
Abstract
Cisplatin (DDP) is a potent chemotherapeutic; however, it can also cause acute kidney injury (AKI). Because of the complexity of the toxicity it induces, few effective methods exist for ameliorating any form of DDP-induced AKI. Recent research has suggested that the complement system is a potential molecular target for such amelioration. In the study here, in vivo (male ICR mice) and in vitro (HK-2 cells) models of DDP-induced AKI were established to investigate the potential therapeutic effects of Vitamin D (VD) against this form of AKI. Endpoints assessed in vivo/in vitro included overall renal function, degree of renal damage, and complement receptor C5aR expression using histology, immunohistochemistry, immunofluorescence, RT-PCR, and Western blots. The data indicated that the use of VD treatment could reduce renal pathological damage along with expression of TNFα, IL-1β, IL-18, and C5aR; however, an over-expression of C5aR weakened the protective effects of VD/VD receptor (VDR) against oxidative damage and inflammatory cell infiltration. Using a luciferase reporter gene assay and ChIP analysis, it was demonstrated that C5aR was transcriptionally inhibited by VDR. In conclusion, VD/VDR could delay DDP-induced AKI by inhibiting the expression of C5aR through transcriptional regulation and reducing the production of downstream pro-inflammatory cytokines. The present study revealed the regulatory mechanism of VD/VDR in acute renal inflammation and provides new insights into its therapeutic function in DDP-induced AKI.
Introduction
Acute kidney injury (AKI) is characterized by a rapid loss of renal function and is a frequent complication in hospitalized patients. The main clinical causes of AKI include renal ischemia–re-perfusion, nephrotoxins, and obstruction (Bucaloiu et al. Citation2012). AKI is classified as pre-renal, post-renal, or parenchymal (also known as intrinsic), depending on the primary site of injury. In general, reversible pre- and post-renal AKI has a relatively small impact on patient survival, whereas irreversible late-stage AKI is associated with an increased risk of morbidity or mortality and represents a global health concern (Kaufman et al. Citation1991; Bucaloiu et al. Citation2012). Even the milder form of AKI has adverse consequences and may develop into renal fibrosis, which is a common irreversible manifestation of late-stage chronic kidney disease (Chawla et al. Citation2011; Chawla and Kimmel Citation2012). Experimental evidence indicates that both the initiation and maintenance of AKI result from immune-mediated damage; the inflammatory response plays an important role in AKI pathogenesis and determining its severity (Nozaki et al. Citation2011; Dellepiane et al. Citation2020). Owing to the complex pathogenesis of AKI, strategies for its prevention or treatment are limited (Kher and Kher Citation2020). Therefore, novel treatment strategies that ameliorate the detrimental effects of AKI are urgently needed.
Cisplatin (DDP), an alkylating agent that most commonly causes acute renal tubular necrosis, is one of the most commonly used and effective chemotherapeutic drugs for various types of solid tumors (Dasari and Tchounwou Citation2014). The nephrotoxicity associated with DDP severely limits its clinical application (Li et al. Citation2019). DDP-induced renal injury primarily occurs in the proximal tubules of the internal cortex and outer end of the medulla (corresponding to the main intake sites of DDP). Renal tubular cells are lost owing to necrosis and apoptosis, followed by inflammatory cell infiltration and fibroproliferative changes (Cummings and Schnellmann Citation2002). Current research reveals that DDP-induced parenchymal AKI is a complex process with mechanisms that include oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction, DNA damage, apoptosis, and inflammatory responses (Peres and da Cunha Citation2013). Unfortunately, the precise molecular/cellular mechanisms that underlay the various DDP- induced nephrotoxicities remain unclear. As such, no effective treatment or preventative agent has been developed to counter these toxicities (Cao et al. Citation2018; Ni et al. Citation2019). Some studies have suggested that inflammatory mechanisms are closely associated with the pathogenesis of DDP-induced nephrotoxicity, including the recruitment of inflammatory cells, such as leukocytes and macrophages (Liu et al. Citation2006; Faubel et al. Citation2007; Thurman et al. Citation2007). This suggests that the inhibition of the inflammatory response may be key to inhibiting further development of DDP-induced renal injury.
The complement system is an ancient host defense system that participates in innate and adaptive immunity. It not only protects the internal environment of the organism by protecting against pathogenic agents but produces various byproducts during its activation that mediate inflammation and promote local acute inflammatory reactions, thereby affecting adaptive immune responses (Ramesh and Reeves Citation2002). The complement cascade is acutely activated during AKI (Farrar et al. Citation2006). Complement activation injury is achieved through the formation of membrane attack complexes, recruitment of immune cells, and activation of complement receptors on various cells (Castellano et al. Citation2019). These findings suggest that the activation of the complement system may contribute to the development of DDP-induced nephrotoxicity.
C5a is an active small-molecule fragment produced during complement system activation. As the most powerful anaphylatoxin, C5a can induce several biological responses, particularly inflammatory reactions. Through specific binding to the C5a anaphylatoxin chemokine receptor (C5aR/C5aR1/CD88) - a G protein-coupled receptor that is expressed on various cells including renal tubular epithelial cells - C5a triggers the release of inflammatory cytokines and other substances that induce local inflammation (Bujko et al. Citation2017). Several studies suggest excessive activation of the complement system leads to the production of proinflammatory C5a, which exacerbates the inflammatory response (Hollmann et al. Citation2008; Girke et al. Citation2014). C5a/C5aR interactions play an important role in the regulation and coordination of many inflammatory and immune processes and are involved in the pathogenesis of a wide range of inflammatory and immune diseases (Pan et al. Citation2009; Peng et al. Citation2019; Li et al. Citation2021). Therefore, C5a/C5aR may be an important pathogenic pathway in DDP-induced renal injury and a novel therapeutic target against DDP-induced nephrotoxicity.
The Vitamin D receptor (VDR) is a protein belonging to the nuclear hormone receptor superfamily. Many biological functions of Vitamin D (VD) are realized through the combination of its active metabolite 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) or other agonists, such as paricalcitol, with VDR to mediate the transcription of target genes (Blanco et al. Citation1995). Moreover, 1,25(OH)2D3 is a pleiotropic hormone with potent immunomodulatory activity in both innate and adaptive immunity (Mora et al. Citation2008; Hart et al. Citation2011). VDR is highly expressed in various organs, including the gastrointestinal tract, skin, lung, and kidney, and has various functions (Yang et al. Citation2018). The activation of VDR by 1,25(OH)2D3 or its active analogs can protect the kidney by inhibiting the renin-angiotensin system (RAS) and inflammatory reactions and can alleviate proteinuria and progression of renal fibrosis in chronic or end-stage renal disease (Zhang et al. Citation2010; Zhu et al. Citation2019; Li et al. Citation2022). However, little is known about the role of the VDR in AKI.
The study reported here was undertaken to investigate the relationship between VD/VDR and C5aR and to determine any potential utility for using VD to attenuate DDP-induced AKI. Based on molecular activities described in previous studies, it was hypothesized here that VD/VDR would alleviate DDP-induced AKI, in part, by acting upon the complement system, thereby inhibiting the activation of C5aR by C5a.
Materials and methods
Animals and treatment
ICR mice (male, 8-wk-old) were purchased from Liaoning Changsheng Biotechnology (Benxi, China). Upon arrival, all mice were housed under specific pathogen-free conditions in the Animal Facility of Shengjing Hospital, China Medical University. The facilities were maintained at 22 °C with a 50% relative humidity and a 12-hr light:dark cycle. All animals had ad libitum access to standard rodent chow and filtered tap water. The diet was confirmed to contain 1000 IU Vitamin D/kg, a level which assured the mice were all not deficient in the vitamin at the time of the study.
After acclimatization for 1 wk, mice were randomly allocated into four groups (n = 5/group): a vehicle, VD (2-wk daily free drinking pretreatment with a solution containing 10 μl CCE [cholecalciterol cholesterol emulsion]/100 ml water), DDP (single intraperitoneal [IP] injection of 20 mg cisplatin [in saline]/kg), and a DDP/VD [pre-treatment] group. Vehicle and DDP-only hosts received 2 wk daily free drinking with water alone. Vehicle and VD-only hosts were IP-injected with saline to mimic the other two regimens. All injections took place within 24 hr following the end of the 2-wk pretreatment.
DDP was purchased from MedChemExpress (MCEHY-17394; Princeton, NJ). CCE, a concentrated Vitamin D3 lotion used in the clinical treatment of VD deficiency was obtained from Sine-Jin Zhu (Shanghai, China). The dose of DDP employed was based upon the earlier study by Nozaki et al. (Citation2011). CCE, also known as vitamin D3 (VD), is metabolized by the liver and kidneys in the body to form 1,25(OH)2D3. The dose of CCE used was based upon body surface area and after conversions that permitted the dose ratio in the mice to be equivalent to that used in humans (Liu et al. Citation2018). Body weight was recorded for 10 d, i.e. starting 1 wk prior to the IP injection to 72 hr after the injection. At 72 hr post-DDP/vehicle treatment, all mice were euthanized by cervical dislocation; at necropsy, blood was recovered by cardiac puncture and immediately processed to generate serum while the kidneys were removed, weighed and then processed for use in the experiments as outlined below.
All protocols involving the use of the mice were approved by the Ethics Committee of China Medical University (Shenyang, China) [2020PS389K(X1)].
Blood urea nitrogen (BUN), serum creatinine (sCr), and malondialdehyde (MDA) levels
After the blood was naturally coagulated and centrifuged (3000 rpm, 10 min), the serum was collected. Aliquots were then evaluated using commercially-available kits for BUN (F9480, Fankew, Shanghai, China) or sCr (BC1535, Solarbio, Beijing, China), according to manufacturer instructions.
Renal MDA levels were evaluated from one of the two organs removed from each host. A representative sample from one kidney (50 mg/host) was homogenized in 500 µl saline (so all samples are in a fixed amount of solution), centrifuged, and the resultant supernatant collected. MDA levels in each supernatant were then evaluated (in triplicate) using a WLA048b MDA assay kit (Wanleibio, Shenyang, China), following manufacturer protocols. In brief, 100 μl kit-provided MDA standard (sample of MDA at fixed total amount), ethanol, or test sample were added to dedicated tubes. Each tube then received 100 μl kit reagent 1, 3 ml kit reagent 2, and 1 ml kit reagent 3. The tubes were then placed in a 95 °C waterbath for 40 min; after cooling to room temperature, each tube was centrifuged, and the absorbance value (OD532 nm) of each supernatant was measured. From the OD value and the known standard concentration for the MDA, the relative concentration of MDA in each original homogenate was calculated (as per kit instructions). Based on the known volume of homogenate, the total MDA present could then be calculated, and outcomes normalized to total MDA/mg kidney. All samples were evaluated in triplicate for each individual kidney.
For studies in which treated cell cultures were evaluated for MDA, all the protocols above were used again. In each experiment, a total of 107 cells were lysed in a fixed volume of 500 μl saline and then, after centrifugation to pellet debris, analyzed as above. Outcomes could then be normalized to total MDA/107 cells.
Histopathology, immunohistochemistry (IHC), and immunofluorescence (IF) analysis of renal tissues
The second kidney from each host was embedded in paraffin and sections (4-μm) were then generated. These, in turn, were subjected to staining with either hematoxylin and eosin (H&E), Masson’s trichome (G1340), or periodic acid-Schiff (PAS, G1285) stains (all from Solarbio). Other sections were used for immunohistochemical analyses (IHC) for IL-1β, MPO, and C5aR, or for immunofluorescence (IF) staining (against F4/80) (WLH2545, Wanlei Biotechnology).
For IHC, kidney sections were de-waxed, placed in 0.01 mM citrate buffer, and boiled for 10 min in a microwave for antigen retrieval. An UltraSensitiveTM SP (Mouse/Rabbit) IHC Kit (KIT-9720, MXB, Fuzhou, China) was then used to stain the sections, according to manufacturer protocols. After rinsing the slides with 0.25% Triton-X 100 in PBS (phosphate-buffered saline, pH 7.4), kit-provided blocking endogenous peroxidase reagent was placed on each slide and the samples incubated at room temperature for 10 min. After a second PBS wash, kit-provided nonspecific blocking agent solution was placed on each slide and the samples were incubated again at room temperature for 10 min. After another gentle PBS wash, the slides were coated with a solution of PBS containing anti-C5aR (1:500, manufacturer-recommended dilution), anti-IL-1β (1:500), or anti-MPO (1:150), and then incubated at 4 °C overnight. After a gentle PBS wash, each slide was coated with biotin-labeled sheep anti-mouse/-rabbit IgG polymer and incubated at room temperature for 20 min. After PBS rinsing, kit streptomycin-avidin peroxidase solution was placed on each slide and the samples were incubated at room temperature for 10 min. After a final gentle PBS rinse, a solution of diaminobenzidine (DAB, Zhongshanjinqiao, Beijing, China) was placed on each slide, and color development was allowed to proceed at room temperature. After 1 min, each slide was gently rinsed with water to stop the reaction, then counterstained with hematoxylin, dehydrated, sealed (neutral gum), and stored for later analyses. Micrographs of the stained sections were captured using a Nikon optical microscope (Tokyo, Japan); intensities of C5aR, IL-1β, or MPO in each section were analyzed via grayscale analysis (ImageJ software; NIH, Bethesda, MD).
For IF, after blocking/washing steps as above, slides were coated with a solution of PBS containing rabbit anti-mouse F4/80 (1:500 dilution) antibody and then incubated overnight at 4 °C. After a gentle rinse with PBS, the slides were coated with a PBS solution bearing fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (Proteintech, Wuhan, China) and incubated in the dark for 2 hr. After a final rinse with PBS, each slide was coated with a solution of PBS spike with DAPI (4′,6-diamidino-2- phenylindole, Beyotime, Shanghai, China) counterstain, incubated for 5 min at room temperature, rinsed, and then sealed with glycerol gelatin. The total number of fluorescing cells (i.e. F4/80+) was then evaluated using a fluorescence microscope (Nikon) and analyzed using ImageJ software.
For all analyses, certified histologists blinded to sample identities examined the stained materials. With the PAS-stained samples, 20 renal tubulointerstitial fields in the cortical area were randomly selected within each slice (in a clockwise direction) and examined using a light microscope (40× magnification) (Nikon). Renal tubule injury was scored as: no obvious pathological changes (0 points), small focal injury (0.5 points), injury < 10% (1 point), 10–25% injury (2 points), 25–75% injury (3 points), and injury ≥ 75% (4 points). The average value for each slice was calculated.
Cell culture and treatment
Human proximal tubular epithelial cells (HK-2, from Benxi Experimental Base of Shengjing Hospital, China Medical University) were cultured in 10-cm culture dishes at 37 °C in 5% CO2 incubators. All cells were maintained in F12/DMEM supplemented with 10% fetal bovine serum (all materials from Procell, Wuhan, China) and passaged at ≈ 90% confluency. For use in the assays here, cells at 90% confluency were harvested using 0.25% trypsin (Servicebio, Wuhan, China), isolated cells were then counted, and cells were seeded into 6-well plates (1.5 × 106/well, 2 ml/well). After allowing 12 hr to adhere, cells were randomly designated as control, VD, DDP, or a DDP/VD groups.
Cells that were to be pre-treated with VD received 40 µl of a 10 µM paricalcitol (MCEHY-50919, MedChemExpress) solution to achieve a final level of 200 nM paricalcitol/well. Control and DDP-only wells received paricalcitol vehicles only. Paricalcitol is a biologically active VD analog that has the same effect as 1,25(OH)2D3 and does not require metabolism. It can directly bind to receptors in cells and exert its effects. After pre-treatment for 12 hr, the cells (as well as all other parallel cultures) were gently rinsed with the clean medium. Thereafter, dedicated wells that were to be treated with DDP received a dose of the DDP (in medium) that resulted in a final well concentration of 20 μg DDP/ml; the control wells and VD-only wells received DDP vehicle only. All cultures were then incubated for 24 hr at 37 °C.
MTT assay
To determine the effects of the various treatment regimens on cell viability, the culture medium from cells in dedicated wells (triplicate wells/regimen) was removed, fresh culture medium was added (100 µl) followed by 20 µl of MTT stock solution (WLA021a, Wanlei Biotechnology), and incubation of the plate at 37 °C for 4 hr. Dimethyl sulfoxide (150 μl; Wanlei Biotechnology) was then added to each well to help dissolve formazan crystals that had formed within viable cells, and the plate was incubated for 10 min at room temperature. Thereafter, the absorbance value (OD570 nm) in each well was measured in a Tecan plate reader (Shanghai, China). From these values, relative cell viability was calculated by comparison of OD values to those of the control cultures.
Reverse-transcription polymerase chain reaction (RT-PCR) analysis
Total RNA from mouse kidney (50 mg/mouse) or the treated cell cultures (2.5 x 106 cells total/treatment group) was extracted using a Varzyme total RNA extraction reagent (Vazyme, Nanjing, China). After confirming the purity and concentration of each collected total RNA sample using spectrophotometric measurements at 280 and 260 nm, HiScript III RT SuperMix for qPCR (Vazyme) was used to reverse transcribe 2 µg of each RNA sample into cDNA. In turn, these materials were then processed using ChamQ Universal SYBR qPCR Master Mix (Vazyme) in the performance of real-time polymerase chain reactions in an ABI PRISM 7500 system (Applied Biosystems, Foster City, CA). Primers used in these experiments are listed in . In all cases, standardized relative mRNA expression levels were determined based on GAPDH expression using the comparative 2−ΔΔCt method.
Table 1. Sequences of the primers used in the RT-PCR.
Western blot analysis
Mouse kidney (50 mg/mouse) or pellets of cell cultures (2.5 x 106 cells total/treatment group) were placed/re-suspended in RIPA lysis buffer (Invent Biotechnologies, Shanghai, China), and proteins were extracted via ultrasonication of the samples while on ice. After determining their total concentration in the post-centrifugation supernatant, extracted proteins were separated via PAGE and the resolved materials then were electrotransferred to polyvinylidene fluoride membranes (Cytiva, Marlborough, MA). Dedicated membranes were generated for each protein of interest; this helped to avoid the issue of repeated inter-analysis stripping which would have compromised protein values.
Each membrane was first blocked in a solution of TBS-T (Tris-buffered saline [pH 7.4] containing 0.1% Triton X-100) supplemented with 5% milk powder. Thereafter, sets of each dedicated membrane were placed in a solution of TBS-T containing primary rabbit/mouse antibody against human or mouse (as appropriate) TNFα, IL-1β, IL-18, VDR, or C5aR. Antibody against GAPDH was included in each incubation to permit monitoring of protein loading. All antibodies used in the experiments are noted in . After incubation overnight at 4 °C, the membranes were gently rinsed with TBS-T and then placed in a solution of TBS-T containing horseradish peroxidase (HRP)-conjugated species- specific secondary antibody. After incubation at room temperature for 2 hr, the membranes were gently rinsed with TBS-T and then treated with Chemiluminescence HRP Substrate Solution (Tanon, Shanghai, China). After a final gentle rinse with TBS-T, signals on each membrane were detected using a Healthcare imaging system (Cytiva). Quantitative analysis of the density of each protein band was then performed using ImageJ software (NIH, Bethesda, MD), and all data were normalized to GAPDH levels prior to analyses. All outcomes were then reported in terms of relative expression in treatment groups vs. their corresponding kidney/cell culture controls.
Table 2. Antibody information.
Construction of C5aR-overexpression plasmid and transfection
The CDS sequence of human C5aR with a total length of 1053 bp was searched for in the National Center for Biotechnology Information database (NCBI, http://www.ncbi.nlm.nih.gov); this information was then imported to SnapGene software (Insightful Science, San Diego, CA) for primer design and amplification of the CDS region. The up- and downstream primers used for amplification were 5′-ATGGACTCCTTCAATTATACCACC-3′ and 5′-AGGTAACACCATCCCACGAAAAGT-3′, respectively. Two restriction endonuclease sites, i.e. KpnI and NotI, were selected on a pcDNA3.1 plasmid (Invitrogen, Carlsbad, CA), and the recognition sequences of these two enzymes were added to the 5′ end of the above primers. The PCR product was then inserted into the pcDNA3.1 vector to construct a C5aR-over-expression plasmid (pcDNA3.1- C5aR).
For this, first, cells were divided into an empty vector and C5aR+/+ groups and then into a control, VD, DDP, and DDP + VD groups (for a total of 8 groups). The plasmids were then transfected into HK-2 cells that were at ≈ 80% confluency using jetPRIME® Transfection Reagent (Polyplus, Illkirch-Graffenstaden, France), following manufacturer protocols. After 24 hr of transfection and drug treatment, proteins were extracted for Western blot analyses as above.
Luciferase reporter gene assay
To explore the potential regulatory effects of VDR on C5aR, a luciferase reporter gene assay was performed. Specifically, the coding region of human chromosome C5aR was searched for in the NCBI database to identify the C5aR promoter sequence. The promoter range was set from +2000 bp to −100 bp of the coding starting point (NC_000019.10:47305477-47307576). A JASPAR database (http://jaspar.genereg.net) was used to predict the VD reaction element (VDRE) site on the C5aR promoter; three putative sequences (+91 bp to +98 bp, −30 bp to −37 bp, and–481 bp to −488 bp) were obtained by setting the relative score threshold to 90%. Primers for the PCR products containing these VDRE sites were designed using SnapGene. The forward and reverse primer sequences used were: Primer 1 [F] 5′-CCTCCCGGGTTCAAGTAATTC- 3′, [R] 5′-CAAGAGTTCAAGACCAGCCTG-3′; Primer 2 [F] 5′-TCCTTGAGGTGGAGTCTTGC- 3′, [R] 5′-ATGCCTGTAGTCCCAGCTAC-3′; and, Primer 3 [F] 5′-TCACAGGTTCAGGGG- ATTAAG-3′, [R] 5′-ATGTCCAGGCTCCTTAGCTCG-3′. Recognition sequences of two endo- nuclease sites, i.e. KpnI and BglII, selected on the pGL3-Basic vector (Promega, Madison, WI) were added to the 5′ end of the up- and downstream primers, respectively. Ultimately, three pGL3-VDRE recombinant plasmids were constructed by inserting PCR products into the vector.
To perform these particular transfection experiments, HEK-293T (human embryonic kidney cells, [Benxi Experimental Base of Shengjing Hospital] commonly used in transfection experiments due to high transfection efficiency and ease of cultivation) was employed in place of the HK-2 cells. The HEK-293T cells were maintained in DMEM/high glucose medium supplemented with 10% fetal bovine serum and cultured in 6-cm culture dishes at 37 °C until needed. At the time of the experiment, aliquots of 3 x 104 cells were placed into 96-well plates for treatments. The HEK-293T cells were designated as a vector group and three VDRE groups (separate sets transfected with one of each of the three pGL3-VDRE plasmids), and each group was then sub-divided into a control group and a VD/paricalcitol group. After treatments as outlined above (12 hr pre-treatment, 24 hr of transfection), both Renilla and firefly luciferase activities were quantified using a Dual-Luciferase® Reporter Assay System (E1910, Promega), according to manufacturer instructions. The firefly/Renilla luciferase activity ratio was then calculated for each well. All data were normalized to the ratio of the control group, obtaining the relative luciferase activity of each group.
Chromatin Immunoprecipitation (ChIP) assay
To verify the binding of VDR to VDRE in the C5aR promoter region, a SimpleChIP® Plus Sonication ChIP Kit 4 °C and RT Reagent system (Cell Signaling Technology, Danvers, MA) was used to assay protein-bound DNA. Sets of HK-2 cells (each containing 107 cells/10-cm culture dish, 10 ml total medium/dish) were divided into a control and VD group, and treated with paricalcitol or medium only for 24 hr. After the medium was removed from each well, the cells were then fixed by the addition of 270 μl 37% formaldehyde and kept at room temperature (20–25 °C) for 10 min, after which 1 ml kit glycine solution was added to stop the cross-linking reaction. After the removal of the formaldehyde/glycine mixture, the cells underwent a gentle wash with PBS. The cells were then isolated from their wells, centrifuged to pellet, and re-suspended in kit ChIP sonication nuclear lysis buffer. Their nuclear chromatin was then “cut” by repeated ultrasonication to produce 200–1000 bp DNA fragments. Samples from each of the paricalcitol or medium groups were then sub-divided into Histone H3 (positive control), IgG (negative control), input (2% input sample control), and VDR groups, according to the subsequent analyses with a given antibody.
For this step, samples of each fragmented material were placed in an Eppendorf tube and then received a solution of kit ChIP buffer containing antibodies against Histone H3, IgG, or VDR. After incubation at 4 °C for 16 hr, immune precipitates were then mixed with a solution of kit-provided Protein G Magnetic Capture Beads for 2 hr at 4 °C. Purified immunoprecipitated products were then eluted with a kit of DNA elution buffer. Determination of the quality and quantification of the amount of DNA bound to the various proteins (with particular focus on the amount associated with VDR which would bind VDRE in the C5aR promoter region) in each yield was then performed using common PCR and RT-PCR analysis with the primers targeting three putative binding sites within the C5aR promoter (the three primer pairs are described above). Analysis of subsequently-obtained RT-PCR results was performed using the Fold-Enrichment (2-ΔΔCt [ChIP/NIS], ΔΔCt [ChIP/NIS] = ΔCt [normalized ChIP] - ΔCt [IgG]) method.
Statistical analysis
All data are expressed as means ± SD. A Student’s t-test was used for paired comparisons of the measured outcomes. Multiple comparisons were performed using a one-way analysis of variance (ANOVA) followed by a Bonferroni test. Statistical significance was set at p < 0.05. All data were analyzed using Prism software (v.8, GraphPad, San Diego, CA).
Results
VD Pre-treatment improves general renal status in DDP-treated AKI mice
Macroscopic analyses of the kidneys of the AKI mouse model showed a pale appearance in the DDP group samples; however, VD/CCE pre-treatment significantly mitigated the overall kidney appearance vs. that with the DDP alone (). Mice treated with DDP had a higher kidney (sum of both kidneys) to body weight ratio than DDP + VD mice (), suggesting that VD exerted a protective effect against the impact of DDP on renal morphology. Overall, body weights were lower in DDP-treated mice than in vehicle controls within 72 hr after injection. However, VD pre-treatment appeared to mitigate this induced weight loss (). The H&E staining of DDP-treated mice kidneys revealed a degraded structure and disorganized arrangement of renal tubules along with unduly expanded lumens, necrotic and exfoliated brush borders, and protein cast formation; however, the glomerulus showed no noticeable variation (). These findings were confirmed by Masson staining, which also showed there were no significant differences in renal fibrosis among the groups (). Notably, VD treatment only moderately ameliorated renal pathological injury in dual therapy mice compared to that in the DDP-alone mice. The PAS staining analysis indicated that the renal tubular epithelial cells in the DDP-only mice were visibly swollen and contained vacuolar degeneration, and an almost- disappeared brush edge; these cells also exhibited many purplish-red substances, suggesting that severe necrosis had taken place.
Figure 1. VD impact on renal status of DDP-induced AKI mouse model. (A) Renal macroscopic morphology. (B) Ratio of kidney weight to body weight. (C) Body weight changes. (D) H&E staining (40×). (E) Masson staining (40×). (F) Histologic analysis of PAS-stained sections (20×). in D-E, bars = 50 μm. (G) sCr assay. (H) BUN assay. (I) MDA levels. Values shown are means ± SD. *p < 0.05, **p < 0.01, ****p < 0.0001. DDP, cisplatin; H&E, hematoxylin and eosin; PAS, periodic acid-Schiff; sCr, serum creatinine; BUN, blood urea nitrogen; MDA, malondialdehyde.
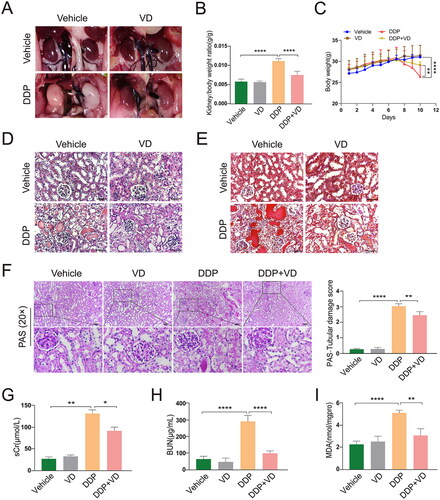
As with the outcomes in the histologic analyses above, it was also seen here that the pathological damage to renal tubules in mice pre-treated with VD was reduced compared with that in DDP-only mice (). Moreover, the sCr and BUN levels in the DDP/VD mice were remarkably lower than those in the DDP-treated mice (), indicating use of the VD could effectively “protect” renal function in DDP-treated mice. Similarly, when the extent of oxidative kidney injury in these hosts was measured by assessments of MDA levels (marker of lipid peroxidation), it was found that VD pre-treatment reduced MDA accumulation in the organ as compared with in the same tissue of DDP mice ().
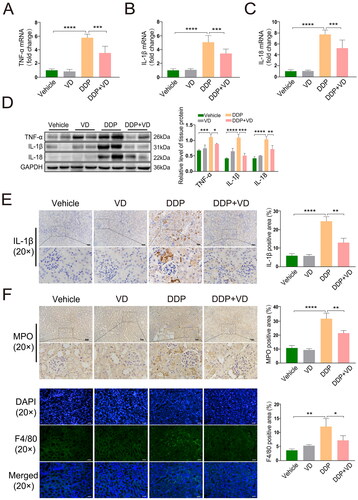
Kidney mRNA and protein expression of pro-inflammatory TNFα, IL-1β, and IL-18 in DDP-treated mice was significantly higher than in tissues from control and DDP/VD mice (). Changes in IL-1β levels - based on IHC analyses - were consistent with these results (). In addition, infiltration of inflammatory cells into the kidney tubules was decreased in the DDP/VD group compared with in the DDP-only hosts, based on IHC and IF analysis of MPO (neutrophil-specific enzyme) and F4/80 (macrophage marker) levels (). When taken together, these findings suggest that VD pre-treatment effectively could lead to an attenuation of subsequent DDP-induced kidney injury in vivo.
Figure 2. VD effect on pro-inflammatory cytokine levels in DDP-induced AKI mouse model. Relative mRNA expression of (A) TNFα, (B) IL-1β, and (C) IL-18 (RT-PCR). (D) Protein expression of TNFα, IL-1β, and IL-18 (Western blot); GAPDH protein levels used as loading control. (E) IL-1β expression (IHC). Bars = 50 μm. (F) Expression of MPO (IHC) and F4/80 (IF). Bars = 50 μm. Values shown are means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. DDP, cisplatin; AKI, acute kidney injury; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IHC, immunohistochemistry; IF, immunofluorescence; MPO, myeloperoxidase; F4/80, mouse EGF-like module-containing mucin-like hormone receptor-like 1.
VD Attenuated injury marker levels in DDP-treated HK-2 cells
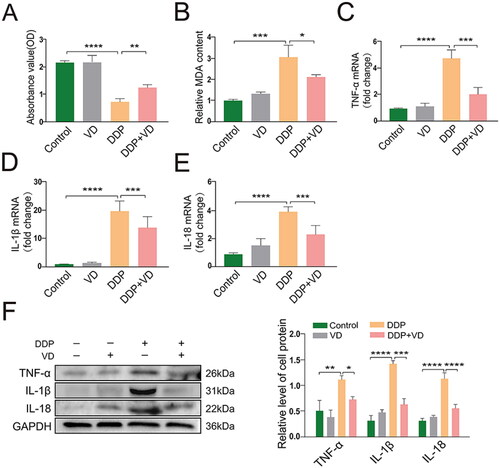
To investigate the potential effects of VD against DDP cytotoxicity in proximal renal tubular epithelial HK-2 cells, an MTT assay was performed. These analyses found that while cell proliferation was significantly inhibited in cells treated with DDP only, the effect was abrogated by VD pre-treatment (). Moreover, the use of VD reduced overall oxidative damage to these cells (). As mRNA and protein levels of TNFα, IL-1β, and IL-18 in the VD/DDP cultures were also significantly lower than in the DDP-only cultures (), this suggested that VD likely acted to protect the HK-2 cells against DDP-induced inflammatory injury by impacting on pro-inflammatory cytokine formation. This could also help to explain why there were similar patterns of reduced inflammatory damage in the kidneys of the DDP vs. VD/DDP-treated mice (see ).
Figure 3. VD impact on injury marker levels in DDP-treated HK-2 cells. (A) Cytotoxicity (MTT assay). (B) Lipid peroxidation (MDA content). relative mRNA expressions of pro-inflammatory (C) TNFα, (D) IL-1β, and (E) IL-18 (RT-PCR). (F) Protein levels of TNFα, IL-1β, and IL-18 (Western blot); GAPDH protein levels used as loading control. Values shown are means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. DDP, cisplatin; MDA, malondialdehyde; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Interaction of VD/VDR and C5aR ex vivo and in vitro
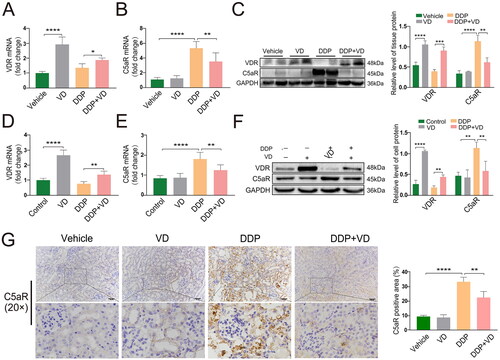
Both ex vivo and in vitro, it was found that mRNA and protein levels of C5aR were significantly lower in the DDP/VD groups than in DDP-only groups (). These outcomes corresponded with an increase in VDR levels in recovered tissues/cultures that had received any VD pre-treatment (). In addition, immunostaining showed that C5aR expression in the proximal renal tubules of DDP/VD mice was significantly lower than that in the DDP mice (), indicating a possible inhibitory effect of VD/VDR on C5aR.
Figure 4. VD/VDR impact on C5aR expression in AKI in vivo and in vitro. Relative mRNA expression of (A) VDR and (B) C5aR in mice (RT-PCR). (C) Protein expression of VDR and C5aR in mice (Western blot); GAPDH protein levels used as a loading control. Relative mRNA expression of (D) VDR and (E) C5aR in HK-2 cells (RT-PCR). (F) HK-2 cell protein expression of VDR and C5aR (Western blot); GAPDH protein levels used as loading control. (G) C5aR expression and distribution (IHC). Bars = 50 μm. Values shown are means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. VDR, Vitamin D receptor; C5aR, C5a anaphylatoxin chemotactic receptor; DDP, cisplatin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IHC, immunohistochemistry.
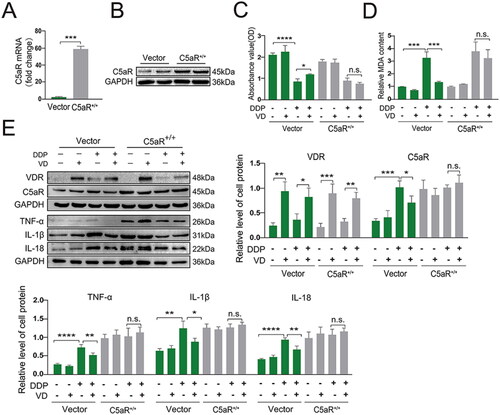
To further explore relationships between VD/VDR and C5aR, HK-2 cells were transfected with plasmid pcDNA3.1-C5aR (C5aR+/+) to over-express C5aR (verified by RT-PCR and Western blot; ). The protective effects from VD on lowered cell proliferation and against oxidative stress (i.e. inhibition of MDA accumulation) inducible by DDP were largely reversed in C5aR+/+ cells (). Interestingly, VD-induced decreases (relative to DDP-only samples) in the expression of downstream pro-inflammatory cytokines TNFα, IL-1β, and IL-18 were no longer noted in DDP/VD-treated C5aR+/+ cells (compared with in DDP-treated C5aR+/+ cells; ). These findings showed that VD reduced inflammatory injury in DDP-treated cells, in part, through changes in C5aR pathway activities.
Figure 5. Cytoprotective effect of VD/VDR in DDP-induced cells with C5aR over-expression. C5aR over-expression was validated using (A) RT-PCR and (B) Western blot. (C) MTT assay. (D) MDA levels. (E) Protein expression of VDR, C5aR, TNFα, IL-1β, and IL-18 (Western blot); GAPDH levels used as a loading control. Values shown are means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s., not significant. VDR, Vitamin D receptor; DDP, cisplatin; C5aR, C5a anaphylatoxin chemotactic receptor; MDA, malondialdehyde; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Vector, empty pcDNA3.1 vector; C5aR+/+, pcDNA3.1-C5aR plasmid.
VD/VDR targets VDRE sites in the C5aR promoter region
To investigate the potential regulatory mechanism by which VDR could impact C5aR, the C5aR sequence was first located in the GRCh38.p14 human genome; Chr19:47307477- 47322066 was found in the coding strand with a positive transcription direction (). As shown in , the C5aR promoter sequence was obtained by setting the range of the potential promoter region to 2000 bp upstream and 100 bp downstream of the gene starting point. The JASPAR database was searched for the VDRE where VDR might bind to the promoter. Three VDRE sites with the highest relative scores were screened using a 90% relative score threshold (). Subsequently, the three potential VDRE sequences were located and labeled on the promoter sequence of C5aR (). The relative luciferase activity of the three VD groups transfected with plasmids containing different VDRE sequences was lower than that of the control group, indicating that VDR binds to the VDREs on the C5aR promoter and acts as a transcription inhibitor ().
Figure 6. VD/VDR targets VDRE sites in C5aR promoter region. (A) Location and transcription direction of C5aR in the human genome. (B) Calculated C5aR promoter region. (C) Three VDRE sites with the highest relative score. (D) Location of VDRE in C5aR promoter region. (E) Relative luciferase activity by Luciferase reporter gene assay; schematic diagram. (F) Binding between VDR and C5aR promoter as confirmed by ChIP-PCR. Values shown are means ± SD. *p < 0.05, **p < 0.01, n.s., not significant. VDR, Vitamin D receptor; VDRE, Vitamin D reaction element; C5aR, C5a anaphylatoxin chemotactic receptor; ChIP, Chromatin Immunoprecipitation. Vector, empty pGL3-basic vector; VDRE-1/2/3, three predicted VDRE sites on C5aR promoter.
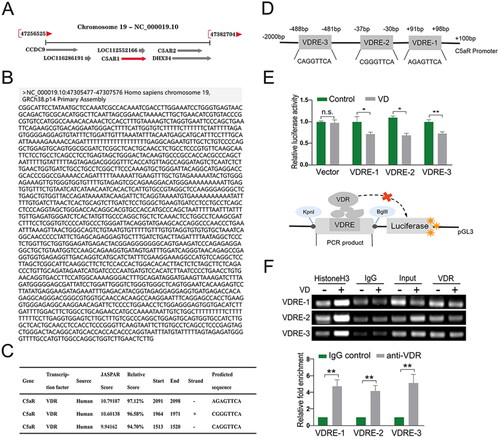
The combination of VDR and VDRE in the C5aR promoter region was also verified by ChIP (). The results show that compared with the negative control (IgG) group, the VDR group revealed a higher expression of common PCR products; that is, in the DNA bound by the VDR antibody, the PCR products containing the predicted VDRE sites were obtained with the three designed primers. In addition, RT-PCR showed that the relative fold-enrichment of the VDR antibody groups with the predicted VDREs was significantly higher than in the negative control group (). Based on these findings, it was concluded VD exerted its negative regulatory effect on C5aR by directly targeting VDRE in the C5aR promoter.
Discussion
Though a common anti-tumor agent, the efficacy of DDP is limited due to its propensity to cause renal injury. To explore the potential therapeutic effects of VD against DDP-induced kidney injury, as in a previous study (Nozaki et al. Citation2011), an in vivo model was established by intraperitoneal injection of mice with DDP (20 mg/kg). Owing to the large dose and short DDP administration period, VD (i.e. CCE) oral pre-treatment was provided daily for a period of 2 wk in advance of the DDP treatment to ensure that bodily concentrations of VD were sufficient at the time of DDP dosing.
At the end of the experiment, it was found that the kidney: body weight ratios were higher in DDP-only mice than in DDP/VD hosts. In addition, mice receiving DDP alone had significant changes in kidney histology, and VD pre-treatment effectively abrogated the development of these alterations. Based on those findings, as expected, the BUN and sCr levels in DDP-only mice were adversely impacted, but “restored” by pre-treatment with VD. Based on the blood parameters, it was concluded that renal microstructure destruction was reduced by pre-treatment with VD in vivo, effectively protecting excretion and reabsorption functions in hosts subsequently treated with cisplatin.
Based on the current findings and those of previous studies, it would seem that VD treatment could effectively promote the preservation of renal morphology and function and mitigates kidney injury in AKI models and potentially other hosts who receive cisplatin treatments for cancer. However, the mechanisms for this protective effect are not clear as yet.
One means by which renal damage is known to occur in AKI hosts is via amplification of inflammatory events in the kidneys. Among the various cytokines known to partake in these events, IL-18 (secreted by activated macrophages and natural killer T-cells) can promote the release of TNFα by local monocytes/macrophages and so stimulate the continued secretion of IL-18 through a positive feedback loop, thereby accelerating inflammatory events in situ. Similar to the findings of previous studies (Azak et al. Citation2013; Hamzawy et al. Citation2019; Hu et al. Citation2020), VD pre-treatment of the mice here effectively reduced DDP-inducible accumulation of tissue MDA and elevated expression of pro-inflammatory TNFα, IL-1β, and IL-18 caused by the DDP treatment. However, unlike in the current experiments, those previous studies typically used transgenic mice (i.e. VDR-/-) or other methods (i.e. ischemia-reperfusion injury) to establish the AKI model. Nevertheless, based on the comparable outcomes from this study and those previous studies, it would seem VD treatments could effectively promote the preservation of renal morphology and function and mitigate kidney injury associated with AKI.
Several immune cell types impart bidirectional effects on renal deterioration during AKI as well as upon any subsequent recovery/improvement (Dellepiane et al. Citation2020). During AKI, local inflammatory reactions typically undergo cascade amplification and often develop into chronic conditions. Some studies have suggested neutrophils infiltrate the kidney and damage renal tissue through the release of granule products and recruitment of natural killer T-cells (Li et al. Citation2007; Lever et al. Citation2019). As noted above, resident macrophages in the kidneys can release pro-inflammatory cytokines that can further promote renal tubular injury (Zhang et al. Citation2012; Arango Duque and Descoteaux Citation2014). It is possible that some of the observed protective effects from the VD pre-treatments are related to impacts on immune cells themselves or their abilities to be recruited to the site of inflammation. Previous studies revealed that VD/VDR affects both innate and adaptive immunity, in great part through the inhibition of the activation/proliferation of immune cells (Rigby et al. Citation1984; von Essen et al. Citation2010; Hart et al. Citation2011). In the current experiments, as indicated by the outcomes of the IHC analyses, DDP-alone caused significant neutrophil\macrophage infiltration into the renal tubules; however, levels of these cells were significantly reduced in the kidneys of the DDP/VD mice. Considering how VD normally functions in the kidney after entering the body, the results here suggest another novel function, i.e. that VD can reduce inflammatory cell infiltration caused by DDP (or potentially other toxicants) and inhibit their activation so as to reduce overall production/release of inflammatory mediators. Further studies are needed to more explicitly verify this novel viewpoint.
Similar outcomes with respect to cytokine formation/release, etc. were noted in the in vitro studies performed here. Specifically, levels of the three pro-inflammatory cytokines in the DDP/VD cultures were significantly lower than those in DDP-only cultures. This suggested to us that activated VDR can act on these specific types of epithelial cells to inhibit inflammatory injury that could be induced by cisplatin or other toxicants that act in a similar manner.
It may also be that other components of the host immune system are activated by DDP treatment to give rise to the above-noted pathologies in the mouse kidneys. Previous studies have revealed that the complement system (in conjunction with affected inflammatory cells) plays an important role in exacerbating - and potentially alleviating - inflammation in AKI (Holditch et al. Citation2019). In an activated complement cascade, C5a binds C5aR receptor expressed on neutrophils, monocytes, and renal tubular epithelial cells to induce the recruitment of neutrophils and macrophages and the production of inflammatory mediators (Arumugam et al. Citation2003; Zhang et al. Citation2017; Peng et al. Citation2019). From this, it could be assumed that blocking or reducing C5aR expression could help inhibit the development of renal inflammation in general and in the case of DDP treatment in particular. However, this approach has yielded contradictory outcomes. For example, one study found no effect of changes in C5aR expression/activation on macrophage activity (Huang et al. Citation2019), while other studies showed that treating macrophages with a large amount of C5a promoted the release of anti-inflammatory mediators (Bosmann et al. Citation2012; Seow et al. Citation2013). In the present study, C5aR expression in the kidneys was seen to be increased in the DDP-induced AKI hosts, but these levels were found to be ‘lowered’ in VD-pre-treated mice. From this, it is possible to conclude that use of VD leads to increased VDR expression/activity and that this, in turn, helps to down-regulate C5aR expression in renal cells, and mitigation of DDP-inducible toxicities.
To confirm whether any protective effect of VD seen here was in fact related to C5aR expression/activity, and because outcomes with the HK-2 cells were similar to those in the AKI mice, studies using over-expression of the C5aR were performed in vitro and the impact of VD again evaluated. In these experiments, as expected, VD pre-treatment could not impart the same protective effects previously seen against cytotoxicity and MDA accumulation in the DDA- treated cells. This led us to believe VD could ultimately alter the expression of downstream pro-inflammatory cytokines normally induced by DDP at least in part through down-regulation of C5aR expression/activity in the renal cells. To determine more clearly if VD could directly impact C5aR expression during DDP-induced toxicities, three potential VD reaction element (VDRE) sites in the C5aR promoter region were first identified, located, and verified as sites where VD/VDR became bound to VDRE on the C5aR promoter. ChIP and PCR assays here confirmed the three VDRE binding sites on the C5aR promoter did in fact recruit VDR, indicating that C5aR may be a direct transcription target of VDR in renal tubular epithelial cells.
Conclusions
The in vivo, ex vivo, and in vitro studies here demonstrated that the use of Vitamin D (VD) could partially protect kidneys from DDP-induced AKI, and that this effect occurred in part by the vitamin-inducing transcriptional inhibition of C5aR. A subsequent decrease in activity of the C5aR pathway then likely led to the mitigation of downstream inflammatory responses normally induced by DDP. Collectively, the current findings provide a deeper understanding of the potential anti-inflammatory effects of Vitamin D and provide new insights into clinical treatments for AKI. Although there was no difference in water consumption measured between the various groups in the animal experiments, due to limited time and experimental conditions, measures of VD levels in animal blood were not performed for this paper. However, the samples are archived and will undergo such analyses in an upcoming study. In addition, whether the observed VD effects are generic to all AKI, or active solely in the case of DDP induction, remains to be investigated.
Authors’ contributions
Conceptualization (ZW); methodology (CS); software (NL); validation (RK, SW); resources (SG); data curation (ZW, CS); writing original draft (ZW); review and editing (YL, JK); supervision (CZ); funding acquisition (JK).
Acknowledgments
The authors sincerely thank M.M. Pan He (Zunyi Medical University, Zunyi, Guizhou) for graciously providing the pcDNA3.1(+) vector
Disclosure statement
The authors declare no conflicts of interest. The authors alone are responsible for the content of this manuscript.
Additional information
Funding
References
- Arango Duque G, Descoteaux A. 2014. Macrophage cytokines: Involvement in immunity and infectious diseases. Front Immunol. 5:491. doi: 10.3389/fimmu.2014.00491.
- Arumugam T, Shiels I, Strachan A, Abbenante G, Fairlie D, Taylor S. 2003. A small molecule C5a receptor antagonist protects kidneys from ischemia/re-perfusion injury in rats. Kidney Int. 63(1):134–142. doi: 10.1046/j.1523-1755.2003.00737.x.
- Azak A, Huddam B, Haberal N, Koçak G, Ortabozkoyun L, Şenes M, Akdoğan MF, Denizli N, Duranay M. 2013. Effect of novel Vitamin D receptor activator paricalcitol on renal ischemia/re-perfusion injury in rats. Ann R Coll Surg Engl. 95(7):489–494. doi: 10.1308/003588413X13629960049117.
- Blanco J, Wang I, Tsai S, Tsai M, O'Malley B, Jurutka P, Haussler M, Ozato K. 1995. Transcription factor TFII-B and Vitamin D receptor cooperatively activate ligand-dependent transcription. Proc Natl Acad Sci U S A. 92(5):1535–1539. doi: 10.1073/pnas.92.5.1535.
- Bosmann M, Sarma J, Atefi G, Zetoune F, Ward P. 2012. Evidence for anti-inflammatory effects of C5a on innate IL-17A/IL-23 axis. FASEB J. 26(4):1640–1651. doi: 10.1096/fj.11-199216.
- Bucaloiu I, Kirchner H, Norfolk E, Hartle J, Perkins R. 2012. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 81(5):477–485. doi: 10.1038/ki.2011.405.
- Bujko K, Rzeszotek S, Hoehlig K, Yan J, Vater A, Ratajczak M. 2017. Signaling of the comple- ment cleavage product anaphylatoxin C5a through C5aR (CD88) contributes to pharmaco-logical hematopoietic stem cell mobilization. Stem Cell Rev Rep. 13(6):793–800. doi: 10.1007/s12015-017-9769-6.
- Cao X, Nie X, Xiong S, Cao L, Wu Z, Moore P, Bian J. 2018. Renal protective effect of polysulfide in DDP-induced nephrotoxicity. Redox Biol. 15:513–521. doi: 10.1016/j.redox.2018.01.012.
- Castellano G, Franzin R, Sallustio F, Stasi A, Banelli B, Romani M, De Palma G, Lucarelli G, Divella C, Battaglia M, et al. 2019. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/β-catenin signaling after ischemia/re-perfusion injury. Aging. 11(13):4382–4406. doi: 10.18632/aging.102059.
- Chawla L, Amdur R, Amodeo S, Kimmel P, Palant C. 2011. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 79(12):1361–1369. doi: 10.1038/ki.2011.42.
- Chawla L, Kimmel P. 2012. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 82(5):516–524. doi: 10.1038/ki.2012.208.
- Cummings B, Schnellmann R. 2002. DDP-induced renal cell apoptosis: Caspase 3-depend-ent and -independent pathways. J Pharmacol Exp Ther. 302(1):8–17. doi: 10.1124/jpet.302.1.8.
- Dasari S, Tchounwou P. 2014. DDP in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 740–364–378. doi: 10.1016/j.ejphar.2014.07.025.
- Dellepiane S, Leventhal J, Cravedi P. 2020. T-Cells and acute kidney injury: A two-way relationship. Front Immunol. 11:1546. doi: 10.3389/fimmu.2020.01546.
- Farrar CA, Zhou W, Lin T, Sacks S. 2006. Local extravascular pool of C3 is a determinant of post-ischemic acute renal failure. FASEB J. 20(2):217–226. doi: 10.1096/fj.05-4747com.
- Faubel S, Lewis E, Reznikov L, Ljubanovic D, Hoke T, Somerset H, Oh D, Lu L, Klein C, Dinarello C, et al. 2007. DDP-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in kidney. J Pharmacol Exp Ther. 322(1):8–15. doi: 10.1124/jpet.107.119792.
- Girke G, Kohl B, Busch C, John T, Godkin O, Ertel W, Schulze-Tanzil G. 2014. Tenocyte activation and regulation of complement factors in response to in vitro cell injury. Mol Immunol. 60(1):14–22. doi: 10.1016/j.molimm.2014.03.008.
- Hamzawy M, Gouda S, Rashed L, Morcos M, Shoukry H, Sharawy N. 2019. 22-Oxacalcitriol prevents acute kidney injury via inhibition of apoptosis and enhancement of autophagy. Clin Exp Nephrol. 23(1):43–55. doi: 10.1007/s10157-018-1614-y.
- Hart P, Gorman S, Finlay-Jones J. 2011. Modulation of the immune system by UV radiation: More than just the effects of Vitamin D? Nat Rev Immunol. 11(9):584–596. doi: 10.1038/nri3045.
- Holditch S, Brown C, Lombardi A, Nguyen K, Edelstein C. 2019. Recent advances in models, mechanisms, biomarkers, and interventions in DDP-induced acute kidney injury. IJMS. 20(12):3011. doi: 10.3390/ijms20123011.
- Hollmann T, Mueller-Ortiz S, Braun M, Wetsel R. 2008. Disruption of the C5a receptor gene increases resistance to acute Gram-negative bacteremia and endotoxic shock: Opposing roles of C3a and C5a. Mol Immunol. 45(7):1907–1915. doi: 10.1016/j.molimm.2007.10.037.
- Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, Wang J, Cao K, Zhang W. 2020. VDR activation attenuate DDP induced AKI by inhibiting ferroptosis. Cell Death Dis. 11(1):73. doi: 10.1038/s41419-020-2256-z.
- Huang S, You J, Wang K, Li Y, Zhang Y, Wei H, Liang X, Liu Y. 2019. N-Acetylcysteine attenuates DDP-induced acute kidney injury by inhibiting the C5a Receptor. Biomed Res Int. 2019:4805853. doi: 10.1155/2019/4805853.
- Kaufman J, Dhakal M, Patel B, Hamburger R. 1991. Community-acquired acute renal failure. Am J Kidney Dis. 17(2):191–198. doi: 10.1016/s0272-6386(12)81128-0.
- Kher A, Kher V. 2020. Prevention and therapy of AKI in Asia: A big challenge. Semin Nephrol. 40(5):477–488. doi: 10.1016/j.semnephrol.2020.08.004.
- Lever J, Hull T, Boddu R, Pepin M, Black L, Adedoyin O, Yang Z, Traylor A, Jiang Y, Li Z, et al. 2019. Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight. 4:e125503. doi: 10.1172/jci.insight.125503.
- Li A, Yi B, Han H, Yang S, Hu Z, Zheng L, Wang J, Liao Q, Zhang H. 2022. Vitamin D-VDR (Vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy. 18(4):877–890. doi: 10.1080/15548627.2021.1962681.
- Li L, Huang L, Sung S, Lobo P, Brown M, Gregg R, Engelhard V, Okusa M. 2007. NKT cell activation mediates neutrophil IFNγ production and renal ischemia-re-perfusion injury. J Immunol. 178(9):5899–5911. doi: 10.4049/jimmunol.178.9.5899.
- Li L, Wei T, Liu S, Wang C, Zhao M, Feng Y, Ma L, Lu Y, Fu P, Liu J. 2021. Complement C5 activation promotes Type 2 diabetic kidney disease via activating STAT3 pathway and disrupting the gut-kidney axis. J Cell Mol Med. 25(2):960–974. doi: 10.1111/jcmm.16157.
- Li X, Wang Q, Deng G, Liu Y, Wei B, Liu X, Bao W, Wang Q, Wu S. 2019. Porous Se:SiO2 nanospheres attenuate DDP-induced acute kidney injury via activation of Sirt1. Toxicol Appl Pharmacol. 380:114704. doi: 10.1016/j.taap.2019.114704.
- Liu M, Chien C, Burne-Taney M, Molls R, Racusen L, Colvin R, Rabb H. 2006. A pathophysiologic role for T-lymphocytes in murine acute DDP nephrotoxicity. J Am Soc Nephrol. 17(3):765–774. doi: 10.1681/ASN.2005010102.
- Liu N, Zhang Y, Su H, Wang J, Liu Z, Kong J. 2018. Effects of cholecalciferol cholesterol emulsion on renal fibrosis and aquaporin-2 and -4 in mice with unilateral ureteral obstruction. Biomed Pharmacother. 102:633–638. doi: 10.1016/j.biopha.2018.03.093.
- Mora J, Iwata M, von Andrian U. 2008. Vitamin effects on the immune system: Vitamins A and D take center stage. Nat Rev Immunol. 8(9):685–698. doi: 10.1038/nri2378.
- Ni J, Hou X, Wang X, Shi Y, Xu L, Zheng X, Liu N, Qiu A, Zhuang S. 2019. Correction: 3-deazaneplanocin A protects against DDP-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression. Cell Death Dis. 10(8):543. doi: 10.1038/s41419-019-1725-8.
- Nozaki Y, Nikolic-Paterson D, Yagita H, Akiba H, Holdsworth S, Kitching A. 2011. Tim-1 promotes DDP nephrotoxicity. Am J Physiol Renal Physiol. 301(5):F1098–1104. doi: 10.1152/ajprenal.00193.2011.
- Pan H, Shen Z, Mukhopadhyay P, Wang H, Pacher P, Qin X, Gao B. 2009. Anaphylatoxin C5a contributes to the pathogenesis of DDP-induced nephrotoxicity. Am J Physiol Renal Physiol. 296(3):F496–504. doi: 10.1152/ajprenal.90443.2008.
- Peng Q, Wu W, Wu K, Cao B, Qiang C, Li K, Sacks SH, Zhou W. 2019. The C5a/C5aR1 axis promotes progression of renal tubulointerstitial fibrosis in a mouse model of renal ischemia- re-perfusion injury. Kidney Int. 96(1):117–128. doi: 10.1016/j.kint.2019.01.039.
- Peres L, da Cunha A. 2013. Acute nephrotoxicity of DDP: Molecular mechanisms. J Bras Nefrol. 35(4):332–340. doi: 10.5935/0101-2800.20130052.
- Ramesh G, Reeves W. 2002. TNFα mediates chemokine and cytokine expression and renal injury in DDP nephrotoxicity. J Clin Invest. 110(6):835–842. doi: 10.1172/JCI200215606.
- Rigby W, Stacy T, Fanger M. 1984. Inhibition of T-lymphocyte mitogenesis by 1,25- dihydroxy- vitamin D3 (calcitriol). J Clin Invest. 74(4):1451–1455. doi: 10.1172/JCI111557.
- Seow V, Lim J, Iyer A, Suen J, Ariffin J, Hohenhaus D, Sweet M, Fairlie D. 2013. Inflammatory responses induced by lipopolysaccharide are amplified in primary human monocytes but suppressed in macrophages by complement protein C5a. J Immunol. 191(8):4308–4316. doi: 10.4049/jimmunol.1301355.
- Thurman J, Lenderink A, Royer P, Coleman K, Zhou J, Lambris J, Nemenoff R, Quigg R, Holers V. 2007. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ischemia/re-perfusion. J Immunol. 178(3):1819–1828. doi: 10.4049/jimmunol.178.3.1819.
- von Essen M, Kongsbak M, Schjerling P, Olgaard K, Odum N, Geisler C. 2010. Vitamin D controls T-cell antigen receptor signaling and activation of human T-cells. Nat Immunol. 11(4):344–349. doi: 10.1038/ni.1851.
- Yang S, Li A, Wang J, Liu J, Han Y, Zhang W, Li Y, Zhang H. 2018. Vitamin D receptor: A novel therapeutic target for kidney diseases. Curr Med Chem. 25(27):3256–3271. doi: 10.2174/0929867325666180214122352.
- Zhang K, Li G, He Q, Li Y, Tang M, Zheng Q, Xu G, Zhang K. 2017. C5a/C5aR pathway accelerates renal ischemia/re-perfusion injury by down-regulating PGRN expression. Int Immunopharmacol. 53:17–23. doi: 10.1016/j.intimp.2017.10.006.
- Zhang M, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, et al. 2012. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 122(12):4519–4532. doi: 10.1172/JCI60363.
- Zhang Y, Kong J, Deb D, Chang A, Li Y. 2010. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J Am Soc Nephrol. 21(6):966–973. doi: 10.1681/ASN.2009080872.
- Zhu X, Wu S, Guo H. 2019. Active Vitamin D and Vitamin D receptor help prevent high glucose induced oxidative stress of renal tubular cells via AKT/UCP2 signaling pathway. Biomed Res Int. 2019:9013904. doi: 10.1155/2019/9013904.