
Abstract
Clinical evidence suggests that most idiosyncratic drug-induced liver injury (IDILI) is immune-mediated. The danger hypothesis suggests that liver injury and inflammation would increase the risk of an immune response leading to IDILI. Therefore, a reasonable hypothesis would be that an underlying chronic liver disease such as non-alcoholic steatohepatitis (NASH) would increase the risk of developing IDILI due to inflammation and release of danger signals from damaged cells. In order to test this hypothesis, mice were fed a methionine-/choline-deficient (MCD) diet that produces a consistent NASH phenotype, along with amodiaquine (AQ) – a drug known to cause IDILI in humans. This study employed both wild-type C57BL/6 mice and PD-1−/− mice co-treated with anti-CTLA-4 antibodies. The PD-1−/− + anti-CTLA-4 model produces an immune-mediated liver injury very similar to the idiosyncratic liver injury observed in humans. The liver injury observed in the present experiment was dominated by the injury caused by the MCD diet; there was no significant difference between mice treated with the MCD diet alone and those also treated with AQ, whether in wild-type mice of the PD-1−/− model. Therefore, the MCD diet, which results in a state that mimics NASH, did not appear to increase the liver injury associated with AQ treatment. Ultimately, an animal model is just that – only a model, and cannot provide a definitive answer to clinical questions. However, given the difficulty of performing clinical studies with appropriate control populations, the present results provide important evidence to support a general clinical finding that underlying liver injury does not usually increase the risk of IDILI.
Introduction
Idiosyncratic drug-induced liver injury (IDILI) is a significant cause of liver failure, and its unpredictable nature increases the risk of drug development. Clinical evidence suggests that most IDILI is immune-mediated (Mak and Uetrecht Citation2017). The danger hypothesis suggests that liver injury and inflammation would increase the risk of an immune response leading to IDILI. If the immune system plays a significant role in IDILI pathogenesis, then an underlying inflammatory condition has the potential to be a risk factor for IDILI. It has been suggested that the risk of IDILI is higher in patients with non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) (Tarantino et al. Citation2007; Massart et al. Citation2017). These are both liver diseases characterized by steatosis, necro-inflammation, and fibrosis, and highly prevalent among obese individuals (Jansen Citation2004). However, unless NAFLD/NASH is associated with a marked increase in the risk of IDILI, the relationship would be difficult to detect because the absolute incidence of IDILI is low, and there are many other confounding factors in the NAFLD/NASH population. Thus, it is very difficult to rigorously test the hypothesis that NASH increases the risk of IDILI in humans.
Amodiaquine (AQ), an anti-malarial drug known to cause IDILI in humans, was used in these studies because it has been shown to provide a good model of IDILI. Unlike most drugs that cause IDILI, AQ not only causes IDILI in humans, it also causes mild liver injury in mice that appears to be mediated by NK/NKT cells and resolves despite continued treatment with the drug (Metushi, Cai et al. Citation2015). In addition, an animal model of IDILI has been developed wherein impairing immune tolerance unmasks the ability of other drugs to cause IDILI. In this model, AQ causes injury very similar to IDILI in humans with histology that is characterized by piece-meal necrosis and a mononuclear leukocyte infiltrate (Metushi, Hayes et al. Citation2015). Unlike in wild-type mice, this injury does not resolve with continued treatment and it was found to be mediated by CD8 T-cells (Mak and Uetrecht Citation2015).
There are also animal models of NASH. One such model involves treating mice with a methionine-/choline-deficient (MCD) diet. Similar to NASH in humans, the MCD diet provides a consistent phenotype of steatosis and severe liver injury (Machado et al. Citation2015). The study here, therefore, combined these models to determine if NASH increased the severity of IDILI.
Materials and methods
Animals and drug treatments
C57BL/6 mice (female, 8–10 weeks of age) were purchased from Charles River Labs (Montreal, Canada). Female PD1−/− mice (between 10–12 weeks of age) were bred and housed as in the Metushi, Cai et al. (Citation2015) study. AQ (Ipca Laboratories Ltd., Mumbai, India) was thoroughly mixed with an amino acid control diet (ACD) or a MCD (TD.94149 and TD.90262, respectively, Envigo, Mississauga, Canada) at 0.2% (w/w) and then provided to the mice in small jars for ad libitum intake. Wild-type C57BL/6 mice were randomly allocated into four groups: control diet (Control ACD), control ACD diet containing 0.2% AQ (ACD/AQ), MCD diet (Control MCD), and MCD diet containing 0.2% AQ (MCD/AQ). PD1−/− mice were split into three groups: anti-CTLA-4 (250 µg intraperitoneally [IP] at 3 days and 1 day before treatment and thereafter once weekly; Clone 9D9; Bio-X-cell, West Lebanon, NH) and MCD diet (PD1−/−/anti-CTLA-4/MCD), anti-CTLA-4 and control ACD diet containing 0.2% AQ (PD1−/−/anti-CTLA-4/AQ), and anti-CTLA-4 and MCD diet containing 0.2% AQ (PD1−/−/anti-CTLA-4/AQ/MCD). Treatments lasted for up to 5 weeks. At designated timepoints, mice were euthanized by CO2 asphyxiation and blood collected from the orbital sinus. At necropsy, the liver and spleen of each host was removed for analyses in the protocols outlined below. All animal protocols used in this study were approved by the University of Toronto Animal Care Committee and conducted in an animal facility accredited by the Canadian Council on Animal Care.
Alanine aminotransferase
Blood was collected weekly as previously described (Mak and Uetrecht Citation2015), serum was prepared, and levels of serum alanine aminotransferase (ALT) were then measured using an InfinityTM ALT kit (TR71121; Thermo Scientific, Middleton, VA), according to manufacturer protocols.
Histology
Identical portions of the liver and spleen samples isolated at necropsy were placed in 10% neutral buffered formalin solution (Sigma, Oakville, Canada). Samples were then embedded in paraffin, sectioned (to 4 µm), stained with H&E, and then analyzed at the CFIBCR Histology/Microscope Core Unit (Toronto, Canada).
Isolation of mononuclear cells and flow cytometry
Mononuclear cells (MNC) were isolated from livers and spleens, stained with antibodies, and then phenotyped by flow cytometry using a previously described protocol (Mak and Uetrecht Citation2015). In these analyses, separate aliquots of isolated MNC were stained using standard protocols and dilutions of conjugated antibodies against macrophages (M1 and M2), myeloid-derived suppressor cells (MDSC), CD8 T-cells, CD4 T-cells, T-helper (TH)-17 cells, T-regulatory (Treg) cells, NK cells, NKT cells, B-cells, or memory T-cells. By these analyses, macrophages were characterized as CD11b+F4/80+ and further characterized as M1 (CD11b+F4/80+ and iNOS+ or IL-12+) or M2 (CD11b+F4/80+ and Arg1+ or IL-10+). MDSC were characterized as CD11b+Gr1+, CD4 T-cells as CD3+CD4+, CD8 T-cells as CD3+CD8+, TH17 cells as CD4+IL-17+, Treg cells as CD4+FoxP3+, NK cells as NK1.1+CD3–, NKT cells as NK1.1+CD3+, B-cells as CD45R+, and memory T-cells as CD4+CD62L+. Antibodies against F4/80, CD11b, iNOS, IL-12, IL-10, Gr1, CD3e, CD4, NK1.1, CD45R, IL-17, CD62L, and Foxp3, as well as Fixable Viability Dye, were all purchased from eBioscience (San Diego, CA), anti-Arg-1 was from R&D (Minneapolis, MN), and anti-CD8a was from BD Biosciences (San Jose, CA). Once stained, the samples were rinsed in phosphate-buffered saline (PBS, pH 7.4) supplemented with 10% fetal bovine serum (FBS; Sigma), and then underwent analysis in a BD LSR II system (BD Biosciences), using system-associated software. A minimum of 10 000 live MNC/sample was acquired. Data were subsequently reported out in terms of percent of total live MNC.
Statistical analysis
Means ± SE were calculated for each experimental group. Data were analyzed using a one- or two-way analysis of variance (ANOVA). A p value < 0.05 was considered significant. All statistical analyses were performed using Prism Software (GraphPad, San Diego, CA).
Results
Combination of the MCD diet and a drug known to cause IDILI in humans does not cause greater liver injury in wild-type or PD1−/− mice compared to the MCD diet alone
Wild-type C57BL/6 mice or PD1−/− mice were allocated into groups and treated for up to 5 weeks. Over the course of the treatments, ALT levels were measured weekly to determine any degree of liver injury. Wild-type mice treated with the control ACD diet developed no liver injury, whereas mice treated with AQ mixed in the ACD diet developed a mild delayed onset liver injury (). Wild-type mice treated with the MCD diet alone developed significantly elevated ALT levels compared with control mice, whereas mice treated with AQ mixed into the MCD diet also developed significantly elevated ALT levels (). ALT levels were not significantly different between Control MCD mice and MCD/AQ mice.
Figure 1. Mice treated with AQ in a MCD diet did not develop significantly greater liver injury than mice treated with MCD alone. (A) Wild-type C57BL/6 mice in four groups; control diet (Control ACD), control ACD diet with 0.2% AQ (ACD/AQ), MCD diet (Control MCD), and MCD diet with 0.2% AQ group (MCD/AQ). (B) PD1−/− mice in three groups; anti-CTLA-4 and MCD diet (PD1−/−/anti-CTLA-4/MCD), anti-CTLA-4 and control ACD diet with 0.2% AQ group (PD1−/−/anti-CTLA-4/AQ), and anti-CTLA-4 and MCD diet with 0.2% AQ group (PD1−/−/anti-CTLA-4/AQ/MCD). Values shown are means ± SE. Values significantly differ from one another at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (analyzed using two-way ANOVA).
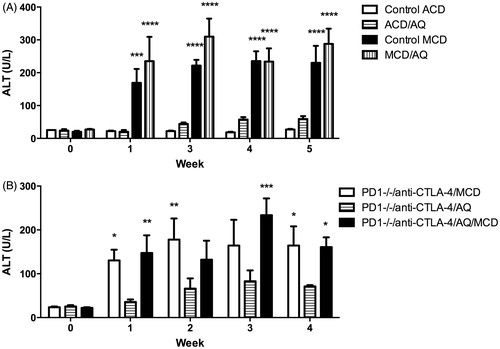
Liver injury for PD1−/− mice was measured in the same manner as the wild-type mice. PD1−/− mice treated with anti-CTLA-4 and AQ developed more severe liver injury, as seen in previous experiments (Mak and Uetrecht Citation2015) (). PD1−/− mice treated with anti-CTLA-4 and the MCD diet alone or with added AQ developed significantly greater ALT levels than counterparts treated with anti-CTLA-4 and AQ in the ACD diet at certain weeks (). However, as in wild-type mice, ALT levels did not significantly differ between treatments with the MCD diet alone and with the MCD + AQ mix diet. The injury caused by the MCD diet alone did not appear to be greater in the PD-1−/− mice, an outcome that suggested us that the immune system did not play an important role in this injury.
Figure 2. Representative H&E-stained liver sections. (A) Control ACD, (B) ACD/AQ, (C) Control MCD, (D) MCD/AQ, (E) PD1−/−/anti-CTLA-4/MCD, (F) PD1−/−/anti-CTLA-4/AQ, and (G) PD1−/−/anti-CTLA-4/AQ/MCD. Group identities fully defined in legend. Mice treated with AQ in the MCD diet and with MCD alone developed similar macro- and microvesicular steatosis. The PD1−/−/anti-CTLA-4/MCD group and the PD1−/−/anti-CTLA-4/AQ/MCD group also developed similar macro- and microvesicular steatosis.
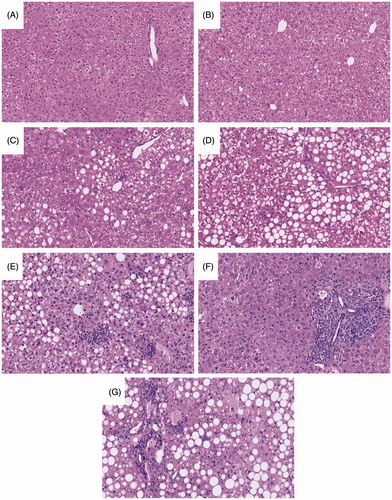
Liver histology was examined to determine gross morphological changes corresponding to the liver injury indicated by the measured ALT levels. The C57BL/6 mice or those treated with AQ alone presented with what appeared to be normal livers with no necro-inflammatory changes (). Both wild-type C57BL/6 groups treated with the MCD diet developed similar macro- and microvesicular steatosis typical for the MCD diet, along with an accumulation of a few neutrophils and macrophages throughout the organ (). The PD1−/−/anti-CTLA-4/MCD group and the PD1−/−/anti-CTLA-4/AQ/MCD group also developed similar macro- and microvesicular steatosis (). Interestingly, although AQ treatment did not lead to a further increase in the ALT of PD1−/−/anti-CTLA-4/MCD mice, there actually appeared to be more necro-inflammatory foci in the PD1−/−/anti-CTLA-4/MCD mice than in the PD1−/−/anti-CTLA-4/AQ/MCD mice. However, there was no greater single-cell hepatocyte injury in the fatty areas ().
Changes in hepatic leukocyte populations associated with the MCD diet
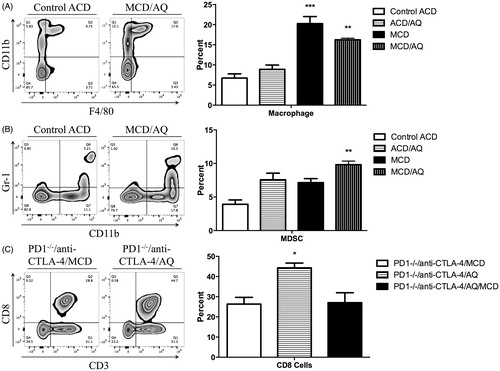
Liver immune cells were phenotyped using flow cytometry to assess any potential role of the immune system in observed liver injury. Only results for immune cell populations with significant changes among cell types are reported here. There were no differences in hepatic leukocyte populations in the wild-type mice, except for macrophages and CD11b+Gr1+ cells, which are presumably MDSC (). PD1−/− mice treated with anti-CTLA-4 and the MCD diet, those treated with anti-CTLA-4 and AQ, and those that received anti-CTLA-4 and AQ in the MCD diet showed no differences in hepatic leukocyte populations, except PD1−/− mice treated with anti-CTLA-4 and AQ had a significantly increased percentage of CD8 T-cells compared with levels in the other PD1−/− mice groups (). Additional immune cell phenotypes that indicated no significant differences between the groups are presented in Supplemental Figures 1 and 2. Overall, inflammation associated with the MCD diet was limited.
Figure 3. Flow cytometric analysis of liver mononuclear leukocytes. A representative zebra plot and average of three animals is shown. Mononuclear cells with significant differences among the treatment groups compared with control are displayed (when the difference was not significant the data are shown in Supplemental Figures 1 and 2). Wild-type C57BL/6 mice in four groups: Control ACD, ACD/AQ, Control MCD, and MCD/AQ. PD1−/− mice in three groups: PD1−/−/anti-CTLA-4/MCD, PD1−/−/anti-CTLA-4/AQ, and PD1−/−/anti-CTLA-4/AQ/MCD. Group identities fully defined in legend. (A) Significantly increased percentage of macrophages in the MCD/AQ group compared with the Control group. (B) Significantly increased percentage of MDSCs in the MCD/AQ group compared with the Control group. (C) Significantly increased percentage of CD8 T cells in the PD1−/−/anti-CTLA-4/AQ group compared with the PD1−/−/anti-CTLA-4/MCD group. Values shown are means ± SE. Values significantly differ from one another at *p < 0.05, **p < 0.01, and ***p < 0.001 (analyzed using one-way ANOVA).
Discussion
As stated earlier, it appears that most IDILI is immune-mediated. Therefore, it would be expected that injury that results in the release of danger-associated molecular pattern molecules (DAMPs) and/or inflammation would help to stimulate an immune response and increase the risk and severity of IDILI. Consistent with this prediction is the report that NAFLD is associated with an increase in the incidence of IDILI (Tarantino et al. Citation2007; Massart et al. Citation2017). Although those authors suggested an increase in risk, they acknowledged that the evidence was weak. In contrast, Zimmerman (Citation1999) famously observed that, in general, preexisting liver disease does not increase the risk of IDILI, although it may increase the consequences because of decreased liver reserve. The work of Russo and Watkins (Citation2004) illustrated how difficult it was to find an appropriate control population to determine if a specific factor is an IDILI risk factor. In the case of NAFLD, among other confounding factors, the use of medication in this population is also likely higher than in the general population. Similarly, it has been observed that stimulation of the immune system through toll-like receptors or cytotoxic drugs (such as acetaminophen) in combination with drugs that cause IDILI in humans did not lead to animal models of IDILI (Cho and Uetrecht Citation2017).
The present study found that AQ-induced liver injury in wild-type or in PD-1−/− mice, wherein in the latter AQ has been characterized to cause cytotoxic T-cell–mediated liver injury, is not significantly increased due to underlying NASH. This outcome here is consistent with the general observation by Zimmerman (Citation1999) that preexisting liver disease is not a risk factor for IDILI. Therefore, the underlying inflammation and release of danger signals from NASH is differentiated by the immune system and unable to augment immune-mediated IDILI. Danger signals due to AQ-induced cell damage is likely still important because the immune system requires Signal 2 of the immune response to verify its activation; however, it is not clear how the immune system differentiates between the two sources of danger signals and what the danger signals are (Cho and Uetrecht Citation2017). In vitro experiments have shown drug-induced hepatocyte danger signal release can activate macrophage inflammasomes to release IL-1β, a plausible first step in activating the immune system in IDILI (Kato and Uetrecht Citation2017; Mak et al. Citation2017). These danger signals are hypothesized to be transported by cell-derived vesicles called exosomes (Holman et al. Citation2016; Church and Watkins Citation2017) that can carry signals from one cell to another. Further research is necessary in identifying the specific danger signals that lead to immune-mediated IDILI.
We are cognizant of several limitations in the present study. First, only one drug was studied; there may be some drugs in which NAFLD does increase the risk of IDILI. For example, with halothane, the risk of IDILI appears to be higher in obese patients (Neuberger Citation1990). The most likely explanation in that case is that halothane is very lipophilic and distributes to fat; correspondingly then, an increased dose would be required to achieve anesthesia in obese patients. A larger limitation in the current study is that it utilized a model that may not reflect IDILI in patients with NAFLD. Nevertheless, while we believe this impaired immune tolerance model reflects one mechanism of IDILI in humans, i.e. CD8 T-cell–mediated liver injury, there may be differences, and certainly most patients with IDILI do not have the degree of impaired immune tolerance as observed in this model.
It is interesting to note that although it was previously shown that the liver injury caused by AQ in PD-1−/− hosts involves CD8 T-cells (Mak and Uetrecht Citation2015), and the number of CD8 T-cells in the AQ-treated PD-1−/− model were increased in the current study, the MCD diet appeared to prevent this increase (). In addition, the MCD diet also appeared to increase the number of myeloid-derived suppressor cells. This is a likely explanation for why the MCD diet did not increase the immune-mediated injury caused by AQ. It seems to be a general pattern that injury can cause inflammation, but it also increases cells involved in immune tolerance, which prevents more severe liver damage. A study of the immune response to interventions such as the MCD diet may be important for understanding possible risk factors for IDILI in humans. Although the MCD model reproduces many of the characteristics of NASH in humans, there are differences. In particular, the animals fed the MCD diet weigh less than animals on a standard diet, which is in contrast to patients with NASH who are generally obese. The current analyses also found that although there was a significant liver injury in the MCD model, the degree of hepatic inflammation in the MCD model was uncharacteristically limited.
Conclusion
The risk of developing IDILI depends on genetic, nongenetic, and environmental factors (Chalasani and Björnsson Citation2010). Our results are consistent with the general finding that underlying liver injury and inflammation do not appear to increase the risk of IDILI. That, however, does not falsify the danger hypothesis; as it may be that the immune system is able to differentiate whether a drug or something else causes the injury. Although the results of the present study suggest that NAFLD is not a risk factor for IDILI, it is a model and as such does not definitively answer the question. However, given that it is extremely difficult to do a clinical study that adequately controls for the multiple confounding factors in patients with NAFLD, it is unlikely that this issue will soon be resolved.
Acknowledgements
We would like to thank Dr M. A. Hayes for reviewing the histology sections.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chalasani N, Björnsson E. 2010. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology. 138:2246–2259.
- Cho T, Uetrecht J. 2017. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem Res Toxicol. 30:295–314.
- Church R, Watkins P. 2017. The transformation in biomarker detection and management of drug-induced liver injury. Liver Intl. 37:1582–1590.
- Holman N, Mosedale M, Wolf K, LeCluyse E, Watkins P. 2016. Subtoxic alterations in hepatocyte-derived exosomes: An early step in drug-induced liver injury? Toxicol Sci. 151:365–375.
- Jansen P. 2004. Non-alcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 16:1079–1085.
- Kato R, Uetrecht J. 2017. Supernatant from hepatocyte cultures with drugs that cause idiosyncratic liver injury activates macrophage inflammasomes. Chem Res Toxicol. 30:1327–1332.
- Machado M, Michelotti G, Xie G, de Almeida T, Boursier J, Bohnic B, Guy C, Diehl A. 2015. Mouse models of diet-induced non-alcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10:e0127991.
- Mak A, Uetrecht J. 2015. The role of CD8 T-cells in amodiaquine-induced liver injury in PD1−/− mice co-treated with anti-CTLA-4. Chem Res Toxicol. 28:1567–1573.
- Mak A, Uetrecht J. 2017. Immune mechanisms of idiosyncratic drug-induced liver injury. J Clin Transl Res. 3:145–156.
- Mak A, Johnston A, Uetrecht J. 2017. Effects of immunization and checkpoint inhibition on amodiaquine-induced liver injury. J Immunotoxicol. 14:9–94.
- Mak A, Kato R, Weston K, Hayes A, Uetrecht J. 2017. An impaired immune tolerance animal model distinguishes the potential of troglitazone/pioglitazone and tolcapone/entacapone to cause IDILI. Toxicol Sci. 161:412–420.
- Massart J, Begriche K, Moreau C, Fromenty B. 2017. Role of non-alcoholic fatty liver disease as risk factor for drug-induced hepatotoxicity. J Clin Transl Res. 3:212–232.
- Metushi I, Cai P, Dervovic D, Liu F, Lobach A, Nakagawa T, Uetrecht J. 2015. Development of a novel mouse model of amodiaquine-induced liver injury with a delayed onset. J Immunotoxicol. 12:247–260.
- Metushi I, Hayes M, Uetrecht J. 2015. Treatment of PD-1−/− mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 61:1332–1342.
- Neuberger J. 1990. Halothane and hepatitis. Incidence, predisposing factors and exposure guidelines. Drug Saf. 5:28–38.
- Russo M, Watkins P. 2004. Are patients with elevated liver tests at increased risk of drug-induced liver injury? Gastroenterology. 126:1477–1480.
- Tarantino G, Conca P, Basile V, Gentile A, Capone D, Polichetti G, Leo E. 2007. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol Res. 37:410–415.
- Zimmerman H, editor. 1999. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver. Philadelphia: Lippincott Williams & Wilkins.