanol (2:1, v/v). The lipid-containing upper phase was aspirated and dried. Triton X-100 (1% in chloroform) was added, and the solvent evaporated. Deionized water was added and vortexed until the solution cleared, and total triacylglycerol quantified by enzymatic assay (L-Type Triglyceride M Assay kit, Wako Diagnostics, Richmond, VA).
Glucose uptake
Glucose uptake by differentiated 3T3-L1 adipocytes was determined using Glucose Uptake Glo Assay kit (Promega, #J1342) and luminescence read with 0.3–1 sec integration on a luminometer (VICTOR3 Multilabel Plate Reader, Perkin-Elmer).
Statistical analysis
The experiments were performed in triplicate unless described otherwise. The data are expressed as mean ± SEM. Statistical analysis was done by Student’s t-test using Prism 9.0 software (GraphPad); P < 0.05 was considered to be statistically significant.
Supplemental Material
Download Zip (7.4 MB)Acknowledgments
We are grateful to all members of the Fox laboratory for helpful discussions and criticism.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2256094.
Additional information
Funding
References
- Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234(2):187–208. doi: 10.1016/S0378-1119(99)00210-3
- Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A. 2009;106(18):7507–7512. doi: 10.1073/pnas.0810916106
- Chew GL, Pauli A, Schier AF. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat Commun. 2016;7(1):11663. doi: 10.1038/ncomms11663
- Churbanov A, Rogozin IB, Babenko VN, et al. Evolutionary conservation suggests a regulatory function of AUG triplets in 5’-UTRs of eukaryotic genes. Nucleic Acids Research. 2005;33(17):5512–5520. doi: 10.1093/nar/gki847
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789–802. doi: 10.1016/j.cell.2011.10.002
- Lee S, Liu B, Lee S, et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A. 2012;109(37):E2424–32. doi: 10.1073/pnas.1207846109
- Gao X, Wan J, Liu B, et al. Quantitative profiling of initiating ribosomes in vivo. Nat Methods. 2015;12(2):147–153. doi: 10.1038/nmeth.3208
- McGillivray P, Ault R, Pawashe M, et al. A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Res. 2018;46(7):3326–3338. doi: 10.1093/nar/gky188
- Barbosa C, Peixeiro I, Romao L, et al. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9(8):e1003529. doi: 10.1371/journal.pgen.1003529
- Pavitt GD, Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol. 2012;4(6):a012278. doi: 10.1101/cshperspect.a012278
- Sidrauski C, McGeachy AM, Ingolia NT, et al. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015;4:e05033. doi: 10.7554/eLife.05033
- Andreev DE, O’Connor PB, Fahey C, et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. Elife. 2015;4:e03971. doi: 10.7554/eLife.03971
- Wethmar K, Smink JJ, Leutz A. Upstream open reading frames: molecular switches in (patho)physiology. BioEssays. 2010;32(10):885–893. doi: 10.1002/bies.201000037
- Young SK, Wek RC. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J Biol Chem. 2016;291(33):16927–16935. doi: 10.1074/jbc.R116.733899
- Trujillo ME, Scherer PE. Adiponectin–journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med. 2005;257(2):167–175. doi: 10.1111/j.1365-2796.2004.01426.x
- Straub LG, Scherer PE. Metabolic messengers: adiponectin. Nat Metab. 2019;1(3):334–339. doi: 10.1038/s42255-019-0041-z
- Guo Q, Cao S, Wang X. Adiponectin intervention to regulate betatrophin expression, attenuate insulin resistance and enhance glucose metabolism in mice and its response to exercise. Int J Mol Sci. 2022;23(18):23. doi: 10.3390/ijms231810630
- Lafontan M, Viguerie N. Role of adipokines in the control of energy metabolism: focus on adiponectin. Curr Opin Pharmacol. 2006;6(6):580–585. doi: 10.1016/j.coph.2006.08.002
- Rasmussen MS, Lihn AS, Pedersen SB, et al. Adiponectin receptors in human adipose tissue: effects of obesity, weight loss, and fat depots. Obesity (Silver Spring). 2006;14:28–35. doi: 10.1038/oby.2006.5
- Fu Y, Luo N, Klein RL, et al. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J Lipid Res. 2005;46(7):1369–1379. doi: 10.1194/jlr.M400373-JLR200
- Maeda N, Funahashi T, Matsuzawa Y, et al. Adiponectin, a unique adipocyte-derived factor beyond hormones. Atherosclerosis. 2020;292:1–9. doi: 10.1016/j.atherosclerosis.2019.10.021
- Kubota N, Yano W, Kubota T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6(1):55–68. doi: 10.1016/j.cmet.2007.06.003
- Turyn J, Korczynska J, Presler M, et al. Up-regulation of rat adipose tissue adiponectin gene expression by long-term but not by short-term food restriction. Mol Cell Biochem. 2008;312(1–2):185–191. doi: 10.1007/s11010-008-9733-5
- Sanchez-Gurmaches J, Cruz-Garcia L, Gutierrez J, et al. Adiponectin effects and gene expression in rainbow trout: an in vivo and in vitro approach. J Exp Biol. 2012;215(8):1373–1383. doi: 10.1242/jeb.061697
- Khalil RB, El Hachem C. Adiponectin in eating disorders. Eat Weight Disord. 2014;19(1):3–10. doi: 10.1007/s40519-013-0094-z
- Monteleone P, Fabrazzo M, Martiadis V, et al. Opposite changes in circulating adiponectin in women with bulimia nervosa or binge eating disorder. J Clin Endocrinol Metab. 2003;88(11):5387–5391. doi: 10.1210/jc.2003-030956
- Miyazaki Y, Mahankali A, Wajcberg E, et al. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89(9):4312–4319. doi: 10.1210/jc.2004-0190
- Yang WS, Jeng CY, Wu TJ, et al. Synthetic peroxisome proliferator-activated receptor-γ agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes Care. 2002;25(2):376–380. doi: 10.2337/diacare.25.2.376
- Bajaj M, Suraamornkul S, Piper P, et al. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89(1):200–206. doi: 10.1210/jc.2003-031315
- Iwaki M, Matsuda M, Maeda N, et al. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52(7):1655–1663. doi: 10.2337/diabetes.52.7.1655
- Maeda N, Takahashi M, Funahashi T, et al. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50(9):2094–2099. doi: 10.2337/diabetes.50.9.2094
- Tonelli J, Li W, Kishore P, et al. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes. 2004;53(6):1621–1629. doi: 10.2337/diabetes.53.6.1621
- Tiikkainen M, Hakkinen AM, Korsheninnikova E, et al. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53(8):2169–2176. doi: 10.2337/diabetes.53.8.2169
- Yu JG, Javorschi S, Hevener AL, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51(10):2968–2974. doi: 10.2337/diabetes.51.10.2968
- Rasouli N, Yao-Borengasser A, Miles LM, et al. Increased plasma adiponectin in response to pioglitazone does not result from increased gene expression. Am J Physiol Endocrinol Metab. 2006;290(1):E42–E6. doi: 10.1152/ajpendo.00240.2005
- Banga A, Unal R, Tripathi P, et al. Adiponectin translation is increased by the PPARγ agonists pioglitazone and ω-3 fatty acids. Am J Physiol Endocrinol Metab. 2009;296(3):E480–9. doi: 10.1152/ajpendo.90892.2008
- Ishida M, Shimabukuro M, Yagi S, et al. MicroRNA-378 regulates adiponectin expression in adipose tissue: a new plausible mechanism. PLoS One. 2014;9(11):e111537. doi: 10.1371/journal.pone.0111537
- Rajan S, Panzade G, Srivastava A, et al. miR-876-3p regulates glucose homeostasis and insulin sensitivity by targeting adiponectin. J Endocrinol. 2018;239(1):1–17. doi: 10.1530/JOE-17-0387
- Hwang JS, Lee WJ, Hur J, et al. Rosiglitazone-dependent dissociation of HuR from PPAR-gamma regulates adiponectin expression at the posttranscriptional level. FASEB J. 2019;33:7707–7720. doi: 10.1096/fj.201802643R
- Matsuda M, Shimomura I. Roles of adiponectin and oxidative stress in obesity-associated metabolic and cardiovascular diseases. Rev Endocr Metab Disord. 2014;15(1):1–10. doi: 10.1007/s11154-013-9271-7
- Liu Z, Gan L, Wu T, et al. Adiponectin reduces ER stress-induced apoptosis through PPARα transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016;7(11):e2487. doi: 10.1038/cddis.2016.388
- Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28(2):91–98. doi: 10.1016/S0968-0004(03)00002-1
- Gebauer F, Preiss T, Hentze MW. From cis-regulatory elements to complex RNPs and back. Cold Spring Harb Perspect Biol. 2012;4(7):a012245. doi: 10.1101/cshperspect.a012245
- Mazumder B, Seshadri V, Imataka H, et al. Translational silencing of ceruloplasmin requires the essential elements of mRNA circularization: poly(A) tail, poly(A)-binding protein, and eukaryotic translation initiation factor 4G. Mol Cell Biol. 2001;21(19):6440–6449. doi: 10.1128/MCB.21.19.6440-6449.2001
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595
- Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite. Nucleic Acids Res. 2008;36(Web Server):W70–4. doi: 10.1093/nar/gkn188
- Reid DW, Xu D, Chen P, et al. Integrative analyses of translatome and transcriptome reveal important translational controls in brown and white adipose regulated by microRNAs. Sci Rep. 2017;7(1):5681. doi: 10.1038/s41598-017-06077-3
- Gleason AC, Ghadge G, Chen J, et al. Machine learning predicts translation initiation sites in neurologic diseases with nucleotide repeat expansions. PLoS One. 2022;17(6):e0256411. doi: 10.1371/journal.pone.0256411
- Wek RC. Role of eIF2alpha kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol. 2018;10(7):a032870. doi: 10.1101/cshperspect.a032870
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6(4):318–327. doi: 10.1038/nrm1618
- Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7(6):1165–1176. doi: 10.1016/S1097-2765(01)00265-9
- Miller RA, Chu Q, Le Lay J, et al. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121(6):2518–2528. doi: 10.1172/JCI45942
- Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Bio. 2004;167(1):27–33. doi: 10.1083/jcb.200408003
- Young SK, Baird TD, Wek RC. Translation regulation of the glutamyl-prolyl-tRNA synthetase gene EPRS through bypass of upstream open reading frames with noncanonical initiation codons. J Biol Chem. 2016;291(20):10824–10835. doi: 10.1074/jbc.M116.722256
- Hussain T, Llacer JL, Fernandez IS, et al. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell. 2014;159(3):597–607. doi: 10.1016/j.cell.2014.10.001
- Dever TE, Ivanov IP, Sachs MS. Conserved upstream open reading frame nascent peptides that control translation. Ann Rev Genet. 2020;54(1):237–264. doi: 10.1146/annurev-genet-112618-043822
- Zhang H, Wang Y, Wu X, et al. Determinants of genome-wide distribution and evolution of uOrfs in eukaryotes. Nat Commun. 2021;12(1):1076. doi: 10.1038/s41467-021-21394-y
- Pakos-Zebrucka K, Koryga I, Mnich K, et al. The integrated stress response. EMBO Rep. 2016;17(10):1374–1395. doi: 10.15252/embr.201642195
- Naimo GD, Gelsomino L, Catalano S, et al. Interfering role of ERalpha on adiponectin action in breast cancer. Front Endocrinol. 2020;11:66. doi: 10.3389/fendo.2020.00066
- Tsankof A, Tziomalos K. Adiponectin: a player in the pathogenesis of hormone-dependent cancers. Front Endocrinol. 2022;13:1018515. doi: 10.3389/fendo.2022.1018515
- Ghaddar N, Wang S, Woodvine B, et al. The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-driven lung cancer. Nat Commun. 2021;12(1):4651. doi: 10.1038/s41467-021-24661-0
- Ren Y, Zhao H, Yin C, et al. Adipokines, hepatokines and myokines: focus on their role and molecular mechanisms in adipose tissue inflammation. Front Endocrinol. 2022;13:873699. doi: 10.3389/fendo.2022.873699
- Zappala G, Rechler MM. IGFBP-3, hypoxia and TNF-α inhibit adiponectin transcription. Biochemical And Biophysical Research Communications. 2009;382(4):785–789. doi: 10.1016/j.bbrc.2009.03.112
- Chandrasekar B, Patel DN, Mummidi S, et al. Interleukin-18 suppresses adiponectin expression in 3T3-L1 adipocytes via a novel signal transduction pathway involving ERK1/2-dependent NFATc4 phosphorylation. J Biol Chem. 2008;283(7):4200–4209. doi: 10.1074/jbc.M708142200
- Simons PJ, van den Pangaart PS, Aerts JM, et al. Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J Endocrinol. 2007;192(2):289–299. doi: 10.1677/JOE-06-0047
- Arif A, Jia J, Willard B, et al. Multisite phosphorylation of S6K1 directs a kinase phospho-code that determines substrate selection. Mol Cell. 2019;73(3):446–457. doi: 10.1016/j.molcel.2018.11.017
- Arif A, Terenzi F, Potdar AA, et al. EPRS is a critical mTORC1–S6K1 effector that influences adiposity in mice. Nature. 2017;542(7641):357–361. doi: 10.1038/nature21380
- Yao P, Potdar AA, Ray PS, et al. The HILDA complex coordinates a conditional switch in the 3′-untranslated region of the VEGFA mRNA. PLoS Biol. 2013;11(8):e1001635. doi: 10.1371/journal.pbio.1001635
- Yao P, Potdar AA, Arif A, et al. Coding region polyadenylation generates a truncated tRNA synthetase that counters translation repression. Cell. 2012;149(1):88–100. doi: 10.1016/j.cell.2012.02.018
- Sampath P, Mazumder B, Seshadri V, et al. Transcript-selective translational silencing by Gamma Interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol. 2003;23(5):1509–1519. doi: 10.1128/MCB.23.5.1509-1519.2003
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143
- Gopalappa R, Suresh B, Ramakrishna S, et al. Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res. 2018;46(12):e71. doi: 10.1093/nar/gky222