Abstract
Asthma has origins in early life. Epidemiological studies show that maternal, more than paternal, asthma significantly increases a child’s risk of developing the disease. Experimental animal models exist which reproduce the increased susceptibility to asthma seen in human studies, and allow analysis of immunotoxic mechanisms that may contribute to neonatal allergy. In addition to maternal asthma, chemically-induced skin contact hypersensitivity or exposure during pregnancy of non-allergic females to certain environmental agents, e.g., air pollution particles, can also result in increased susceptibility to asthma in their offspring. We review here experimental models of maternal transmission of asthma risk, the progress to date in identifying mechanisms, and potential directions for future research.
| Abbreviations: | ||
| AHR – | = | airway hyperresponsiveness, |
| AI – | = | airway inflammation, |
| CAS – | = | bovine casein, |
| CpG – | = | Cyt-phosphate-Guo oligonucleotides, |
| DCs – | = | dendritic cells, |
| OVA – | = | chicken ovalbumin, |
| Rcp – | = | recipient |
Maternal asthma
Approximately 20 million Americans have asthma (American Lung Association, Citation2005), with prevalence increasing (Centers for Disease Control, Citation1998), especially in children. Currently asthma is the most common serious chronic disease of childhood (Asthma and Allergy Foundation, Citation2000).
Asthma has origins in early life (Gern et al., 1999; Holt and Jones, 2000; Vance and Holloway, 2002; Wright, 2004; Saglani and Bush, 2007) and the first exposure to allergen at a young age may be a critical factor in development of the disease (Gelfand et al., 2004). While human susceptibility to asthma has clearly both genetic predisposition and environmental factors contributing to onset of the disease (Holloway et al., 1999; Hammad and Lambrecht, 2006), epidemiological studies show that maternal asthma, rather than paternal, significantly increases the risk of developing the disease during childhood (Ruiz et al., 1992; Litonjua et al., 1998; Liu et al., 2003). In an especially extensive epidemiologic study from Arizona, Martinez et al. (1995) showed that maternal asthma is a major risk factor for persistent wheezing and for late onset wheezing even after 6 years of age. Similar findings were made in the United Kingdom, indicating that the problem is not geographically restricted (Kurukulaaratchy et al., Citation2005). This ‘maternal effect’ suggests that perinatal events dramatically influence the early susceptibility to allergic airway disease (Prescott, 2006). Notably, current therapeutic approaches are unable to change the natural course of the disease early in life (Guilbert et al., 2006). Hence, progress in under-standing the mechanisms of the maternal effect may allow for better prevention and therapy.
This review will focus on studies of neonatal allergy as a consequence of maternal exposures. More general efforts to develop and improve developmental immunotoxicity testing models have been extensively reviewed by Dietert and Holsapple (Dietert and Holsapple, 2007).
Experimental models
Existing experimental models of maternal transmission of asthma and allergy risk can be subdivided into allergen-induced maternal asthma and maternal toxic exposure models. For both types, maternal exposures can occur either premating or during pregnancy.
Allergen models
In a dog model, Barrett et al. (2003) were able to show that the offspring from allergic ragweed-sensitized parents, when exposed to inhaled allergen, develop allergic sensitization and an asthmatic phenotype, whereas the offspring from non-allergic parents do not. Since both parents were allergic, data from these studies does not specify whether a parental effect was maternal or paternal. One valuable advantage of the dog model is the potential to obtain larger amounts of fluids or tissues for mechanistic analysis. While the development of the immune system in dogs is more similar to human biology than seen with rodents, there remain differences in placental structure in both canines and rodents compared to human that may prove relevant (Holsapple et al., 2003).
A mouse model of maternal asthma risk was suggested by Herz et al. (2001). In this model BALB/c mice were sensitized to OVA before mating followed by allergen aerosol exposure during pregnancy. T- and B-lymphocyte responses in offspring were followed up until Day 60 postpartum. The offspring of OVA-sensitized mothers had decreased interferon (IFN)-γ production in splenic mononuclear cells as compared to offspring from normal mothers. These offspring also had higher total (but not OVA-specific) IgE levels at 4 wk. A group of the offspring from OVA-sensitized mothers was immunized with β-lactoglobulin (BLG). These mice were noted to have higher levels of anti-BLG IgG1. A large percentage of the OVA-sensitized groups developed skin hypersensitivity reactions consistent with a higher response to allergic stimuli (Herz et al., 2001).
Similar findings were reported by Uthoff et al. (Citation2003), however in their study development of an IgE response against the same allergen was completely prevented early in life. This effect was mediated by diaplacental transfer of allergen-specific IgG1 antibodies (Abs). In contrast, allergic sensitization against a different allergen early in life was accelerated in these mice. This effect was mediated by maternal CD4 and OVA-specific TH2 cells induced by allergic sensitization during pregnancy.
In a mouse model developed in the authors’ laboratory by Hamada et al. (2003) offspring of asthmatic mother mice (sensitized and repeatedly exposed to OVA antigen) showed airway hyperresponsiveness and allergic pulmonary inflammation after an intentionally suboptimal OVA sensitization and exposure protocol that had little effect on normal offspring. Interestingly, the maternal effect was allergen independent since similar results were obtained when offspring of OVA-allergic mothers were exposed to an unrelated allergen, casein. This indicates that the maternal transmission phenomenon cannot be attributed to simple carryover of OVA-specific antibodies. In this model pre-mating treatment with neutralizing anti-interleukin (IL)-4 Ab or reduction of maternal allergen exposure abrogated the maternal effect, showing a critical mechanistic role for IL-4 and suggesting an additional benefit of allergen avoidance. Additionally, allergen exposure during pregnancy is not required to cause the maternal effect, which also helps to exclude carryover of antigen as a possible mechanism. Although the precise cell and molecular basis of maternal transmission remains yet to be identified, this model has opened opportunities for interesting insights regarding potential therapeutic interventions to prevent allergy origin (e.g., with CpG oligos) (Fedulov et al., Citation2005), and provides a convenient tool especially since the vertically-transmitted increased susceptibility is quite durable. Offspring of asthmatic (but not normal) mice develop a full asthma phenotype (airway hyperrespon-siveness [AHR] and allergic inflammation [AI]) even if their initial sensitization occurs as late as 6 wk-of-age. Some changes in the immune response are still evident when sensitization occurs at 10 wk after birth, although the magnitude of the maternal effect gradually declines (Fedulov et al., 2007, 2008).
Our group has used these models to identify specific cellular components of the immune system in the maternal transfer of asthma risk. Dendritic cells in the neonates born to asthmatic mothers are altered from birth and play a central role in the induction of increased asthma susceptibility. Adoptive transfer of dendritic cells from allergic mothers to normal neonates recreates increased asthma susceptibility in the recipients; more surprisingly splenic dendritic cells from allergen-naïve offspring of asthmatic mothers have similar effect, while genetically and “environmentally” identical cells from control donors do not (Fedulov and Kobzik, submitted). Other studies show role of T-lymphocytes in maternal asthma transmission: adoptive transfer of allergen- specific T-lymphocytes to normal (non-asthmatic) females can cause increased susceptibility to asthma is offspring. Hubeau et al. (Citation2006) adoptively transferred allergen-specific T-lymphocytes from ovalbumin- specific T-cell receptor transgenic DO11.10 donors prior to mating. Following intraperitoneal injection of the DO11.10 T-lymphocytes and mating, the females showed no signs of an asthma phenotype, however their offspring had increased susceptibility to asthma when subject to the “suboptimal protocol” (Hubeau et al., 2006).
While abundant evidence exists that maternal allergy can be a strong risk factor for neonatal allergy/asthma, it is worth noting that some studies report dampening or inhibition of allergy in offspring of sensitized mothers. Maternal immunization to D pteronyssinus seems to protect offspring from the development of allergy (Victor et al., 2003). Following sensitization with D pteronyssinus, the offspring of immunized mothers had decreased specific IgE compared to control normal offspring. A more recent study from the same group (Fusaro et al., Citation2007) demonstrated that ‘immunized’ mothers exposed to antigen during pregnancy or breastfeeding underwent intense vertical transmission of antibodies, including IgG in complex with ovalbumin and IgG1 antibody with “anaphylactic function.”
The contradictory data may be explained by differences in the timing and mode of maternal exposure to allergens, and by considering evidence obtained by Uthoff et al. (Citation2003) and others that transplacental transfer of IgE but not IgG is minimal, and that transfer of allergen generally freely occurs. In a recent paper, Matson et al. (2007) showed that the conditions surrounding allergy in the mother during gestation can influence the development of tolerance to that antigen later in life. The authors demonstrate that offspring of mothers with TH1-biased immunity to OVA subjected to ‘recall’ aerosol challenge during pregnancy had reduced levels of Ag-specific IgE and airway eosinophilia compared to neonates of mothers with TH2-biased immunity to OVA or naive controls. This transferred tolerance was allergen-specific.
These observations highlight the importance of evaluating specific pregnancy periods, strain-specific differences (e.g., BALB/c mice [used in the Uthoff et al. study] are known to develop AHR more easily than C57BL/6J mice [used in the Matson et al. study] and which may develop tolerance more easily), as well as models for mechanistic studies of maternal influences and exposures.
Maternal environmental exposures: chemicals
Maternal transmission of asthma risk initiated by chemical-mediated skin contact hypersensitivity has been observed, and provides a potential link between toxic environmental exposures of non-asthmatic mothers and asthma in their offspring. Lim et al. (2007) show that cutaneous application of toluene diisocyanate (TDI) to mothers can lead to increased susceptibility to OVA-induced respiratory allergy in the offspring. In this study BALB/c female mice received two topical applications of vehicle, dinitrochlorobenzene, or TDI before mating with untreated males. Dinitrochlorobenzene is a skin-sensitizer only and known to induce a TH1 response, while toluene diisocyanate is both a skin and respiratory sensitizer that causes a TH2 response. Both cause allergic contact dermatitis. Offspring underwent an intentionally sub-optimal protocol of allergen sensitization and aerosol challenge, followed by evaluation of airway hyperresponsiveness, allergic airway inflammation, and cytokine production. Offspring of TDI but not control mothers developed an asthmatic phenotype following allergen sensitization and challenge, seen as increased Penh values, airway inflammation, broncho-alveolar lavage total cell counts and eosinophilia, and TH2 cytokine imbalance in the lung. These data indicate that maternal non-respiratory allergy can result in the maternal transmission of asthma risk in mice. Interestingly, in contrast to findings in the OVA model, IL-4 was not required for the effect (Lim et al., 2007). A potential role for IL-13 is suggested by its requirement for TH2-skewed contact hypersensitivity in IL-4 deficient mice (Herrick et al., 2000).
This model may prove useful to investigate other indications from the literature that maternal exposures to chemicals can influence offspring immune responses. For example, it has been shown that chemical household products are associated with persistent wheezing (Sherriff et al., Citation2005). Inhaled exposure to toilet bowl cleaners, one of the most dangerous cleaning products that can contain chlorine and hydrochloric acid is harmful for the respiratory tract (American Association of Poison Control Centers, Citation2005). Domestic exposure to volatile organic compounds (VOC) at levels even below currently accepted recommendations may increase the risk of childhood asthma (Rumchev et al., 2004; Rumchev et al., 2007). Of particular interest is the hazardous role of low levels of formaldehyde exposure. Some types of furniture made of chipboard (sometimes call particleboard) may release formaldehyde. As shown, domestic exposure to formaldehyde significantly increases the risk of asthma in young children (Rumchev et al., 2002). In experimental setting exposure to formaldehyde also enhanced contact hypersensitivity (Fujii et al., Citation2005).
Maternal intake of certain medications may result in increased neonatal allergic susceptibility. For example, frequent use of paracetamol in late pregnancy increases the risk of wheezing in the neonates, although the effect does not persist later in life (Shaheen et al., Citation2002). Maternal exposure to antibiotics is suggested to similarly increase asthma risk for the neonate: Rusconi et al. (2007) show that use of antibiotics for urinary tract infections was associated with transient early wheezing, whereas antibiotic administration at delivery was associated with both transient early wheezing and persistent wheezing. Modification of the murine maternal asthma transmission model may prove useful to further analyze and test these and other exposures for immunotoxic effects.
Maternal environmental exposures: inhaled particles
Certain air particulates, especially those of urban environment, contribute to pulmonary disease, including asthma (as remarkably reviewed in (D’Amato, 2002). Interestingly, some particles like the diesel exhaust particles (DEP) have direct pro-allergic actions, seen as adjuvant activity to protein allergens both in humans (Diaz-Sanchez et al., 1997) and in rat and mouse models. In a study by Takano et al the animals were randomized into four experimental groups that received intratracheal instillation with vehicle, OVA, DEP, or the combination of OVA and DEP on a weekly basis for 6 weeks. Respiratory resistance (Rrs) was measured 24 hr after the last instillation. An increase in Rrs in animals that inhaled acetylcholine was significantly greater in the combined treatment with OVA and DEP than in the other treatments (Takano et al., 1998). In a more recent study Matsutomo et al. (2006) used similar ovalbumin sensitization and intranasal challenge model to test diesel exhaust adjuvant properties dynamically. Mice were exposed to low-dose DEP for 7 hr/d, 5 d/wk, for up to 12 wk. AHR to methacholine was evaluated by whole-body plethysmography as well as bronchoalveolar lavage cell analysis and cytokine gene expression in lungs. Repeated exposure of asthmatic mice to low-dose DEP resulted in increased AHR and gene expression of several pro-asthmatic cytokines/chemokines, but these effects rapidly subsided with continued exposure to DEP. The Authors concluded that DEP effects are not prolonged with continuous toxic exposure (Matsumoto et al., 2006). Earlier studies indicated similar effects (Fujimaki et al., 1997; Miyabara et al., 1998; Dong et al., Citation2005).
Our laboratory recently reported that maternal exposure to DEP during pregnancy increases offspring allergic susceptibility (Fedulov et al., 2007, 2008). This is perhaps not surprising considering earlier data that pyrene, an abundant component in DEP, can cause specific and robust induction of IL-4 gene expression by T-lymphocytes acting as a co-factor in the presence of antigen (Bommel et al., 2000). What is unexpected and intriguing about this study, is that even some immunologically ‘inert’ control particles with no soluble components, like the titanium dioxide (TiO2) and the carbon black (CB) particles were also shown to cause maternal transmission of asthma risk (Fedulov et al., 2007, 2008). In this study pregnant BALB/c mice (or non-pregnant controls) received particle suspensions intranasally at Day 14 of pregnancy. Lung inflammatory responses were evaluated 48 h after exposure and were found to be more profound in pregnant, than in normal mice. Offspring of particle or buffer-treated mothers were sensitized and aerosolized with OVA, followed by assays of AHR and AI and showed increased asthma susceptibility. The data indicate that pregnancy enhances lung inflammatory responses to otherwise relatively innocuous inert particles; and that exposures of non-allergic pregnant females to inert or toxic environmental air particles can cause increased allergic susceptibility in offspring.
TiO2 (and CB) particles are prototypical ‘inert’ particles in pulmonary toxicology studies because of the minimal inflammatory response usually seen in vivo in animal models; they do not have soluble components. However, they are not completely innocuous. For example, specially coated TiO2 particles were shown to cause pulmonary inflammation (Warheit et al., Citation2005). Moreover, TiO2 particles were shown to cause pulmonary inflammation with activation of antigen-presenting cells and production of certain chemokines (Drumm et al., 1999; Renwick et al., 2004). They were also associated with increased production of IL-13 by mast cells (Ahn et al., Citation2005) and to increase IL-25 and IL-13 production by lung antigen-presenting cells (Kang et al., Citation2005). Similarly, there are a few studies showing that another generally ‘inert’ particle type, CB particles may have also minor immune system effects (van Zijverden et al., 2000). One of the limitations of the discussed study was use of a single bolus dose of particles via intranasal insufflation.
While this strategy provides proof-of-principle, is less technically cumbersome and saves time and labor (especially true for DEP because of its clumpy, sticky properties), additional studies using aerosol exposures would allow more realistic comparison to actual human exposures. One recent model of inhaled toxicity employs aerosols of residual oil fly ash (ROFA) leachate from combustion power plants as a surrogate for ambient particulate air-pollutants. The ROFA particles were earlier shown to have adjuvant pro-allergic activity in a simple (non-maternal) protocol (Hamada et al., 1999). In a new study, maternal ROFA exposure increased offspring allergic susceptibility, this is seen as increased AHR, AI and evidence of TH2 polarization (Hamada et al., 2007). One could point out that real-life ROFA exposure is rather limited; however for a proof of principle and one using the inhalation route, the model is an excellent tool. It may also be worth mentioning a common drawback for most animal models: they typically use one strain of mice (mostly BALB/c). Additional studies are needed to determine if similar findings occur in other mouse strains.
A recent report by Liu et al. (2007) provides epidemiologic confirmation that maternal exposure to various particulate pollutants, even at low levels, has deleterious effect on fetal health. These authors have examined the association between intrauterine growth restriction (IUGR) among singleton term live births and sulfur dioxide (SO2), nitrogen dioxide (NO2), carbon monoxide (CO), ozone (O3), and fine particles (PM2.5) present in ambient air in the Canadian cities of Calgary, Edmonton, and Montreal for the period 1985-2000. Results show that 10 ug/m3 increase in PM2.5 exposure resulted in significantly increased IUGR. For our purpose a study of similar magnitude focused on allergy status of families exposed to varying levels of air particulates would be invaluable.
While the complete mechanism of the phenomenon remains undeciphered, mechanistic studies indicate that activation of dendritic cells (Porter et al., 2007) and/or T-lymphocytes (Ohtani et al., Citation2005) may be involved. It seems especially compelling that two very different types of maternal exposures (allergen or environmental particles) have the common outcome of increased offspring allergic susceptibility. It is of interest with regard to toxic maternal models, that normal pregnancy is generally a TH2-skewed milieu (Lin et al., 1993; Wegmann et al., 1993). It was shown earlier that pulmonary immune response to certain environmental factors (e.g., ozone) can be enhanced in such setting (Gunnison et al., 1992; Huffman et al., 2004). One could hypothesize that the mechanism may involve shared steps, where the mild TH2 skew of pregnancy is similar to a TH2 skew achieved by allergen exposure, thus ozone or particle exposure complete the adjuvant/stimulus pair and result in fetal TH2 skew. It is however possible that such shared mechanism is entirely different: for example, some recent findings regarding maternal stress and possible transmission of glucocorticoids may be substantially involved, as detailed below.
Maternal environmental exposures: maternal stress
Multiple studies suggest that chronic maternal stress plays deleterious role in fetal and neonatal immune functions. Repeated maternal stress significantly alters offspring leukocyte function (Coe et al., 2002), affects placental transfer of maternal antibodies in a sex-dependent manner (Coe and Crispen, 2000) and can increase susceptibility to infection (Kay et al., 1998). One likely mechanism for maternal stress - induced immune changes is transplacental transmis-sion of maternal glucocorticoids (GCs). It is generally believed that fetus is protected from maternal bioactive GCs by the intracellular enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD), which is abundant in placenta and is responsible for the conversion of cortisol and corticosterone into inactive metabolites (cortisone and 11-dehydrocorticosterone). This mechanism was extensively reviewed in (Yang, 1997). However in some settings stress was shown to reduce the activity of this enzyme by indirect and direct mechanisms (Welberg et al., Citation2005), as well as impair placental circulation causing a hypoxic state (Ohkawa et al., 1991) which reduces 11β-HSD activity.
Decreases in anti-GC protection may result in increased fetal exposure to these hormones. While they are known to generally have anti-inflammatory activity and used as treat-ment against allergy and asthma, under certain experimental conditions stress-induced GCs can contribute to TH2-skewed immune responses. Joachim et al. (2003) were able to show that exogenously applied stress dramatically enhances airway reactivity and airway inflammation in OVA-sensitized and challenged mice. This is a convenient model to study direct (not maternal) stress influences on airway allergic response. A recent paper from the same group indicates that prenatal stress increased vulnerability to airway hyperresponsiveness and inflammation in adult offspring seen as BAL eosinophilia and plethysmography-based airway hyperresponsiveness. Especially interesting is their finding that APCs derived from prenatally stressed offspring triggered preferential expansion of TH2 cells in vitro (Pincus-Knackstedt et al., 2006).
Preliminary findings in our laboratory show that maternal immobilization stress leads to increased allergic susceptibility of the offspring (Lim et al., unpublished). Apparently, these pro-allergic effects of GCs are mediated by their ability to up-regulate IL-4 and IL-13 production in TH2 cells (Elenkov, 2004), although much is still unclear abut specific mechanisms. Some maternal glucocorticoids, such as cortisol, can escape through the placenta and affect fetal cortisol levels, as reviewed in (von Hertzen, 2002). The aforementioned reduction of 11β-HSD activity might also play role in other maternal models, especially those with more prominent maternal exposure protocols (e.g., to particles) where chronic maternal hypoxia is significant.
Summary
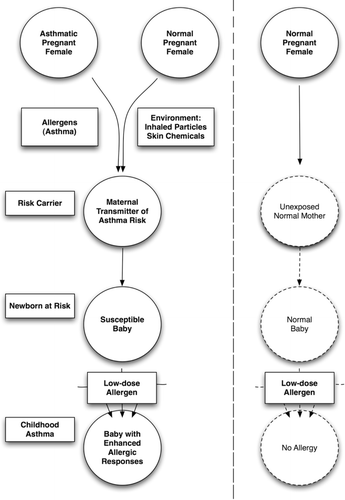
Robust animal models of maternal transmission of asthma risk exist. These models reveal increased asthma susceptibility in neonatal mice after maternal exposures to allergens and environmental pollutants and chemicals. provides a schematic summary of observations made in these models. Precise mechanisms remain under study, but important roles for maternal inflammation and influences on neonatal dendritic cells have been identified. The murine models will allow further analysis of basic mechanisms, but also provide a platform for testing of potential immunotoxicants for effects on maternal transmission of asthma risk.
Figure 1. Immunotoxic exposures of normal mothers or allergy in asthmatic mothers results in increased neonatal allergic susceptibility, suggesting a common pathway for multiple exposures.
References
- Ahn, M. H., Kang, C. M., Park, C. S., Park, S. J., Rhim, T., Yoon, P. O., Chang, H. S., Kim, S. H., Kyono, H., Kim, K. C. 2005. Titanium dioxide particle-induced goblet cell hyperplasia: Association with mast cells and IL-13. Respir. Res. 6:34.
- American Lung Association. 2005. Trends in Asthma Morbidity and Mortality. Epidemiology & Statistics Unit, Research and Program Services.
- American Association of Poison Control Centers, 2005 Annual Report of the American Association of Poison Control Centers’ National Poisoning and Exposure Database. 2005.
- Asthma and Allergy Foundation of America. 2000. Chronic Conditions: A Challenge for the 21st Century. Asthma Facts and Figures. 2000. National Academy on an Aging Society.
- Barrett, E. G., Rudolph, K., Bowen, L. E., Bice, D. E. 2005. Parental allergic status influences the risk of developing allergic sensitization and an asthmatic-like phenotype in canine offspring. Immunology 110:493–500.
- Bommel, H., Li-Weber, M., Serfling, E., Duschl, A. 2005. The environmental pollutant pyrene induces the production of IL-4. J. Allergy Clin. Immunol. 105:796–802.
- Centers for Disease Control. 1998. Surveillance for Asthma, United States, 1960–1995. MMWR 47(SS-1).
- Coe, C. L., Crispen, H. R. 2005. Social stress in pregnant squirrel monkeys (Saimiri boliviensis peruviensis) differentially affects placental transfer of maternal antibody to male and female infants. Health Psychol. 19:554–559.
- Coe, C. L., Kramer, M., Kirschbaum, C., Netter, P., Fuchs, E. 2005. Prenatal stress diminishes the cytokine response of leukocytes to endotoxin stimulation in juvenile rhesus monkeys. J. Clin. Endocrinol. Metab. 87:675–681.
- D’Amato, G. 2005. Urban air pollution and respiratory allergy. Monaldi Arch. Chest Dis. 57:136–140.
- Devereux, G., Litonjua, A. A., Turner, S. W., Craig, L. C., McNeill, G., Martindale, S., Helms, P. J., Seaton, A., Weiss, S. T. 2005. Maternal Vitamin D intake during pregnancy and early childhood wheezing. Am. J. Clin. Nutr. 85:853–859.
- Diaz-Sanchez, D., Tsien, A., Fleming, J., Saxon, A. 2005. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T-helper cell 2-type pattern. J. Immunol. 158:2406–2413.
- Dietert, R. R., Holsapple, M. P. 2005. Methodologies for developmental immunotoxicity (DIT) testing. Methods 41:123–131.
- Dong, C. C., Yin, X. J., Ma, J. Y., Millecchia, L., Wu, Z. X., Barger, M. W., Roberts, J. R., Antonini, J. M., Dey, R. D., Ma, J. K. 2005. Effect of diesel exhaust particles on allergic reactions and airway responsiveness in ovalbumin-sensitized brown Norway rats. Toxicol. Sci. 88:202–212.
- Douwes, J., Pearce, N. 2005. Invited commentary: Is indoor mold xposure a risk factor for asthma? Am. J. Epidemiol. 158:203–206.
- Drumm, K., Schindler, H., Buhl, R., Kustner, E., Smolarski, R., Kienast, K. 2005. Indoor air pollutants stimulate interleukin-8-specific mRNA expression and protein secretion of alveolar macrophages. Lung 177:9–19.
- Elenkov, I. J. 2005. Glucocorticoids and the TH1/TH2 balance. Ann. N. Y. Acad. Sci. 1024:138–146.
- Fedulov, A. V., Leme, A. S., Kobzik, L. 2005. Duration of allergic susceptibility in maternal transmission of asthma risk. Am. J. Reprod. Immunol. 58:120–128.
- Fedulov, A. V., Silverman, E., Xiang, Y., Leme, A., Kobzik, L. 2005. Immunostimulatory CpG oligonucleotides abrogate allergic susceptibility in a murine model of maternal asthma transmission. J. Immunol. 175:4292–4300.
- Fedulov, A. V., Leme, A., Yang, Z., Dahl, M., Lim, R., Mariani, T. J., and Kobzik, L. 2005. Pulmonary exposure to particles during pregnancy causes increased neonatal asthma susceptibility. Am. J. Respir. Cell. Mol. Biol. 38:57–67.
- Fujii, K., Tsuji, K., Matsuura, H., Okazaki, F., Takahashi, S., Arata, J., Iwatsuki, K. 2005. Effect of formaldehyde gas exposure in a murine allergic contact hypersensitivity model. Immunopharmacol. Immunotoxicol. 27:163–175.
- Fujimaki, H., Saneyoshi, K., Shiraishi, F., Imai, T., Endo, T. 2005. Inhalation of diesel exhaust enhances antigen-specific IgE antibody production in mice. Toxicology 116:227–233.
- Fusaro, A. E., Brito, C. A., Victor, J. R., Rigato, P. O., Goldoni, A. L., Duarte, A. J., Sato M. N. 2007. Maternal-fetal interaction: Pre-conception immunization in mice prevents neonatal sensitization induced by allergen exposure during pregnancy and breastfeeding. Immunology 122:107–115.
- Gelfand, E. W., A. Joetham, A, Cui, Z. H., Balhorn, A., Takeda, K., Taube, C., Dakhama, A. 2005. Induction and maintenance of airway responsiveness to allergen challenge are determined at the age of initial sensitization. J. Immunol. 173:1298–1306.
- Gern, J. E., Lemanske, R. F., Jr., Busse, W. W. 2005. Early life origins of asthma. J. Clin. Invest. 104:837–843.
- Guilbert, T. W., Morgan, W. J., Zeiger, R. S., Mauger, D. T., Boehmer, S. J., Szefler, S. J., Bacharier, L. B., Lemanske, R. F., Jr., Strunk, R. C., Allen, D. B., Bloomberg, G. R., Heldt, G., Krawiec, M., Larsen, G., Liu, A. H., Chinchilli, V. M., Sorkness, C. A., Taussig, L. M., Martinez, F. D. 2005. Long-term inhaled corticosteroids in preschool children at high risk for asthma. New Engl. J. Med. 354:1985–1997.
- Gunnison, A. F., Weideman, P. A., Sobo, M. 2005. Enhanced inflammatory response to acute ozone exposure in rats during pregnancy and lactation. Fundam. Appl. Toxicol. 19:607–612.
- Hamada, K., Goldsmith, C. A., Kobzik, L. 2005. Increased airway hyperresponsiveness and inflammation in a juvenile mouse model of asthma exposed to air-pollutant aerosol. J. Toxicol. Environ. Health 58:129–143.
- Hamada, K., Suzaki, Y., Goldman, A., Ning, Y. Y., Goldsmith, C., Palecanda, A., Coull, B., Hubeau, C., Kobzik, L. 2005. Allergen-independent maternal transmission of asthma susceptibility. J. Immunol. 170:1683–1689.
- Hamada, K., Suzaki, Y., Leme, A., Ito, T., Miyamoto, K., Kobzik, L., Kimura, H. 2005. Exposure of pregnant mice to an air pollutant aerosol increases asthma susceptibility in offspring. J. Toxicol. Environ. Health 70:688–695.
- Hammad, H., Lambrecht, B. N. 2005. Recent progress in the biology of airway dendritic cells and implications for understanding the regulation of asthmatic inflammation. J. Allergy Clin. Immunol. 118:331–336.
- Herrick, C. A., MacLeod, H., Glusac, E., Tigelaar, R. E., Bottomly, K. 2005. Th2 responses induced by epicutaneous or inhalational protein exposure are differentially dependent on IL-4. J. Clin. Invest. 105:765–775.
- Herz, U., Joachim, R., Ahrens, B., Scheffold, A., Radbruch, A., Renz, H. 2005. Allergic sensitization and allergen exposure during pregnancy favor the development of atopy in the neonate. Int. Arch. Allergy Immunol. 124:193–196.
- Holloway, J. W., Beghe, B., Holgate, S. T. 2005. The genetic basis of atopic asthma. Clin. Exp. Allergy 29:1023–1032.
- Holsapple, M. P., West, L. J., Landreth, K. S. 2005. Species comparison of anatomical and functional immune system development. Birth Defects Res. B Dev. Reprod. Toxicol. 68:321–334.
- Holt, P. G., Jones, C. A. 2005. The development of the immune system during pregnancy and early life. Allergy 55:688–697.
- Hubeau, C., Apostolou, I., Kobzik L. 2006. Adoptively transferred allergen-specific T-cells cause maternal transmission of asthma risk. Am. J. Pathol. 168:1931–1939.
- Huffman, L. J., Frazer, D. G., Prugh, D. J., Brumbaugh, K., Platania, C., Reynolds, J. S., Goldsmith, W. T. 2005. Enhanced pulmonary inflammatory response to inhaled endotoxin in pregnant rats. J. Toxicol. Environ. Health 67:125–144.
- Joachim, R. A., Quarcoo, D., Arck, P. C., Herz, U., Renz, H., Klapp, B. F. 2005. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom. Med. 65:811–815.
- Kang, C. M., Jang, A. S., Ahn, M. H., Shin, J. A., Kim, J. H., Choi, Y. S., Rhim, T. Y., Park, C. S. 2005. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am. J. Respir. Cell. Mol. Biol. 33:290–296.
- Kay, G., Tarcic, N., Poltyrev, T., Weinstock, M. 2005. Prenatal stress depresses immune function in rats. Physiol. Behav. 63:397–402.
- Kurukulaaratchy, R. J., Waterhouse, L., Matthews, S. M., Arshad, S. H. 2005. Are influences during pregnancy associated with wheezing phenotypes during the first decade of life? Acta Paediatr. 94:553–558.
- Lim, R. H., Arredouani, M. S., Fedulov, A., Kobzik, L., Hubeau, C. 2005. Maternal allergic contact dermatitis causes increased asthma risk in offspring. Respir. Res. 8:56.
- Lin, H., Mosmann, T. R, Guilbert, L., Tuntipopipat, S., Wegmann, T. G. 2005. Synthesis of T-helper 2-type cytokines at the maternal-fetal interface. J. Immunol. 151:4562–4573.
- Litonjua, A. A., Carey, V. J., Burge, H. A., Weiss, S. T., Gold, D. R. 2005. Parental history and the risk for childhood asthma. Does mother confer more risk than father? Am. J. Respir. Crit. Care Med. 158:176–181.
- Litonjua, A. A., Rifas-Shiman, S. L., Ly, N. P., Tantisira, K. G., Rich-Edwards, J. W., Camargo, C. A., Jr., Weiss, S. T., Gillman, M. W., Gold, D. R. 2005. Maternal antioxidant intake in pregnancy and wheezing illnesses in children at 2 yr-of-age. Am. J. Clin. Nutr. 84:903–911.
- Liu, C. A., Wang, C. L., Chuang, H., Ou, C. Y., Hsu, T. Y., Yang, K. D. 2005. Prenatal prediction of infant atopy by maternal but not paternal total IgE levels. J. Allergy Clin. Immunol. 112:899–904.
- Liu, S., Krewski, D., Shi, Y., Chen, Y., Burnett, R. T. 2005. Association between maternal exposure to ambient air pollutants during pregnancy and fetal growth restriction. J. Expo. Sci. Environ. Epidemiol. 17:426–432.
- Martinez, F. D., Wright, A. L., Taussig, L. M., Holberg, C. J., Halonen, M., Morgan, W. J. 2005. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. New Engl. J. Med. 332:133–138.
- Matson, A. P., Zhu, L., Lingenheld, E. G., Schramm, C. M., Clark, R. B., Selander, D. M., Thrall, R. S., Breen, E., Puddington, L. 2005. Maternal transmission of resistance to development of allergic airway disease. J. Immunol. 179:1282–1291.
- Matsumoto, A., Hiramatsu, K., Li, Y., Azuma, A., Kudoh, S., Takizawa, H., Sugawara, I. 2005. Repeated exposure to low-dose diesel exhaust after allergen challenge exaggerates asthmatic responses in mice. Clin. Immunol. 121:227–235.
- Miyabara, Y., Takano, H., Ichinose, T., Lim, H. B., Sagai, M. 2005. Diesel exhaust enhances allergic airway inflammation and hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 157:1138–1144.
- National Center for Health Statistics (NCHS). 2003. Morbidity and Mortality Report, U.S. CDC. EPA Asthma Facts.
- National Center for Health Statistics (NCHS). 2001. CDC. New Asthma Estimates: Tracking Prevalence, Health Care and Mortality.
- Ohkawa, T., Takeshita, S., Murase, T., Okinaga, S., Arai, K. 2005. The effect of an acute stress in late pregnancy on hypothalamic catecholamines of the rat fetus. Acta Obstet. Gynaecol. Japon. 43:783–787.
- Ohtani, T., Nakagawa, S., Kurosawa, M., Mizuashi, M., Ozawa, M., Aiba, S. 2005. Cellular basis of the role of diesel exhaust particles in inducing TH2-dominant response. J. Immunol. 174:2412–2419.
- Pincus-Knackstedt, M. K., Joachim, R. A., Blois, S. M., Douglas, A. J., Orsal, A. S., Klapp, B. F., Wahn, U., Hamelmann, E., Arck, P. C. 2005. Prenatal stress enhances susceptibility of murine adult offspring toward airway inflammation. J. Immunol. 177:8484–8492.
- Porter, M., Karp, M., Killedar, S., Bauer, S. M., Guo, J., Williams, D., Breysse, P., Georas, S. N., Williams, M. A. 2005. Diesel-enriched particulate matter functionally activates human dendritic cells. Am. J. Respir. Cell. Mol. Biol. 37:706–719.
- Prescott, S. L. 2005. Maternal allergen exposure as a risk factor for childhood asthma. Curr. Allergy Asthma Rep. 6:75–80.
- Renwick, L. C., Brown, D., Clouter, A., Donaldson, K. 2005. Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types. Occup. Environ. Med. 61:442–447.
- Romieu, I., Torrent, M., Garcia-Esteban, R., Ferrer, C., Ribas-Fito, N., Anto, J. M., Sunyer, J. 2005. Maternal fish intake during pregnancy and atopy and asthma in infancy. Clin. Exp. Allergy 37:518–525.
- Ruiz, R. G., Kemeny, D. M., Price, J. F. 2005. Higher risk of infantile atopic dermatitis from maternal atopy than from paternal atopy. Clin. Exp. Allergy 22:762–766.
- Rumchev, K., Brown, H., Spickett, J. 2005. Volatile organic compounds: Do they present a risk to our health? Rev. Environ. Health 22:39–55.
- Rumchev, K., Spickett, J., Bulsara, M., Phillips, M., Stick, S. 2005. Association of domestic exposure to volatile organic compounds with asthma in young children. Thorax 59:746–751.
- Rumchev, K. B., Spickett, J. T., Bulsara, M. K., Phillips, M. R., Stick, S. M. 2005. Domestic exposure to formaldehyde significantly increases the risk of asthma in young children. Eur. Respir. J. 20:403–408.
- Rusconi, F., Galassi, C., Forastiere, F., Bellasio, M., De Sario, M., Ciccone, G., Brunetti, L., Chellini, E., Corbo, G., La Grutta, S., Lombardi, E., Piffer, S., Talassi, F., Biggeri, A., Pearce, N. 2005. Maternal complications and procedures in pregnancy and at birth and wheezing phenotypes in children. Am. J. Respir. Crit. Care Med. 175:16–21.
- Saglani, S., Bush, A. 2005. The early-life origins of asthma. Curr. Opin. Allergy Clin. Immunol. 7:83–90.
- Shaheen, S. O., Newson, R. B., Henderson, A. J., Emmett, P. M., Sherriff, A., Cooke, M., the ALSPAC Study Team. 2004. Umbilical cord trace elements and minerals and risk of early childhood wheezing and eczema. Eur. Respir. J. 24:292–297.
- Shaheen, S. O., Newson, R. B., Sherriff, A., Henderson, A. J., Heron, J. E., Burney, P. G., Golding, J., the ALSPAC Study Team. 2002. Paracetamol use in pregnancy and wheezing in early childhood. Thorax 57:958–963.
- Sherriff, A., Farrow, A., Golding, J., Henderson, J. 2005. Frequent use of chemical household products is associated with persistent wheezing in pre-school age children. Thorax 60:45–49.
- Takano, H., Ichinose, T., Miyabara, Y., Yoshikawa, T., Sagai, M. 2005. Diesel exhaust particles enhance airway responsiveness following allergen exposure in mice. Immunopharmacol. Immunotoxicol. 20:329–336.
- Uthoff, H., Spenner, A., Reckelkamm, W., Ahrens, B., Wolk, G., Hackler, R., Hardung, F., Schaefer, J., Scheffold, A., Renz, H., Herz, U. 2003. Critical role of preconceptional immunization for protective and nonpathological specific immunity in murine neonates. J. Immunol. 171:3485–3492.
- van Zijverden, M., van der Pijl, A., Bol, M., van Pinxteren, F. A., de Haar, C., Penninks, A. H., van Loveren, H., Pieters, R. 2005. Diesel exhaust, carbon black, and silica particles display distinct TH1/TH2 modulating activity. Toxicol. Appl. Pharmacol. 168:131–139.
- Vance, G. H., Holloway, J. A. 2005. Early life exposure to dietary and inhalant allergens. Pediatr. Allergy Immunol. 13(S15):14–18.
- Victor, J. R., Jr., Fusaro, A. E., Duarte, A. J., Sato, M. N. 2005. Preconception maternal immunization to dust mite inhibits the Type I hypersensitivity response of offspring. J. Allergy Clin. Immunol. 111:269–277.
- von Hertzen, L. C. 2005. Maternal stress and T-cell differentiation of the developing immune system: Possible implications for the development of asthma and atopy. J. Allergy Clin. Immunol. 109:923–928.
- Ward, L. M., Gaboury, I., Ladhani, M., Zlotkin, S. 2005. Vitamin D-deficiency rickets among children in Canada. Canad. Med. Assoc. J. 177:161–166.
- Warheit, D. B., Brock, W. J., Lee, K. P., Webb, T. R., Reed, K. L. 2005. Comparative pulmonary toxicity inhalation and instillation studies with different TiO2 particle formulations: Impact of surface treatments on particle toxicity. Toxicol. Sci. 88:514–524.
- Wegmann, T. G., Lin, H., Guilbert, L., Mosmann, T. R. 2005. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol. Today 14:353–356.
- Weiss, S. T., Litonjua, A. A. 2005. Maternal diet vs. lack of exposure to sunlight as the cause of the epidemic of asthma, allergies and other autoimmune diseases. Thorax 62:746–748.
- Welberg, L. A., Thrivikraman, K. V., Plotsky, P. M. 2005. Chronic maternal stress inhibits the capacity to up-regulate placental 11β-hydroxysteroid dehydrogenase type 2 activity. J. Endocrinol. 186:R7–12.
- Wright, L. A. 2005. The epidemiology of the atopic child: Who is at risk for what? J. Allergy Clin. Immunol.113(S1): S2–7.
- Yang, K. 2005. Placental 11β-hydroxysteroid dehydrogenase: Barrier to maternal glucocorticoids. Rev. Reprod. 2:129–132.