
Abstract
Immunostimulatory effects of monoclonal antibodies (mAb) through binding to Fcγ receptors (FcγR) on immune cells are a likely cause of cytokine release syndrome. However, it is difficult to detect the potential risk of FcγR-dependent cytokine release associated with mAb in the current standard cytokine release assays (CRA), including the air-drying solid-phase method using human peripheral blood mononuclear cells (PBMC). To increase the sensitivity to detect FcγR-dependent cytokine release due to mAb, a high-density preculture (HDC) method was incorporated into the air-drying solid-phase CRA. Here, PBMC were exposed to panitumumab, trastuzumab, rituximab, or alemtuzumab at 0.1, 0.3, 1, and 3 μg/well for 24 or 48 hr under both non-HDC and HDC conditions. T-cell agonists (anti-CD3 mAb, anti-CD28 super-agonist [SA] mAb) were used as reference mAb. Panitumumab, trastuzumab, rituximab, or alemtuzumab induced cytokine release under both non-HDC and HDC conditions, and cytokine release caused by alemtuzumab was more pronounced under HDC conditions. To investigate FcγR involvement in cytokine release associated with panitumumab, trastuzumab, rituximab, and alemtuzumab, CRA of these four mAb were conducted with anti-FcγRI, -FcγRII, or -FcγRIII F(ab’)2 fragments. The results showed cytokine release caused by trastuzumab, rituximab, and alemtuzumab was significantly suppressed by anti-FcγRIII F(ab’)2 pretreatment, and slightly reduced by anti-FcγRI or anti-FcγRII pretreatment, indicating these mAb induced FcγR (especially FcγRIII)-dependent cytokine release from PBMC. Cytokine release caused by panitumumab was slightly suppressed by anti-FcγRIII F(ab’)2 pretreatment. Anti-CD3 mAb and anti-CD28 SA mAb also induced significant release of cytokines under HDC conditions compared with that under non-HDC conditions. In conclusion, CRA incorporating HDC into the air-drying solid-phase method using human PBMC could sensitively capture the FcγR-dependent cytokine release potential of mAb.
Introduction
Cytokine release syndrome (CRS), which occurred in the Phase-I clinical trial of the anti-CD28 super-agonist (SA) monoclonal antibody (mAb) TGN1412, is one of the most serious adverse effects associated with parenteral administration of therapeutic mAb (Suntharalingam et al. Citation2006). CRS is typically characterized by the release of pro-inflammatory cytokines including interleukins (IL), interferon (IFN)-γ, and tumor necrosis factor (TNF)-α into the blood, resulting in fever, chills, hypotension, and multiple organ failure immediately after treatment with a therapeutic mAb (Finco et al. Citation2014). The cytokine release caused by therapeutic mAb is considered to be mediated by two mechanisms: the binding of the fragment antigen-binding region (Fab) of mAb to the cell surface target on immune cells and the binding of the fragment crystallizable region (Fc) of mAb to Fcγ receptors (FcγR) on effector cells (Wing et al. Citation1996). The former (i.e., binding of Fab) is primarily associated with CRS associated with T-cell agonists such as OKT3 [an anti-CD3 mAb] and TGN1412; the latter (i.e., binding of Fc) is mainly associated with CRS that arises with alemtuzumab, trastuzumab, and rituximab and is due to antibody-dependent cellular cytotoxicity (ADCC) activity (Kirton et al. Citation2011; Paul and Cartron Citation2019).
Cytokine release assays (CRA) using human peripheral blood mononuclear cells (PBMC) have become common in vitro assay systems to predict the risk of clinically relevant CRS because in vivo and in vitro animal studies failed to predict this risk (Stebbings et al. Citation2007; Eastwood et al. Citation2010; Hansel et al. Citation2010; Pallardy and Hunig Citation2010). Several different assay platforms have been introduced as nonclinical risk assessment systems, including liquid-phase, wet-coating solid-phase, and air-drying solid-phase methods (Findlay et al. Citation2010; Römer et al. Citation2011; Eastwood et al. Citation2013). The solid-phase method is commonly used to detect the risk of CRS due to mAb (Finco et al. Citation2014). In fact, TGN1412 caused profound cytokine release from PBMC in the solid-phase method, but not in the liquid-phase method (Findlay et al. Citation2010, Citation2011). However, the cytokine release inducible by TGN1412 was reported to be detected using high-density precultured (HDC) PBMC even in the liquid-phase method because the HDC method enhanced T-cell reactivity via contact with monocytes (Bartholomaeus et al. Citation2014; Hussain et al. Citation2015).
Recently, CRA have been applied to evaluate the mAb that act on FcγR in addition to the mAb having agonistic activity. However, unlike the case with TGN1412, alemtuzumab caused TNFα and IFNγ in the liquid-phase, but not in the solid-phase, method when compared with levels of each induced with an isotype control antibody (Vessillier et al. Citation2015). Further, the increase in TNFα due to alemtuzumab in the solid-phase method was comparable to that attained with trastuzumab or bevacizumab which have been associated with a moderate/low CRS incidence in humans (Chung Citation2008). Such results indicate the disadvantage of the solid-phase method. Thus, it is difficult to detect potential risks of CRS associated with mAb such as alemtuzumab that act on FcγR in the solid-phase method (Findlay et al. Citation2010; Eastwood et al. Citation2013; Grimaldi et al. Citation2016).
To establish a solid-phase assay system for detecting the potential risk of CRS through the binding of mAb to FcγR on effector cells, this study attempted to incorporate HDC-PBMC into the air-drying solid-phase method. In the studies here, several mAb, for example, trastuzumab, rituximab, and alemtuzumab were evaluated as each was reported to cause FcγR-dependent cytokine release (Kirton et al. Citation2011; Paul and Cartron Citation2019). In addition, anti-CD3 mAb and anti-CD28 SA mAb with T-cell agonistic activity were evaluated. Panitumumab was used as a negative control because its risk of CRS has been reported to be low (Bugelski et al. Citation2009). Lastly, the current study also examined the types of FcγR that contribute to the cytokine release caused by these mAb in this assay system.
Materials and methods
Antibodies and reagents
The following mAb were used in the present study: panitumumab (Vectibix®, Amgen, Thousand Oaks, CA, USA; anti-epidermal growth factor receptor (EGFR) human IgG2κ), rituximab (Rituxan®, Genentech, South San Francisco, CA, USA; anti-CD20 chimeric murine/human IgG1κ), trastuzumab (Herceptin®, Genentech; anti-HER2 humanized IgG1κ), and alemtuzumab (Campath®, Genzyme, Cambridge, MA, USA; anti-CD52 humanized IgG1κ). Anti-CD3 murine IgG2a mAb (clone OKT3; BioLegend, San Diego, CA, USA) and anti-CD28 SA murine IgG1κ mAb (clone ANC28.1/5D10; Ancell Corp., Bayport, MN, USA) were used as reference mAb. Mouse IgG1 antibody (BioLegend), mouse IgG2a antibody (BioLegend), human IgG1 antibody (Eureka Therapeutics, Emeryville, CA, USA), and human IgG2 antibody (Sigma, St. Louis, MO, USA) were used as isotype controls. Lipopolysaccharide (LPS; from Escherichia coli Type 055:B5) and phytohemagglutinin (PHA) (both Sigma) were used as positive controls.
RPMI-1640 medium, penicillin–streptomycin, fetal bovine serum (FBS), and phosphate-buffered saline (PBS, pH 7.2) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). CTL anti-aggregate wash supplement 20× was obtained from Cellular Technology Limited (Cleveland, OH, USA). The F(ab’)2 fragments purchased were: anti-human CD64 F(ab’)2 fragment (clone 10.1), anti-human CD32 F(ab’)2 fragment (clone 7.3), anti-human CD16 F(ab’)2 fragment (clone 3G8), and mouse IgG1 F(ab’)2 fragment (clone MOPC 31 C) (all Ancell Corporation).
Immobilization of antibodies
Antibody solutions diluted in sterile PBS were added at 5 μl/well and air-dried onto the surface of wells of 96-well polypropylene plates (untreated 96-well U-bottom microtiter plates; Corning Inc., Corning, NY, USA) by overnight incubation at room temperature. The concentrations of antibodies were set at 0.1, 0.3, 1, and 3 μg/well based on preliminary in-house data and previous publications (Findlay et al. Citation2010, Citation2011; Wolf et al. Citation2012; Vessillier et al. Citation2015). Each coated well was then washed twice with 200 μl sterile PBS. LPS and PHA at a 5 μg/ml final concentration then added to the wells just before the assay without immobilization.
PBMC
Human PBMC were purchased from Cellular Technology Limited. Use of human-derived test materials was approved by the Research Ethics Committee of Daiichi-Sankyo Co., Ltd. (Tokyo, Japan). The requirement for consent was waived by the Ethics Committee.
Cell culture and stimulation
Frozen PBMC from six donors were each thawed at 37 °C and washed with CTL-Thaw Solution (CTL anti-aggregate wash supplement [20×] diluted 20-fold with RPMI-1640 supplemented with 2 mM L-glutamine, 100 U penicillin/ml, and 100 μg streptomycin/ml). The PBMC were then re-suspended in 10% FBS-RPMI1640 (RPMI-1640 with 10% FBS, 2 mM L-glutamine, 100 U penicillin/ml, and 100 μg streptomycin/ml), seeded at 2 × 105 cells in 200 μl volumes into antibody-immobilized wells of 96-well polypropylene plates (see “immobilization of antibodies” in Materials and Methods), and incubated at 37 °C under 5% CO2 for 24 or 48 hr, in duplicate (non-HDC method). For HDC, 1.5 × 107 PBMC (in 1.5 ml 10% FBS-RPMI-1640) were placed into the wells of a 24-well polystyrene plate (Greiner Bio-One International GmbH, Kremsmünster, Austria) and then incubated at 37 °C under 5% CO2 for 24 hr to enhance cell reactivity. Culture conditions for the HDC were based on previous publications (Römer et al. Citation2011; Bartholomaeus et al. Citation2014; Hussain et al. Citation2015, Citation2019). After HDC, the PBMC were inoculated into antibody-immobilized wells as described in the non-HDC method.
For the FcγR blockade experiments, HDC-PBMC were pretreated with each F(ab’)2 fragment (at 10 μg/ml) for 45 min at 4 °C before stimulation in antibody-coated wells, in duplicate. Based on preliminary data, the test antibody concentration was set at 3 μg/well, at which the maximum cytokine release responses induced by the antibodies were expected.
Cytokine measurements
At the end of the incubations, culture supernatant from each well was collected after centrifugation (500 × g, room temperature, 10 min). Concentrations of TNFα, IFNγ, IL-2, IL-6, IL-8, and IL-10, and macrophage inflammatory protein (MIP)-1α in each supernatant were then measured using a Milliplex MAP Human Magnetic Cytokine/Chemokine Bead Panel (Merck, Darmstadt, Germany) in a BioPlex® system (BioPlex 100 array reader with BioPlex Manager software; BioRad, Hercules, CA, USA), according to manufacturer instructions.
Measurement of cell proliferation by WST-8
Three hours prior to the end of incubation, all of the PBMC suspension (i.e., 200 μl) in each well was transferred to a polystyrene flat-bottomed 96-well plate. A total of 20 μl Cell Counting Kit-8 solution containing water-soluble tetrazolium salts-8 (WST-8) (Dojindo Labs, Kumamoto, Japan) was added to each well, and the plate was incubated for 3 hr at 37 °C under 5% CO2. At the end of incubation, formazan production was assessed by measuring the optical density (OD) at 450 nm (reference wavelength 650 nm) using a SPECTRAmaxPLUS microplate reader with SOFTmaxPRO software (Nihon Molecular Devices Co., Tokyo, Japan).
Statistical analysis
Mean values of maximum responses to 0.1, 0.3, 1, or 3 μg mAb/well for each donor were calculated using Excel software (Office 2013; Microsoft, Redmond, WA, USA) (Findlay et al. Citation2010, Citation2011). The maximum responses of mAb were statistically compared to those for panitumumab using a Student’s t-test (homogenous) or Welch’s t-test (not homogenous). In addition, differences in maximum response between HDC and non-HDC conditions were analyzed using these same tests. All data were analyzed using SAS System Release 8.2 software (SAS Institute, Cary, NC, USA). A p-value < 0.05 was considered significant.
In the FcγR blockade experiments, the percent cytokine release induced by immobilized antibodies from F(ab’)2 pretreated PBMC relative to that of mouse IgG1 F(ab’)2 pretreated PBMC was also calculated. For comparisons of F(ab’)2 fragment treatment, cytokine concentrations were analyzed using a Student’s (homogeneous) or Welch’s t-test (not homogeneous).
Results
Enhanced response of cytokine release from PBMC by various mAb under HDC conditions
With PBMC from more than half the donors, trastuzumab, rituximab, and alemtuzumab apparently increased (≥ 3-fold) the release of almost all the measured cytokines except for IL-2 under both non-HDC and HDC conditions when compared with effects from PBS. Panitumumab (negative control) also increased release of almost all the cytokines except IL-2 under HDC conditions and except IL-2 and IL-10 under non-HDC conditions (vs. PBS). However, levels of cytokine release induced by panitumumab were clearly lower than those induced by the other three mAb under both non-HDC and HDC conditions ().
Table 1. Cytokine release from human PBMC induced by various immobilized antibodies.
TNFα, IL-8, and IL-10 release due to alemtuzumab under HDC conditions were significantly higher vs. those under non-HDC conditions. In contrast, neither trastuzumab, rituximab, nor panitumumab caused a difference in cytokine release levels between conditions (). In addition, when compared with panitumumab, all cytokines except IL-2 showed a statistically significant increase or apparent (≥ 3-fold) increase in more than half the donor PBMC tested by alemtuzumab under HDC conditions; significant or apparent increases were seen only for TNFα, IFNγ, IL-6, and IL-8 under non-HDC conditions. On the other hand, no increases were found in the number of increased cytokines induced by rituximab and trastuzumab under HDC conditions when compared with that under non-HDC conditions. Specifically, cytokines that gave rise to a statistically-significant increase or apparent (≥ 3-fold) increase in more than half the donor PBMC tested with rituximab under HDC conditions were TNFα, IFNγ, and IL-8; under non-HDC conditions, the cytokines displaying such outcomes were TNFα, IFNγ, IL-6, IL-8, and MIP-1α. With trastuzumab, under HDC conditions, the cytokines that displayed a statistically significant or apparent (≥ 3-fold) increase in more than half the donor PBMC tested were TNFα, IFNγ, IL-8, and MIP-1α; under non-HDC conditions, TNFα, IFNγ, IL-6, IL-8, and IL-10 displayed these outcomes ( and ). As for other changes, there was a tendency to lower IL-6 release under HDC conditions as a result of PBMC treatment with panitumumab, trastuzumab, rituximab, or alemtuzumab exposure compared with the levels noted under non-HDC conditions ().
Table 2. Cytokine release response to various immobilized antibodies by donor PBMC.
Compared with PBS, anti-CD3 mAb induced significant increases in release of all the cytokines measured under both non-HDC and HDC conditions. There was also a significant increase in TNFα, IL-10, and MIP-1α release caused by anti-CD3 mAb under HDC conditions compared with levels under non-HDC conditions (). Similarly, anti-CD28 SA mAb induced significant increases in release of all measured cytokines compared with that due to PBS under non-HDC and HDC conditions. Lastly, compared to findings under non-HDC conditions, there were significant increases in TNFα and IL-10 release induced by anti-CD28 SA mAb under HDC conditions.
Four isotype controls (human IgG1 and IgG2, mouse IgG1 and IgG2a) also caused increased release of almost all the measured cytokines (except IL-2) compared with PBS under both non-HDC and HDC conditions. As with the other mAb tested, there was a tendency to lower IL-6 release under HDC conditions compared with under non-HDC conditions (). Both LPS- and PHA-positive controls induced marked cytokine release under both non-HDC and HDC conditions, with the increase higher under HDC vs. non-HDC conditions.
For Readers interested in examining cytokine release at the level of each individual donor, these data are provided here in a Supplementary file.
Cell proliferation in PBMC by mAb under HDC conditions
Anti-CD3 mAb and anti-CD28 SA mAb increased cell proliferation under both non-HDC and HDC conditions. Furthermore, the proliferative response due to anti-CD3 mAb was higher under HDC than under non-HDC conditions (). Panitumumab, trastuzumab, rituximab, and alemtuzumab did not induce cellular growth in either condition ().
Table 3. Cell proliferation induced in donor PBMC by various immobilized antibodies.
Time course of cytokine release from/cell proliferation in PBMC induced by various mAb
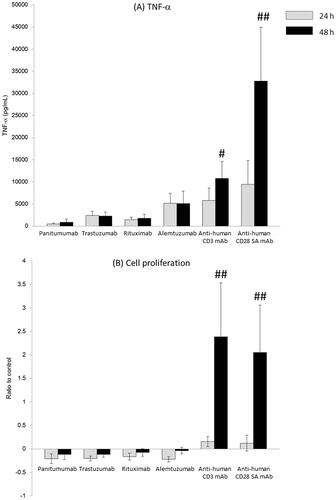
Almost all the releases of cytokines and cell proliferation induced by anti-CD3 and anti-CD28 SA mAb under HDC conditions were time-dependent and more pronounced at 48 than at 24 hr. The results for the release of TNFα (as a representative cytokine) are shown in . In contrast, the release of cytokines due to panitumumab, trastuzumab, rituximab, and alemtuzumab was not affected by the length of the incubation period ().
Figure 1. TNFα release and cell proliferation induced by various immobilized antibodies from human peripheral blood mononuclear cells for 24 or 48 hr. mAb: monoclonal antibody; SA: super-agonist. A 0.2 ml suspension of human PBMC (2 × 105 cells/well) following high-density pre-culture (107 cells/ml) was incubated with immobilized Ab (0.1, 0.3, 1, and 3 μg/well) for 24 or 48 hr. (A) TNFα levels in the supernatants after 24 or 48 hr of exposure. (B) Cell proliferation determined by WST-8 assay over last 3 hr of 24 or 48 hr incubations. Values shown are means ± SD of 5–6 donors from six independent experiments. *p < 0.05, **p < 0.01 (Student’s t-test), #p < 0.05, ##p < 0.01 (Welch’s test): compared with values at 24 hr.
Inhibition of cytokine release by FcγR blockade
Cytokine release induced by alemtuzumab was reduced by pretreatment with anti-FcγRI, -FcγRII, and -FcγRIII F(ab’)2 fragments. Further, the suppressive effect of anti-FcγRIII F(ab’)2 on alemtuzumab-induced cytokine release was more pronounced than that of anti-FcγRI and anti-FcγRII (). The release of multiple cytokines due to trastuzumab was also suppressed by FcγRIII blockade, but not affected by FcγRI or FcγRII. Interestingly, though release of cytokines induced by rituximab was also suppressed most by FcγRIII blockade, IL-10 release by rituximab was only suppressed by FcγRII blockade. For the release of cytokines caused by panitumumab, the IL-6 release was only suppressed by FcγRIII blockade. The cytokine release due to isotype control antibodies was also reduced by FcγRI, FcγRII, or FcγRIII blockade. Release of cytokines due to LPS and PHA was not affected by the inhibition of the FcγR (data not shown).
Table 4. Cytokine release induced by various immobilized antibodies from F(ab’)2 fragment pretreated human PBMC.
Discussion
In this study, cytokine release due to trastuzumab, rituximab, or alemtuzumab in the air-drying solid-phase method was higher than that due to panitumumab under both non-HDC and HDC conditions. Further, unlike for trastuzumab, rituximab, and panitumumab, the cytokine release caused by alemtuzumab was more pronounced under HDC conditions. Consistent with the levels of released cytokines under HDC conditions in the present study, the incidence of infusion-related reaction (IRR; a disorder characterized by adverse reaction [such as chills, fever, nausea, headache, etc.] that typically develops during or several hours after the infusion) in a clinical context was reported to be 40% for trastuzumab (Genentech Citation2018), 77% for rituximab (Genentech Citation2019), and 97% for alemtuzumab (Sanofi Citation2015), whereas that for panitumumab was reported to be very low (4%) (Chung Citation2008). Therefore, the new CRA system developed here that incorporated HDC into an air-drying solid-phase CRA could detect the potential risk of CRS associated with the various types of mAb that induce cytokine release via FcγR in humans.
Here, alemtuzumab caused more pronounced release of TNFα, IL-8, and IL-10 from PBMC under HDC conditions than that under non-HDC conditions. It has been suggested that the release of TNFα, IFNγ, and IL-6 into the blood circulation that is associated with alemtuzumab originates from NK cells, monocytes, and neutrophils (Wing et al. Citation1996; Hu et al. Citation2009; Siders et al. Citation2010; Stebbings et al. Citation2013). In addition, Hussain et al. (Citation2015, Citation2019) reported that expression levels of FcγRIIb and FcγRIIIa on monocytes increased under HDC conditions, but those of FcγRI or FcγRIIa on monocytes under these conditions only slightly increased or remained unchanged, respectively. Further, these authors reported that expression of FcγRIIIa on NK cells under HDC were comparable to that under non-HDC conditions. It was also reported that immobilized anti-FcγRIII antibody induced cellular activation accompanied by TNFα and IFNγ production from NK cells (Romee et al. Citation2013). Moreover, monocytes expressing FcγRIII were reported to show ADCC activity depending on the affinity to FcγRIII, similar to the case for NK cells (Yeap et al. Citation2016), and monocytes were further reported to release TNFα by crosslinking to FcγRIII (Abrahams et al. Citation2000; Kramer et al. Citation2004).
Taking together this information and the present results showing that alemtuzumab-induced release of cytokines under HDC conditions was strongly inhibited by FcγRIII blockade, it was suggested that alemtuzumab induced cytokine release from NK cells and monocytes, and that the levels of release under HDC conditions were more pronounced due to the contributions from cytokines that were being released from monocytes with increased FcγRIIIa expression (due to HDC). Similarly, release of cytokines caused by trastuzumab and rituximab was suppressed by FcγRIII blockade, indicating these mAb also cause release of cytokines via FcγRIII under HDC conditions. However, unlike alemtuzumab, neither trastuzumab nor rituximab enhanced cytokine release under HDC conditions, implying that increased FcγRIII expression on monocytes and cytokine release from monocytes might not be mainly attributable to the cytokine release induced by these mAb under HDC conditions.
T-cell agonists anti-CD3 and anti-CD28 SA mAb induced significant release of cytokines and cell proliferation, especially under HDC conditions. Similar cytokine release and cell proliferation were reported in CRA under HDC conditions for TGN1412, another anti-CD28 SA mAb, or OKT3 (Römer et al. Citation2011; Bartholomaeus et al. Citation2014; Hussain et al. Citation2015). Römer et al. (Citation2011) suggested that co-localization of CD3 and phosphorylated tyrosine on T-cells increased T-cell reactivity under HDC conditions. Lühder et al. (Citation2003) proposed that anti-CD28 SA mAb, unlike conventional anti-CD28 mAb, recognized the proximal epitope of the CD28 antigen (capable of oligomerizing CD28 on T-cell surface), resulting in T-cell receptor (TCR)- independent T-cell activation. It was also postulated that increased FcγRIIb expression on monocytes under HDC conditions (Hussain et al. Citation2015, Citation2019) caused clustering of CD28 on the T-cell surface, leading to T-cell activation. This would be along the lines of interactions between CD80/CD86 on antigen-presenting cells and CD28 on T-cells.
In the present study, compared with PBS, panitumumab associated with a low risk of CRS caused mild increases in cytokine release in both HDC and non-HDC conditions. In solid-phase CRA, it is well known that antibodies induce nonspecific cytokine release via the binding of their Fc section to effector cell FcγR (Findlay et al. Citation2011; Eastwood et al. Citation2013; Vessillier et al. Citation2015; Grimaldi et al. Citation2016). Cytokine release by panitumumab was inhibited by FcγRIII blockade under HDC conditions in this study, suggesting this class of cytokine release was an FcγR- dependent nonspecific effect. In addition, the order of intensity of cytokine release caused by the mAb tested was “trastuzumab (humanized IgG1) ≈ rituximab (humanized IgG1) > panitumumab (human IgG2), and human IgG1 isotype control antibody > human IgG2 isotype control antibody” under both HDC and non-HDC conditions. Moreover, the degree of inhibition by FcγRIII blockade was small for panitumumab (humanized IgG2) compared with the findings for alemtuzumab, rituximab, or trastuzumab (each a humanized IgG1). Taking into account that the affinity of human IgG1 for FcγR is higher than that of human IgG2 for all of FcγRI, FcγRII, and FcγRIII (Brennan et al. Citation2010), the intensity of cytokine release and the effect of FcγR blockade in this study might be related to the affinity of each isotype to FcγR as described above.
In conclusion, it would seem that incorporating HDC into air-drying solid-phase CRA could sensitively capture cytokine-releasing effects induced via by the binding of the Fc region of mAb (such as alemtuzumab) with FcγR on immune cells.
Supplemental Material
Download MS Word (965 KB)Acknowledgments
The authors thank Hitomi Terada for excellent support in the experiments.
Disclosure statement
The authors declare no conflicts of interest. The authors alone are responsible for the content of this paper.
Related Research Data
References
- Abrahams V, Cambridge G, Lydyard P, Edwards J. 2000. Induction of TNFα production by adhered human monocytes: Key role for FcγRIIIa in rheumatoid arthritis. Arthritis Rheum. 43(3):608–616.
- Bartholomaeus P, Semmler L, Bukur T, Boisguerin V, Römer P, Tabares P, Chuvpilo S, Tyrsin D, Matskevich A, Hengel H, et al. 2014. Cell contact-dependent priming and Fc interaction with CD32+ immune cells contribute to the TGN1412-triggered cytokine response. J Immunol. 192(5):2091–2098.
- Brennan F, Morton L, Spindeldreher S, Kiessling A, Allenspach R, Hey A, Muller P, Frings W, Sims J. 2010. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. Mabs. 2(3):233–255.
- Bugelski P, Achuthanandam R, Capocasale RJ, Treacy G, Bouman-Thio E. 2009. Monoclonal antibody-induced cytokine-release syndrome. Expert Rev Clin Immunol. 5(5):499–521.
- Chung C. 2008. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist. 13(6):725–732.
- Eastwood D, Bird C, Dilger P, Hockley J, Findlay L, Poole S, Thorpe S, Wadhwa M, Thorpe R, Stebbings R. 2013. Severity of the TGN1412 trial disaster cytokine storm correlated with IL-2 release. Br J Clin Pharmacol. 76(2):299–315.
- Eastwood D, Findlay L, Poole S, Bird C, Wadhwa M, Moore M, Burns C, Thorpe R, Stebbings R. 2010. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br J Pharmacol. 161(3):512–526.
- Finco D, Grimaldi C, Fort M, Walker M, Kiessling A, Wolf B, Salcedo T, Faggioni R, Schneider A, Ibraghimov A, et al. 2014. Cytokine release assays: Current practices and future directions. Cytokine. 66(2):143–155.
- Findlay L, Eastwood D, Ball C, Robinson C, Bird C, Wadhwa M, Thorpe S, Thorpe R, Stebbings R, Poole S. 2011. Comparison of novel methods for predicting the risk of pro-inflammatory clinical infusion reactions during monoclonal antibody therapy. J Immunol Meth. 371(1-2):134–142.
- Findlay L, Eastwood D, Stebbings R, Sharp G, Mistry Y, Ball C, Hood J, Thorpe R, Poole S. 2010. Improved in vitro methods to predict the in vivo toxicity in man of therapeutic monoclonal antibodies including TGN1412. J Immunol Methods. 352(1-2):1–12.
- Genentech. 2018. Herceptin (trastuzumab) [package insert]. South San Francisco, (CA): Genentech, Inc.
- Genentech. 2019. Rituxan (rituximab) [package insert]. South San Francisco, (CA): Genentech, Inc.
- Grimaldi C, Finco D, Fort MM, Gliddon D, Harper K, Helms WS, Mitchell JA, O'Lone R, Parish ST, Piche M-S, et al. 2016. Cytokine release: A workshop proceedings on the state-of-the-science, current challenges and future directions. Cytokine. 85:101–108.
- Hansel T, Kropshofer H, Singer T, Mitchell J, George A. 2010. The safety and side-effects of monoclonal antibodies. Nat Rev Drug Discov. 9(4):325–338.
- Hu Y, Turner M, Shields J, Gale M, Hutto E, Roberts B, Siders W, Kaplan J. 2009. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology. 128(2):260–270.
- Hussain K, Hargreaves C, Roghanian A, Oldham R, Chan H, Mockridge C, Chowdhury F, Frendeus B, Harper K, Strefford J, et al. 2015. Upregulation of FcγRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood. 125(1):102–110.
- Hussain K, Hargreaves C, Rowley T, Sopp J, Latham K, Bhatta P, Sherington J, Cutler R, Humphreys D, Giennie M, et al. 2019. Impact of human FcγR gene polymorphisms on IgG-triggered cytokine release: Critical importance of cell assay format. Front Immunol. 10:390.
- Kirton C, Gliddon D, Bannish G, Bembridge G, Coney L. 2011. In vitro cytokine release assays: Reducing the risk of adverse events in man. Bioanalysis. 3(23):2657–2663.
- Kramer P, Kramer S, Guan G. 2004. 17β-Extradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 50(6):1967–1975.
- Luhder F, Huang Y, Dennehy K, Guntermann C, Muller I, Winkler E, Kerkau T, Ikemizu S, Davis S, Hanke T, et al. 2003. Topological requirements and signaling properties of T-cell-activating, anti-CD28 antibody superagonists. J Exp Med. 197(8):955–966.
- Pallardy M, Hunig T. 2010. Primate testing of TGN1412: Right target, wrong cell. Br J Pharmacol. 161(3):509–511.
- Paul F, Cartron G. 2019. Infusion-related reactions to rituximab: Frequency, mechanisms and predictors. Expert Rev Clin Immunol. 15(4):383–389.
- Römee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B, et al. 2013. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 121(18):3599–3608.
- Römer P, Berr S, Avota E, Na S, Battaglia M, ten Berge I, Einsele H, Hunig T. 2011. Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood. 118(26):6772–6782.
- Sanofi KK. 2015. MabCampath (alemtuzumab) [package insert]. Sanofi K.K., Tokyo, Japan.
- Siders W, Shields J, Garron C, Hu Y, Boutin P, Shankara S, Weber W, Roberts B, Kaplan J. 2010. Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk Lymphoma. 51(7):1293–1304.
- Stebbings R, Eastwood D, Poole S, Thorpe R. 2013. After TGN1412: Recent developments in cytokine release assays. J Immunotoxicol. 10(1):75–82.
- Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, et al. 2007. "Cytokine storm" in the phase I trial of monoclonal antibody TGN1412: Better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 179(5):3325–3331.
- Suntharalingam G, Perry M, Ward S, Brett S, Castello-Cortes A, Brunner M, Panoskaltsis N. 2006. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 355(10):1018–1028.
- Vessillier S, Eastwood D, Fox B, Sathish J, Sethu S, Dougall T, Thorpe S, Thorpe R, Stebbings R. 2015. Cytokine release assays for the prediction of therapeutic mAb safety in first-in man trials-Whole blood cytokine release assays are poorly predictive for TGN1412 cytokine storm. J Immunol Methods. 424:43–52.
- Wing M, Moreau T, Greenwood J, Smith R, Hale G, Isaacs J, Waldmann H, Lachmann P, Compston A. 1996. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. 98(12):2819–2826.
- Wolf B, Morgan H, Krieg J, Gani Z, Milicov A, Warncke M, Brennan F, Jones S, Sims J, Kiessling A. 2012. A whole blood in vitro cytokine release assay with aqueous monoclonal antibody presentation for the prediction of therapeutic protein induced cytokine release syndrome in humans. Cytokine. 60(3):828–837.
- Yeap W, Wong K, Shimasaki N, Teo E, Quek J, Yong H, Diong C, Bertoletti A, Linn Y, Wong S. 2016. CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci Rep. 6:34310.