
Abstract
Flow cytometry using fluorescent antibodies (FC) is the method of choice for the quantitation of proteins expressed at the surface or inside the cell, but, however, does not allow to selectively measure nuclear expression. We therefore sought to develop a method for the extraction of intact cell nuclei, which can be used for their subsequent immunofluorescent analysis by FC. The studied protein was vascular endothelial growth factor-receptor-type 1 (VEGFR-1) which is important in tumor survival and metastasis. Two human cell lines, A431 (epidermoid carcinoma of skin with low invasive and metastatic potential) and BRO (highly aggressive amelanotic melanoma), were used as examples for tumor cells, and normal human fibroblasts PHF served as a control line. The quality of the extracted nuclei was assessed by their intactness and purity from cytoplasm. The high content of the nuclear markers (PCNA = proliferating cell nuclear antigen, lamin A/C) in the extracted nuclei with almost complete absence of the cytoplasmic β-tubulin demonstrated that the protocol can be used to obtain a pure suspension of single intact cell nuclei. The measurement of the nuclear VEGFR-1 content revealed that it was present only in tumor cell nuclei and that in more malignant BRO cells the receptor content was 1.75 times higher than in A431 (p = 0.014). Thus, the developed method of extraction of cell nuclei for subsequent FC analysis is suitable for the quantitative evaluation of protein content in the native nucleus.
Introduction
The detection and quantification of cellular proteins is important in many fields of biology. In some cases, this goal may be successfully attained by flow cytometry (FC), using fluorescent antibodies. There are many advantages associated with this method, including high levels of accuracy and reproducibility. Moreover, basic FC analysis allows for the detection of a given antigen either on the outer surface of a cell or inside a cell after permeabilization. Nevertheless, even if a protein is found inside a cell, basic FC analysis does not provide information about its distribution in various cellular compartments.
To determine a more exact intracellular localization of proteins, confocal laser microscopy is usually used. However, this technique is semiquantitative, and its results depend to a large extent on subjective perceptions. Conversely, basic FC analysis is free from this disadvantage, but, as noted, this method – in its classical form of analysis of cells – measures only total intracellular level of a protein of interest and is unable to resolve differences in its content in the nucleus versus in the cytoplasm. Optimally, the use of cell nuclei in FC analysis could allow investigators to exclude any signal of a given protein that might be in the cytoplasm. This, in turn, would permit to measure the protein content only in the nucleus. Accordingly, the study reported here was undertaken to optimize protocols for the extraction of purified nuclei from cells for their subsequent use in FC investigations.
Since nuclear expression of proteins involved in regulation of cell growth is important for the study and diagnosis of cancer, to better assess the utility of the new FC-based sensitive-quantitative method for selective protein detection in nuclei, two tumor cell lines of distinct aggressiveness were selected for analysis. As a specific example of a key protein, VEGFR-1 (a growth regulating protein) was ultimately selected for analysis. This choice was vindicated by the findings of a number of studies indicated the important role of VEGFR-1 in increased survival rates and enhanced invasive/metastatic potential of neoplastic cells (Fan et al. Citation2005; Lee et al. Citation2007; Bhattacharya et al. Citation2016).
More importantly, this assay can be used in the context of studies of cells of the immune system. For example, while VEGF is primarily known as a pro-angiogenic factor, it is also plays a vital role in immune functions in that it can inhibit the differentiation and function of immune cells during hematopoiesis (reviewed in Li et al. Citation2016). Among the various major types of immune cells, macrophages, lymphocytes, and dendritic cells (DC) express certain types of VEGF receptors (Barleon et al. Citation1996; Zhang et al. Citation2010, Citation2014).
Ultimately, using VEGFR-1 as the target protein of interest, it was expected that the new method detailed here would permit a selective detection and quantification of its content (as model for other proteins) in cell nuclei. Once such information is in hand, analyses of comparative expression of proteins in nucleus, as opposed to on cell surface, might be performed. Although the study here focused on cancer cells, this type of approach could be very beneficial in subsequent studies of immunotoxicity of a variety of environmental/occupational agents and could provide important mechanistic information to explain how such agents act to modify immune functions in an exposed host.
Materials and methods
Cells and cell culture
The human adherent tumor cell lines A431 (epidermoid carcinoma of skin with low invasive and metastatic potential – obtained from 85-year-old female patient) and BRO (highly aggressive amelanotic melanoma – derived from 34-year-old white male patient) (Price et al. Citation1988; Kao et al. Citation2008; Lockshin et al. Citation1985) were obtained from the Russian Collection of Vertebrate Cell Cultures (Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia). An adherent cell line of telomerized postnatal human fibroblasts (PHF) was used as control line (Yegorov et al. Citation2007). All cells were grown in DMEM medium containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, and gentamicin (40 μg/ml) at 37 °C in a humidified atmosphere containing 5% CO2. All medium/additional culture reagents were purchased from Invitrogen (Waltham, MA). The cells were passaged every 3 days at the density 105/ml. In all experiments, cells of 6th passage were used (logarithmic growth phase, confluence ∼60–70%). Cells were either grown in Costar flasks (Corning Inc., Corning, NY) or on microscope slides (Menzel-Glaser, Germany), depending on the particular protocol requirements. In the case of the flasks, to harvest the cells, a 50 mM EDTA/PBS (phosphate-buffered saline, pH 7.4) was applied to the cultures to allow the cells to detach.
Immunocytochemistry (ICC)
To perform immunocytochemical analyses, polylysine slides with growing cells were divided into two groups. The cells in the first group were immediately fixed in 96% ethanol (5 min) and then in acetone (3 min) at −20 °C. The cells in the second group were treated with 0.05% PBST (0.05% [v/v] Triton-X-100 in PBS) prior to this same fixation. Incubation in the detergent was performed under strict microscopic observation until a clear manifestation of integral nuclei with visually-intact membranes was evident against a background of the washed-out cytoplasm. The above procedure enabled the elimination of cytosolic VEGFR-1 and improved the potential access of antibodies to nuclear antigens.
After fixation, cells in both groups were coated with a solution of 5% bovine serum albumin (BSA) in PBS, then incubated at room temperature for 10 min to prevent nonspecific binding of antibodies. Primary polyclonal rabbit antibody to the intracellular domain of VEGFR-1 (recognizing only membrane-bound isoform of receptor; #ab2350, Abcam, Cambridge, UK) in PBS was then applied (at 5 μg/ml, 40 μl/slide) and the slides were incubated at 4 °C overnight. After gentle washing in PBS, secondary fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody (#ab6717, Abcam) was then applied (1:2000 dilution, 40 μl/slide) and the slides were incubated at 4 °C for 45 min. Cells treated with only secondary antibody served as negative controls. After gentle rinsing in PBS, each slide was analyzed using an Axio Scope A1 fluorescence microscope (Zeiss, Oberkochen, Germany). The presence/magnitude of FITC fluorescence indicated the presence/level of the studied protein in a specific cell compartment.
Flow cytometric analysis with intact and permeabilized cells
Cells from culture flasks were harvested using 50 mM EDTA/PBS, counted (hemocytometer), and the final concentration adjusted to 107/ml PBS. Aliquots (100 μl, 106 cells/sample) were then removed for analysis. One aliquot of cells was kept intact while a second aliquot was permeabilized in 90% cold methanol for 10 min. Each cell set was then treated with a solution of PBS containing primary antibody to the extracellular domain of VEGFR-1 (i.e. recognizing both membrane-bound and soluble isoforms of receptor; #MAB321, R&D Systems, Minneapolis, MN). The level of antibody used was the manufacturer-recommended level of 50 μg/ml PBS (40 μl/sample). The cells were then incubated at room temperature for 60 min. After washing the cells by centrifugation using PBS-5% BSA solution, the cells were then treated with secondary FITC-goat anti-mouse IgG (#STAR70, Serotec, Hercules, CA) at a level of 10 μg/ml PBS (20 μl/sample). The cells were incubated at 4 °C for 30 min, then washed twice in PBS-5% BSA, and re-suspended in 300 μl of 1% formaldehyde–PBS solution. Samples treated with only secondary antibody served as negative controls.
The fluorescence intensity of labeled VEGFR-1 was then evaluated in a BD FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA). An argon laser was used for excitation (λ = 488 nm). The gate to be analyzed was based on a combination of light scattering and cell size using the BD FACSDiva software. A minimum of 100 000 events/sample was acquired.
Extraction of nuclei and labeling
A hypo-osmotic solution containing HEPES (10 mM, pH 7.9), KCl (10 mM), and EDTA (0.1 mM) was selected as the nuclear extraction buffer. To 50 ml of this stock buffer, 2.0 ml of 10% Triton-X100 was added; immediately before use, dithiothreitol (DTT; 1 μl/ml buffer) and protease inhibitor mixture (1 tablet/50 ml extraction solution) were added based on manufacturer instructions (Complete/Mini, Roche, Mannheim, Germany).
Among all the studied nuclear extraction protocols used for obtaining materials for Western blotting, the method described in Chuvpilo et al. (Citation1999) and Tyrsin (Citation2007) was selected as the most gentle. In brief, cells were isolated from the flasks and counted. Aliquots containing 107 cells were then centrifuged (1000 rpm, 5 min, room temperature). The pellet was re-suspended in 1.0 ml cold PBS, transferred to a clean tube, and re-washed in PBS (2 min, 2000 rpm). The resulting pellet was gently re-suspended in 150 μl hypo-osmotic buffer and allowed to incubate for 60 min at 4 °C. All further manipulations were performed on ice. To lyse the swollen cells, the resulting suspension was processed ≈10 times through a syringe with 26-G needle. The cytosolic fraction was then isolated by centrifugation (3 min, 7000 rpm, 4 °C) of the processed suspension. The supernatant (i.e. cytoplasmic fraction) was removed and the pellet (nuclear fraction) re-washed in 1.0 ml buffer. Extracted nuclei were then incubated/stabilized in 1 ml of 5 mM MgCl2–PBS solution for 24 hr at 4 °C. Success of the extraction was confirmed by staining of methanol-fixed smears from the nuclear suspension with both Romanovsky’s Azur-Eosin and Mayer’s hematoxylin solutions (both Abris+, St. Petersburg, Russia). The Azur-Eosin stain was used to assess nuclei purity from cytoplasmic residues (i.e., by absence of pink color of eosin). Mayer's hematoxylin was used to confirm nuclear membrane integrity.
For labeling, a 500 μl aliquot of nuclear suspension (predicted to roughly correspond to ≈5 × 106 nuclei) was transferred to a tube. Preservation of nuclear proteins was determined by evaluation of levels of two specific nuclear markers, PCNA and lamin A/C. For the analysis, a dedicated aliquot received primary mouse antibody against PCNA (#29, Abcam) at a manufacturer-recommended level of 50 μg/ml (50 μl/sample); the parallel sample received mouse antibody against lamin A/C (sc-7292, Santa Cruz, Santa Cruz, CA) at 1 μg/sample. The nuclei were then incubated at room temperature for 60 min. After washing by centrifugation using PBS-5% BSA solution, the nuclear suspensions were then treated with secondary FITC-anti-mouse IgG (#STAR70) at 10 μg/ml PBS (30 μl/sample). The nuclei were then incubated at 4 °C for 30 min, washed twice in PBS before finally being re-suspended in 1% formaldehyde–PBS solution for FC analysis.
To test for cytoplasmic residues in the preparations, a third aliquot of nuclei was treated as above using rabbit primary antibody to human β-tubulin (#2146, Cell Signaling Technology, Danvers, MA) at 1:100 dilution (50 μl/sample). In this case, the secondary antibody used was FITC-anti-rabbit IgG (#STAR121F, Serotec) diluted at 1:200 (30 μl/sample). To measure VEGFR-1, a fourth aliquot of nuclei was labeled using mouse #MAB321 primary antibody (see above) at 50 μg/ml (50 μl/sample). After incubation, FITC-anti-mouse IgG (#STAR70, 10 μg/ml, 30 μl/sample) was employed. In all cases, samples treated only with secondary antibody served as negative controls.
Nuclear analysis by flow cytometry
Flow cytometric analysis of the stained nuclei was carried out in the BD FACSCanto II system using associated BD FACSDiva software. The area of gating was determined in the logarithmic graph DotPlot FSC/SSC. To select a gate, a final aliquot of nuclear suspension was stained with 500 μl propidium iodide (PI, 50 μg/ml; Becton Dickinson) for 30 min in the dark. The quantitative criterion for the content of nuclear proteins was the difference in mean fluorescence intensity (ΔMFI) between experimental and control (i.e. treated with only secondary antibody) samples.
Statistical analysis
The data obtained in at least five independent experiments with three measurements/point being analyzed. All data (means) were then compared for significance of any differences using two nonparametric tests: Mann–Whitney U-test and Kolmogorov–Smirnov test. A p-values < 0.05 was accepted as statistically significant. All data were analyzed using Statistica 6.0 software (Dell, Round Rock, TX).
Results
Detection of VEGFR-1 in tumor and normal cells by immunocytochemistry (ICC)
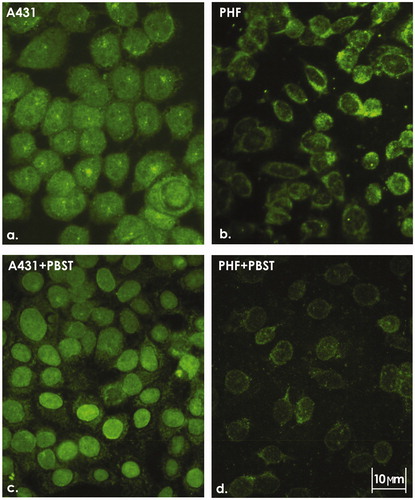
Based on the traditional understanding of tyrosine kinase receptors as membrane-localized receptors, we assumed that the growth-promoting effects of VEGFR-1 are mediated by stimulating VEGFR-1 at the cell surface. However, ICC staining of the BRO (data not shown, see Supplemental data) and A431 human tumor cell lines revealed predominant intracellular localization of the receptor (). In normal fibroblasts (PHF; control), VEGFR-1 was poorly visualized (). To test the possibility that the nucleus represented a major site of VEGFR-1 expression, the cells were treated with Triton X-100 prior to their fixation for ICC experiments. Indeed, this approach, which strongly reduced the cytoplasmic signal, allowed to establish the presence of VEGFR-1 in the nuclei of tumor, but not of normal, cells (). This raises the possibility that it is the nuclear receptor that is involved in carcinogenesis and that VEGFR-1 content in the nucleus may correlate with the degree of cell malignancy. To provide a tool for testing this assumption, the level of the receptor in the nuclei of two neoplastic cell lines with different degrees of aggressiveness was compared.
Figure 1. ICC staining of A431 cells and PHF normal fibroblasts. Intracellular localization of VEGFR-1 in (a) A431 and (b) PHF cells. (c) Nuclear localization of VEGFR-1 in A431 cells treated with 0.05% PBST prior to fixation. (d) Almost complete absence of nuclear VEGFR-1 in PHF cells treated with 0.05% PBST prior to fixation. Representative images are shown. 400X magnification.
Detection of cell surface/intracellular VEGFR-1 in tumor and normal cells by FC analysis
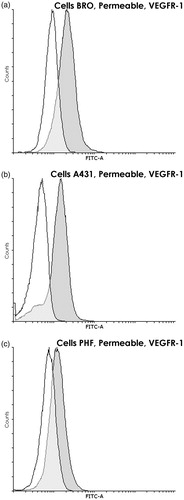
Before directly addressing nuclear VEGFR-1 expression by FC analysis, the study here first compared the amounts of VEGFR-1 expressed on the cell surface to those found in intracellular compartments. This was done by FC analysis comparing intact versus permeabilized cells. Intact cells, where only VEGFR-1 located on cytoplasmic membrane can be detected, displayed very little VEGFR-1 in all three cell lines (ΔMFI = 14.4 [±7.4] for BRO, 21.4 [±8.3] for A431, and 15.8 [±7.2] for PHF (, respectively]). After permeabilization, the measured signal increased sharply (ΔMFI = 425 [±92] for BRO, 302 [±81] for A431, and to 123 [±40] for PHF [, respectively]) as the intracellular receptor became accessible to antibodies. Thus, while VEGFR-1 level on the surface of tumor cells did not statistically differ from that of normal cells and approached values seen with the negative control, the intracellular VEGFR-1 content in malignant cells was almost three times higher than in PHF fibroblasts (p < 0.02). These results showed that the intracellular content of this receptor may be evaluated by FC analysis of fixed and permeabilized cells using the antibody combination chosen.
Figure 2. VEGFR-1 presence on cytoplasmic membrane of BRO and A431 tumor cell lines and PHF normal human fibroblasts. Flow cytometry using nonpermeabilized cells. Staining with anti-VEGFR-1 antibodies (dark gray) and only with control antibodies (white). Graph overlapping area is light gray. VEGFR-1 was almost absent on the cytoplasmic membrane of all three cell lines. Histogram of fluorescence intensity of nonpermeabilized human (a) BRO (ΔMFI = 14.4 ± 7.4), (b) A431 (ΔMFI = 21.4 ± 8.3), and (c) PHF (ΔMFI = 15.8 ± 7.2) cells. Representative histograms are shown.
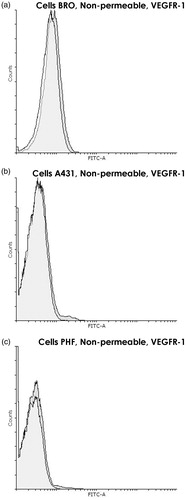
Figure 3. VEGFR-1 within BRO and A431 tumor cell lines and PHF normal human fibroblasts. Flow cytometry using permeabilized cells (90% methanol). Staining with anti-VEGFR-1 (dark gray) and only with control (white) antibodies. Graph overlapping area is light gray. Intracellular VEGFR-1 is found in all three lines; however, in PHF, its content is ≈3 times lower than in both tumor lines (p < 0.02). Histogram of fluorescence intensity of methanol-permeabilized human (a) BRO (ΔMFI = 425 ± 92), (b) A431 (ΔMFI = 302 ± 81), and (c) PHF (ΔMFI = 123 ± 40) cells. Representative histograms are shown.
Preparation and characterization of nuclear suspension suitable for flow cytometry
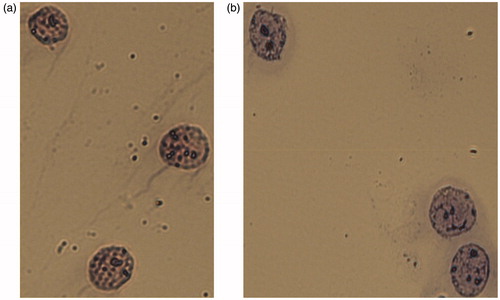
To optimize the protocol above, extracted intact nuclei were prepared for FC analysis. Initially, various published protocols were tested for the isolation of intact nuclei for subsequent use in Western blot analysis (Chuvpilo et al. Citation1999; Fan et al. Citation2005; Lee et al. Citation2007; Tyrsin Citation2007). These pilot analyses found most were unsuitable because either nuclear integrity was not maintained or nuclear proteins were denatured, outcomes incompatible with staining by antibodies directed at conformational determinants. By modifying several parameters of these existing protocols to secure a milder extraction of the nuclei, the procedures detailed in the Methods were arrived upon. Here, isolated nuclei had hardly any damage, there were few coalesced nuclei, and microscopic analysis confirmed the integrity of nuclear membranes and absence of cytoplasmic residues ().
Figure 4. Nuclei of A431 cells. Integrity of nuclear membrane and absence of cytoplasmic residues confirmed by microscopic analysis after staining. (a) Mayer’s hematoxylin. (b) Romanovsky’s Azur-Eosin. Representative microphotographs are shown. 400X magnification.
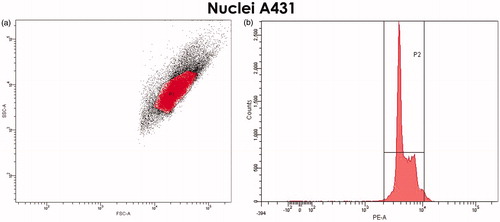
FC analysis of the PI-stained nuclei revealed that nuclear conglomerates were practically absent in the established gate. In the FSC/SSC DotPlot graph, intact nuclei formed a separate “cloud” in the upper right section); this was confirmed with a cell-cycle histogram of PI-stained nuclei: peak to the right of the G2-phase peak was almost not detected ().
Figure 5. Selection of the gate for nuclei of A431 cells. (a) FSC/SSC DotPlot graph. Single, cytoplasm-free nuclei are highlighted in red. (b) The cell-cycle histogram (propidium iodide-staining) confirms the correctness of selected gate.
As a quality control for the nuclear suspension used in the FC analysis, sets of nuclei were stained with antibodies specific for cytoplasmic and nuclear marker proteins. As seen in , cytoplasm-specific β-tubulin was barely detectable in the gate established in concordance with cell-cycle histogram for the extracted nuclei (ΔMFI = 7.6 [±2.6]). It should be stressed that, when the gate was shifted toward higher FSC and SSC values (where unpurified nuclei and conglomerates locate), the β-tubulin content increased to ΔMFI of ≈250. This confirmed that single nuclei without cytoplasmic residues were indeed located in the gate chosen for nuclear analysis.
Figure 6. Analysis of proteins in nuclei of tumor and normal cells. (a) Absence of specific cytoplasmic marker β-tubulin in the nuclei of BRO tumor cells. Flow cytometry. Staining with anti-β-tubulin (dark gray) and only with control (white) antibodies. Graph overlapping area is light gray. β-Tubulin is almost absent in extracted nuclei (ΔMFI = 7.6 ± 2.6). (b) High content of nuclear marker lamin A/C in nuclei of BRO tumor cells. Flow cytometry after staining with anti-LamA/C (dark gray) and only control (white) antibodies. Graph overlapping area is light gray. An extremely high content of lamin A/C was found in extracted nuclei (ΔMFI = 4109 ± 1226). (c–e) VEGFR-1 in nuclei of BRO and A431 cell lines and PHF normal human fibroblasts. Flow cytometry. Staining with anti-VEGFR-1 (dark gray) and only control (white) antibodies. Graph overlapping area is light gray. VEGFR-1 is found in nuclei of BRO and A431 tumor cells and not in nuclei of normal fibroblasts. The highest nuclear VEGFR-1 content was found in the most malignant cell line, that is, ΔMFI for BRO was 1.75 times higher than that for A431 (p = 0.014). Representative histogram of fluorescence intensity of nuclei of human (c) A431 (ΔMFI = 171 ± 49), (d) BRO (ΔMFI = 300 ± 59), and (e) PHF (ΔMFI = 12.7 ± 6.9) cells.
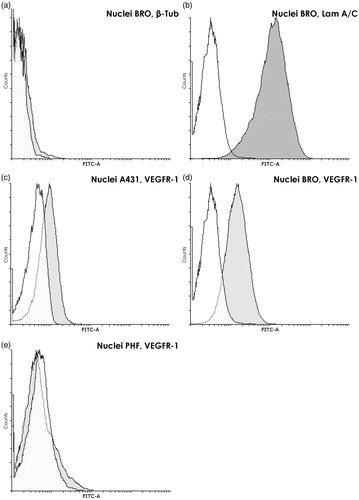
The presence of a high level of specific nuclear markers, that is, PCNA (data not shown) and lamin A/C, ΔMFI = 800 [±176] and 4109 [±1226], respectively (), demonstrated the preservation of proteins in isolated nuclei. Moreover, the results showed the extraction procedure rendered the nuclear membrane antibody-permeable, making additional membrane permeabilization unnecessary. Thus, the recorded ΔMFI reflected total protein content on the surface and inside the nucleus.
Nuclear detection of VEGFR-1 by FC analysis
Since the experiments described above convinced us that nuclei extracted using the new protocol met quality requirements for further FC analysis, nuclear VEGFR-1 expression was then also quantified by FC analysis. Measurements here of nuclear VEGFR-1 content confirmed previously-obtained ICC data on the presence of the receptor in the nuclei of only tumor cells. While a high nuclear VEGFR-1 content was registered in both malignant lines (ΔMFI of 171 [±49] for A431 [] and 300 [±59] for BRO []), the receptor's level in nuclei of normal cells was close to values seen with the negative control (ΔMFI of 12.7 [±6.9] for PHF []). These results also showed that the highest nuclear VEGFR-1 content was found in the most malignant cell line, that is, ΔMFI for BRO was 1.75 times higher than that for the A431 cells (p = 0.014).
Discussion
The results obtained show that the selected protocol for extraction, purification, and stabilization of nuclei was suitable for identifying and measuring the content of nuclear proteins by flow cytometry (FC). The clear advantages of this novel applied method are: (1) the ability to examine a large number of nuclei in one experiment which increases the reliability of results. In addition, the gating used in the FC allows analyzing the proteins of interest only in the region of single, well-purified nuclei; (2) the opportunity to compare the level of a given nuclear protein in different cell lines; and, (3) the ability to monitor the nuclear content of any protein before and after any exposure of the cells to a toxicant/drug.
The results of this paper differ from the traditional understanding of tyrosine kinase receptors being located only on the cell surface. The two methods – ICC with PBST treatment and FC-analysis of cellular nuclei – made it possible to detect the presence of VEGFR-1 in nuclei of tumor cells but not in nuclei of normal cells. The revealed trend indirectly indicates that the nuclear receptor can somehow participate in the process of malignancy. This assumption is supported by the established fact that the highest content of VEGFR-1 was found in the nuclei of the most malignant cell line (i.e. ΔMFI for BRO was 1.75 times higher than that for A431; p = 0.014). Nevertheless, such a hypothesis still requires experimental confirmation.
The current data were consistent with conclusions from Lee et al. (Citation2007) who also found VEGFR-1 in the nucleus. In that study – performed on human breast carcinoma lines by ICC using confocal microscopy – the authors observed co-localization of the receptor with nuclear lamina. Lamins are known to be responsible for nuclear envelope formation and chromatin organization, and their degradation leads to apoptosis (Broers and Ramaekers Citation2014). According to Lee et al. (Citation2007), VEGFR-1 seems to provide physical stabilization of the nuclear lamina and therefore inhibits apoptosis of tumor cells.
However, other noncanonical mechanisms of action by the receptor should not be excluded. In particular, according to Bhattacharya et al. (Citation2016), intracrine VEGFR-1 signaling mediates the activity of pro-survival pathways via phosphorylation of certain proteins by the kinase domain of the receptor. In addition, given the low tyrosine kinase potential of VEGFR-1, the signaling cascade can be also initiated by different protein–protein interactions. Moreover, the latter scenario may be implemented even by means of receptor fragments that influence gene expression (Chen and Hung Citation2015).
The absence of the receptor in nuclei of normal cells on the one hand, and the highest nuclear VEGFR-1 content in highly malignant BRO cells on the other, suggest that level of the nuclear receptor might be used for prognostic purposes. These same findings also could suggest that VEGFR-1 itself might be considered as a potential target in anti-cancer therapy. Of course, the above reasoning still needs to be verified in a series of experiments with different tumor cell lines and primary tumor samples of common histogenesis with varying degrees of aggressiveness. Further, as a therapeutic target, the intracellular (including nuclear) localization of VEGFR-1 would require use of novel methods to suppress its activity. While the use of monoclonal antibodies will not produce the desired effect, genetically engineered approaches might prove effective. Indeed, in Owen et al. (Citation2012), synthetic Morpholino oligonucleotide (blocking the formation of the membrane-bound isoform of VEGFR-1) administered to athymic mice with human breast adenocarcinoma resulted in a decrease in tumor volume by 88.9% and vascular density by 50% (each relative to control). Such impressive results seem to have been achieved by an influence of Morpholino oligomer on the total cellular pool of VEGFR-1, and not only on its surface portion.
Certainly, accurate data about the localization and functions of any protein can only be obtained through combining several methods of investigation. Therefore, it is hoped that the approach here for the detection of proteins in native nuclei using flow cytometry will expand the knowledge of the mechanisms of action of the proteins under study and will prove useful both for fundamental science and for practical medicine.
References
Supplemental Material
Download PDF (543 KB)Acknowledgements
Authors E.G.T., S.I.N., A.N.I., O.O.R. performed all experiments. All authors analyzed the data and contributed to writing/editing of the paper. The authors would also like to acknowledge Prof. Thomas Hünig (Institute for Virology and Immunobiology, University of Würzburg, Germany) for excellent advices and critical manuscript editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. 1996. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 87:3336–3343.
- Bhattacharya R, Ye X, Wang R, Ling X, McManus M, Fan F, Boulbes D, Ellis L. 2016. Intra-crine VEGF signaling mediates the activity of pro-survival pathways in human colorectal cancer cells. Cancer. Res. 76:3014–3024.
- Broers J, Ramaekers F. 2014. The role of the nuclear lamina in cancer and apoptosis. Adv Exp Med Biol. 773:27–48.
- Chen M, Hung M. 2015. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. Febs J. 282:3693–3721.
- Chuvpilo S, Avots A, Berberich-Siebelt F, Glöckner J, Fischer C, Kerstan A, Escher C, Inashkina I, Hlubek F, Jankevics E. 1999. Multiple NF-ATc isoforms with individual transcriptional properties are synthesized in T-lymphocytes. J Immunol. 162:7294–7301.
- Fan F, Wey J, McCarty M, Belcheva A, Liu W, Bauer T, Somcio R, Wu Y, Hooper A, Hicklin D, et al. 2005. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 24:2647–2653.
- Kao W, Lin C, Lee L, Lee P, Hung C, Lin Y, Chen S, Ke F, Hwang J, Lee M. 2008. Investigation of MMP-2 and -9 in a highly invasive A431 tumor cell sub-line selected from a Boyden chamber assay. Anticancer Res. 28:2109–2120.
- Lee T, Seng S, Sekine M, Hinton C, Fu Y, Avraham H, Avraham S. 2007. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally-expressed VEGFR1/FLT1. PLoS Med. 4:e186.
- Li YL, Zhao H, Ren XB, Li YL, Zhao H, Ren XB. 2016. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol Med. 13:206–214.
- Lockshin A, Giovanella B, de Ipolyi P, Williams L, Mendoza J, Yim S, Stehlin J. 1985. Exceptional lethality for nude mice of cells derived from a primary human melanoma. Cancer Res. 45:345–350.
- Owen L, Uehara H, Cahoon J, Huang W, Simonis J, Ambati B. 2012. Morpholino-mediated increase in soluble Flt-1 expression results in decreased ocular and tumor neovascularization. PLoS One. 7:e33576.
- Price J, Sauder D, Fidler I. 1988. Tumorigenicity and metastatic behavior in nude mice of two human squamous cell carcinoma lines that differ in production of the cytokine ETAF/IL-1. J Invest Dermatol. 91:258–262.
- Tyrsin D. 2007. Autoregulation of NFATc1 gene. Dissertation for the completion of Doctorate in Natural Sciences (Bayerische Julius-Maximilians-Universitat, Wurzburg, 2007).
- Yegorov E, Moldaver M, Vishiniakova K, Terekhov S, Dashinimaev E, Cheglakov I, Toropygin I, Iarygin K, Chumakov P, Korochkin L. 2007. Enhanced control of proliferation in telomerized cells. Ontogenez. 38:105–119.
- Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen J, Jiao S, Gao Y, Liu C, Duan Z, et al. 2014. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity. 40:501–514.
- Zhang J, Silva T, Yarovinsky T, Manes TD, Tavakoli S, Nie L, Tellides G, Pober JS, Bender JR, Sadeghi MM. 2010. VEGF blockade inhibits lymphocyte recruitment and ameliorates immune-mediated vascular remodeling. Circ Res. 107:408–417.