
Abstract
Objective: To investigate the clinicopathological features of children with Sturge–Weber syndrome and to analyze the correlation between the distribution area of leptomeningeal angiomatosis, the degree of cerebral cortical calcification, and the degree of cerebral atrophy associated with epileptic seizures. Methods: 10 children were diagnosed with SWS with FCD IIIc by histopathology and immunohistochemistry. Spearman correlation analysis was used to calculate the association of SWS with FCD IIIc and seizures in children. Results: The leptomeningeal angiomatosis area was markedly positively correlated with the degree of brain atrophy in 10 children with SWS (r = 0.783, p = 0.007). The distribution of leptomeningeal hemangiomatosis, the degree of cortical calcification, and brain atrophy were not significantly correlated with epilepsy. Conclusion: SWS may be accompanied by FCD IIIc. The more extensive the cerebral lobes of leptomeningeal angiomatosis in SWS, the more pronounced the brain atrophy.
Introduction
Sturge–Weber syndrome (SWS), also known as cerebral facial hemangioma syndrome or trigeminal neuroangiomatosis, is a congenital neurocutaneous syndrome. Without population-based evidence, estimates of frequency range from one in 20,000 to 50,000 live births [Citation1]. Typical pathological changes are facial port-wine birthmark (PWB), intracranial leptomeningeal angiomatosis distributed along the trigeminal nerve, and eye choroidal hemangioma [Citation2]. Epilepsy occurs in almost all infants with SWS, and focal cortical dysplasia (FCD) may be one of the findings in patients with SWS with refractory epilepsy [Citation3]. In this study, SWS with FCD IIIc was first diagnosed histopathologically and then the association between SWS with FCD IIIc and seizures was investigated.
Material and methods
Inclusion and exclusion criteria
Inclusion criteria were as follows: (1) The children with SWS had PWB, eye choroidal hemangioma, and intracranial leptomeningeal angiomatosis [Citation4]; (2) The children had intracranial leptomeningeal angiomatosis without PWB or eye choroidal hemangioma; (3) The children presented with refractory epilepsy, and the surgical method was focal epilepsy resection or functional cerebral hemispherectomy [Citation5]; (4) Histopathology confirmed FCD IIIc.
Exclusion criteria were as follows: (1) The children with SWS had PWB and eye choroidal hemangioma, without central nervous system vascular malformation; (2) The children had no seizure, and no seizure-related surgical treatments were performed; (3) The children had extensive and severe bilateral cerebral hemispherical leptomeningeal angiomatosis, and were treated conservatively with drugs to control seizures; (4) The cerebral cortex tissue of the children were not sent for histopathological diagnosis.
Clinical data
Children with secondary epilepsy and surgical treatment of SWS syndrome were collected from the electronic medical record system from January 2015 to June 2022. The score of seizure frequency (SSF) was calculated as follows: Referring to the previous scoring criteria for epileptic seizures [Citation6], the score was 0 for no seizures, 1 point for occasional or 1–2 seizures per year, 2 points for several seizures per year or less than once a month, 3 points for once a month or several times a month, 4 points for once a week, and 5 points for at least once a day. All clinical data evaluated in this study were approved by the hospital’s Human Rights Investigation Committee and informed consent was obtained from the children’s parents or legal guardians.
Magnetic resonance imaging
The patient was sedated with 0.5% chloral hydrate (0.5 mL/kg, maximum dosage < 10 mL) before scanning. A Siemens Skyra 3.0 T MR scanner with a head-neck phased array coil was used. The child was supine and a routine plain scan was performed after the localization scan was completed. Scanning sequence and parameters were as follows: Transverse T2WI, TSE sequence, TR 2300 ms, TE 108 ms, scan matrix 228 × 384, FOV 194 mm × 230 mm, slice thickness 6 mm; Transection T1WI was performed using TSE inversion recovery sequence, TR 1800 ms, TE 42 ms, TI 800 ms, scan matrix 182 × 320, FOV 187 mm × 230 mm, slice thickness 6 mm. Transection T2 flair, TSE flip recovery sequence, TR 9000 ms, TE 134 ms, TI 2600 ms, scan matrix 190 × 320, FOV 194 mm × 230 mm, slice thickness 6 mm; Sagittal T1WI was obtained using GRE sequence, TR 200 ms, TE 2.5 ms, scan matrix 256 × 320, FOV 250 mm × 250 mm, slice thickness 5 mm. Enhanced scan: GD-DTPA meglumine was injected through the cubital vein at 0.2 mmol/kg body weight at a flow rate of 2 mL/s. Transverse and sagittal T1WI and transverse T2 flair sequences were acquired. The parameters were as follows: Transverse T1WI, GRE sequence, TR 200 ms, TE 2.5 ms, scanning matrix 216 × 320, FOV 194 mm × 230 mm, slice thickness 6 mm were used. Sagittal T1WI was obtained using GRE sequence, TR 200 ms, TE 2.5 ms, scan matrix 248 × 320, FOV 242 mm × 250 mm, slice thickness 5 mm. The T2 flair sequence parameters in the transverse view are the same as in the plain scan.
Intracranial hemangioma area: MRV can exhibit multiple tortuous vascular shadows on the surface of the cerebral hemisphere. The area of leptomeningeal angiomatosis was calculated as no leptomeningeal angiomatosis in all brain lobes (0 point), leptomeningeal angiomatosis was limited to one brain lobe (1 point), limited to three brain lobes (2 points), limited to one hemisphere (3 points), and diffused whole brain (4 points).
Assessment of brain atrophy
Two pediatric radiologists with more than a decade of experience independently reviewed the images to observe the lesion location, shape, extent, signal intensity characteristics, enhancement characteristics, and degree of brain atrophy. In case of disagreement, a consensus was reached after discussion.
Global cortical atrophy (GCA) was assessed by preoperative magnetic resonance imaging (MRI) in all patients [Citation7]. The fraction of atrophy in the frontal, parietal, temporal, occipital, and insula lobes in both hemispheres was summarized in this research. A score of 0 to 3 was given to each assessed area. No cortical atrophy equals a GCA grade of 0. Mild atrophy (opening of sulci) was a GCA grade 1. Moderate atrophy (volume loss of gyri) was a GCA grade 2, and severe atrophy (knife blade atrophy) was a GCA grade 3. The GCA score ranges from 0 to 15, from no atrophy to maximum atrophy.
Neurosurgical procedures
Resection of epileptic foci: This was appropriate for a relatively limited range of lesions. According to the pathological changes, the part to determine incision, the lesional area was exposed layer by layer, and the electroencephalogram (EEG) monitoring was used to determine the scope of the epileptogenic zone, and then the atrophic cortex and leptomeningeal hemangioma were resected under the microscope, and the cortical EEG monitoring was used to confirm that there was no abnormal discharge, and the incision was closed layer by layer.
Functional hemispherectomy: for a wide range of lesions involving one hemisphere. The lesion was exposed layer by layer through a large frontotemporal parietal incision on the affected side. First, the central cortex was excised, the lateral fissures were dissected, the annular sulcus was exposed, and the temporal horn was entered. The amygdala, head and body of the hippocampus were removed, the umbrella of the hippocampus was severed and the corpus callosum was completely disconnected. The wound was filled with a gelatin sponge for compression and hemostasis, and the incision was closed layer by layer. Subdural drainage tubes were retained and removed after 3 to 5 days.
Pathology
After resection of the epileptic foci, brain tissue samples were obtained. The tissue was fixed in 4% neutral formaldehyde, and the paraffin sections were 4 μm thick and stained with HE. Focal cortical dysplasia (FCD) was diagnosed and classified according to the classification criteria of the International League Against Epilepsy (ILAE) in 2017 [Citation8].
Immunohistochemical staining
Applying the MaxVisionTM method, ready-to-utilize antibodies including CD31, CD34, SMA, glial fibrillary acidic protein (GFAP), S-100, neurofibrofilament protein (NF), neuron-specific nuclear protein (NeuN), OLig-2, and vimentin were purchased from Fuzhou Maixin Biotechnology Co., Ltd. (Fuzhou, China). The known positive sections were used as the positive control and PBS was used as the negative control instead of the primary antibody. The positive results were brownish-yellow without background staining.
Follow-up
Post-operative follow-up ranged from 2 to 55 months. Postoperative epileptic remission was evaluated by referring to Engel classification criteria [Citation9]: This was defined as Engel Class I (seizure-free or free from disabling seizures), Engel Class II (rare disabling seizures), Engel Class III (seizure reduction), and Engel Class IV (No worthwhile improvement).
Statistical methods
SPSS20.0 was used for data analysis and a normality test was performed on the data measurements. If the normal distribution was observed, mean ± standard deviation was used for description; if not, median (quad spacing) was used for description. Pearson correlation (normal distribution) or Spearman rank correlation analysis (non-normal distribution) was performed on the distribution of leptomeningeal angiomatosis, the degree of calcification in the cortex, the degree of brain atrophy, and the status of epilepsy. The test level α = 0.05, p < 0.05 was considered statistically significant. Pairwise correlation analysis was performed between the distribution area of leptomeningeal angiomatosis, the degree of calcification in the cortex, and the degree of brain atrophy. Pearson correlation (normal distribution) or Spearman rank correlation analysis (non-normal distribution) were applied. The test level α was adjusted to 0.025, p < 0.025 is a statistically significant difference.
Results
Clinical data
A total of 10 children had epilepsy secondary to SWS, including three males and seven females. The age ranged from 8 months to 48 months, with a mean age of 23.1 ± 14.2 months. The age of the first onset of epilepsy ranged from 1 month to 17 months, with an average of 6.5 ± 5.3 months. The onset of secondary seizures was clinically presented, including nine focal seizures and one generalized seizure. There were nine infants accompanied by facial PWB (), with four cases on the left and five cases on the right, with a maximum diameter of 7–15 cm, involving the trigeminal nerve distribution region and not crossing the midline. One patient did not have facial PWB. The score of seizure frequency was 4.4 ± 1.3, which was about once a week. All of the children had moderate atrophy, with reduced cerebral gyrus volume. The cortical calcification score was 1.9 ± 0.7, indicating that there were about three cortical and subcortical calcifications. The leptomeningeal angiomatosis score was 2.9 ± 1.0, and about all the children had leptomeningeal angiomatosis in one hemisphere. The specific conditions for all children are shown in .
Figure 1. (A) The infant’s left forehead and upper eyelid were wine colored, which was light red and smooth. (B) The sulci and fissure of the bilateral cerebral hemispheres were widened and deepened, and the gyri were narrowed. The left cerebral hemisphere was more evidently atrophied than the right, and bilateral lateral ventricles and third ventricles were widened. (C) Diffuse gyri-like enhancement was observed on the brain surface of the left cerebral hemisphere, and linear enhancement was observed on the brain surface of the right parietal lobe.
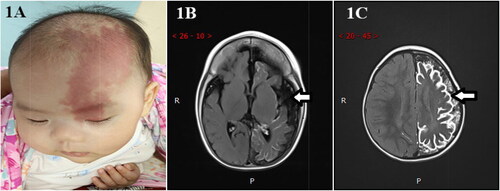
Table 1. Clinicopathological data of 10 children with Sturge–Weber syndrome.
Imaging features
MRI of the brain revealed intracranial structural abnormalities, differing degrees of abnormal leptomeningeal angiomatosis enhancement, varying degrees of brain atrophy, and cortical calcification on the surface of the involved brain lobes. Compared with the contralateral area, the gyri of the lesion atrophied markedly, the sulci and fissure widened and deepened, and the lateral ventricle and third ventricle widened (). The enhancement scan disclosed thickened and tortuous vessels on the surface of the cerebral hemispheres and typical gyrus enhancement of leptomeningeal angiomatosis ().
Clinical symptoms analysis
In this study, all 10 children had a history of refractory epilepsy, and the area of leptomeningeal angiomatosis, the degree of calcification in the lesion cortex, and the degree of brain atrophy were different in different cases. Spearman correlation analysis indicated that the area of leptomeningeal angiomatosis was markedly positively correlated with the degree of brain atrophy (r = 0.783, p = 0.007). There was no significant correlation between the leptomeningeal angiomatosis area and cortical calcification (r = 0.583, p = 0.077). The degree of brain atrophy was not significantly correlated with the degree of cortical calcification (r = 0.116, p = 0.750). There was no significant correlation between leptomeningeal angiomatosis area and age, age of initial epileptic seizure, epileptic seizure type, and seizure frequency (rage = –0.526, page = 0.118; r initial age = –0.478, pinitial age = 0.163; repileptic seizure type = 0.000, pepileptic seizure type= 1.000; rseizure frequency = 0.274, pseizure frequency = 0.444). There was no significant correlation between cortical calcification and age, age of initial epileptic seizure, epileptic seizure type, and seizure frequency (rage = –0.178, page = 0.622; rinitial age = –0.354, pinitial age = 0.315; repileptic seizure type = 0.504, pepileptic seizure type = 0.137; rseizure frequency = –0.094, pseizure frequency = 0.795). There was no significant correlation between the degree of brain atrophy and age, age of initial epileptic seizure, epileptic seizure type, and seizure frequency (rage = –0.535, page = 0.111; rinitial age = –0.325, pinitial age = 0.360; repileptic seizure type = –0.272, pepileptic seizure type = 0.447; rseizure frequency = 0.408, pseizure frequency = 0.242).
Surgical results
All ten children underwent surgery, including one resection of the epilepsy area and nine functional hemispherectomies. The procedure takes 3–5 h and results in 50–300 mL of blood loss. A post-operative re-examination revealed that the lesion had been completely resected and the connections between the affected cerebral cortex and the basal ganglia and lateral cerebral cortex had been severed. Two children had reduced limb activity and low muscle tension after surgery, but recovered to pre-surgery levels three months after surgery. One child had several non-routine seizures during the perioperative period.
Pathological features
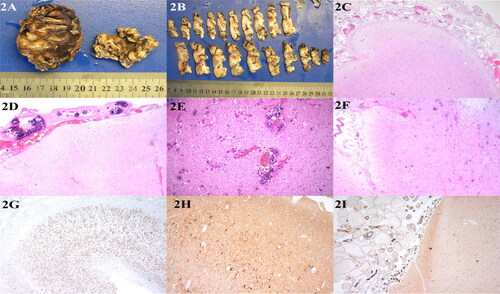
A gross examination of the meningeal surface indicated a network of small gray-red blood vessels, and the boundary between cortex and white matter was unclear in the section of brain parenchyma. The cortex was gray-red and rough, and there were numerous hard brown-yellow calcifications (). Microscopic examination demonstrated that the surface of the SWS brain tissue was thickened by leptomeningeal angiogenesis, with irregularly dilated capillary lumens widely distributed in the subarachnoid space, mostly arranged in reticular or honeycomb shape, and presented a cavernous hemangiomatous change. The malformed vessels had thin walls, with a small number of fibrous thickened vessels and calcification in some walls (). Extensive calcification occurred in the superficial parenchyma along the gyrus in a zonal pattern (). Cortical atrophy led to smaller and narrower gyri, and small neurons in the gray matter were vertically arranged into micro-columnar structures. Locally abnormal neurons were observed scattered (). Balloon-like cells were absent throughout the cortex (both gray and white matter). In this study, ten SWS children with leptomeningeal angiomatosis on the surface of the cerebral cortex were treated with surgical resection for refractory epilepsy, and all were diagnosed with SWS with FCD IIIc by postoperative histopathology.
Figure 2. (A) Gross examination of the meningeal surface reveals a network of small grayish-red vessels. (B) The boundary between the cortex and white matter was not clear on the parenchymal section of the brain, and the cortex was gray-red and tough, with many brown-yellow hard calcifications. (C) The leptomeningeal angiomatosis were thickened on the surface of brain tissue, and irregular dilated capillary lumens were widely distributed in the subarachnoid space, presenting a cavernous hemangiomatous change, H&E 10 × 4. (D) Malformed vessel wall showed obvious calcification, HE 10 × 4. (E) Neuronal cells proliferation are seen around calcification in the superficial brain parenchyma, H&E 10 × 10. (F) Cerebral cortical atrophy H&E 10 × 4. (G) NeuN showed disordered cortical structure around leptomeningeal angiomatosis and partial neuron loss, IHC 10 × 4. (H) NF showed that the cytoplasm of individual neurons was highly stained, IHC 10 × 4. (I) Endothelial cell CD31 positive expression of IHC 10 × 4
Immunophenotype
Immunohistochemical staining revealed partial neuron loss of NeuN in brain tissue and disordered arrangement of the surrounding cortex of the calcified foci (). No balloon-like cells were identified by vimentin staining in the gray or white matter throughout the cortex. GFAP and S-100 staining revealed astrogliosis, and NF was cytosolically positive in abnormal neurons (). Vascular wall SMA was positive, and CD31 and CD34 were strongly expressed in endothelial cells (). Olig-2 was positively expressed in the nuclei of white matter cells in the cortex.
Follow-up
All patients were observed from 2 months to 55 months. According to the Engel grading, 7 of 10 patients achieved Engel I, 2 of 10 patients achieved Engel II, and 1 of 10 patients achieved Engel III. The postoperative follow-up time of three cases was very short (2–6 months), which was not sufficient to determine the exact surgical results.
Discussion
At present, the etiology of SWS is not explicit. From the perspective of development, studies believe that typical SWS is caused by abnormal initial vascular development, which is caused by somatic mutations originating from vascular blast subsets [Citation10]. Recent studies indicate that somatic G protein subunit alpha Q (GNAQ) gene expression of SWS is frequently upregulated through the over-activation of RAS-MEK-ERK, HIPO-YAP, and indirect mTOR. It promoted the proliferation of vascular endothelial cells and inhibited apoptosis, resulting in vascular malformation [Citation11]. Subsequently, the research recognized that mutations in GNA11, a gene highly homologous to GNAQ, can also lead to SWS [Citation12], and the seriousness of SWS depends on the developmental point at which the mutation occurs.
In this study, SWS was associated with FCD type IIIc. In the early stages of embryonic development, the primitive venous plexus diverges into three branches, which supply blood to different parts. The outer branch supplies the face and scalp, the middle branch supplies the meninges, and the inner branch supplies the brain tissue. The origin of SWS is thought to be a developmental disorder of the primitive venous plexus of the head, which persists without regression and causes dysmorphic hemangiomas on the skin and meninges. Children with SWS have vascular malformations in the cerebral cortex that cause hypoperfusion, ischemia, and hypoxia in brain tissue, as well as neuronal migration disorders, a reduction in the number of neurons, and a disorder in the arrangement of neurons, which may contribute to SWS with FCD IIIc. SWS can be accompanied by FCD, while FCD is the epileptogenic focus of partial SWS.
In terms of clinical presentation, nine patients presented with facial PWB and leptomeningeal angiomatosis, two of which were accompanied by impaired mental development, and three with impaired muscle strength in one limb. One case had only leptomeningeal angiomatosis and no facial PWB. In this group, all 10 children had intractable epilepsy, and 7 patients had Engel grade I after the operation. The treatment effect was significant, with no post-operative convulsions, and the prognosis for the head wound was favorable. There were two cases of Engel II, with reduced limb activity, poor muscle tone, and no disabling seizures. There was a patient with Engel grade III who had several perioperative non-routine episodes. Since the number of cases in this group was still insufficient, further determination of the long-term effects was required by increasing the number of cases and the observation time.
Cortical dysplasia in SWS is considered to be the origin of epilepsy, so the complete resection of the lesion site of FCD is one of the indicators for the prognosis of epilepsy control effect in this type of surgery [Citation13]. The efficacy of postoperative seizure control may not be optimal if only the leptomeningeal angiomatosis and its subcortical tissue are removed. According to the new classification of FCD proposed by the ILAE in 2017, FCD with vascular malformations, such as cavernous hemangioma, arteriovenous malformations, and leptomeningeal angiomatosis, but without balloon-like cells, was defined as FCD IIIc. Immunohistochemical staining of NF and vimentin is more specific in identifying abnormal morphologies of neurons and balloon-like cells in FCD lesions and can help pathologists diagnose FCD. MRI showed focal cortical thickening, gray matter disambiguation, and focal cortical signal enhancement on both the T2 and the fluid attenuation-reversal recovery sequences. The nonspecific nature of the FCD imaging findings may also be one of the reasons why only a few SWS with FCD have been reported. Although these findings are not specific, they are indicative of cortical dysplasia and are consistent with histopathological findings. These abnormal signals are frequently disregarded before surgery, which may be contributed to by the physicians focus only on the typical characteristics of SWS, and not considering the possibility of FCD. The number of reported cases of SWS in combination with cortical developmental malformations should increase once physician understanding of these disorders improves.
In this research, Spearman correlation analysis demonstrated a significant positive correlation between the area of leptomeningeal angiomatosis and the degree of brain atrophy in infants with SWS. The more extensive the cerebral lobe involved in leptomeningeal angiomatosis, the more obvious the brain atrophy. There was no significant correlation between the area of leptomeningeal angiomatosis and the degree of cortical calcification, and there was no significant correlation between the degree of cerebral atrophy and the degree of cortical calcification. The distribution area of leptomeningeal angiomatosis, the degree of cortical calcification, and brain atrophy were not significantly correlated with the age of the infant, the age of the initial epileptic seizure, epileptic seizure type, and seizure frequency, further indicating that cortical dysplasia is the pathological basis of epileptic seizure in children. The lack of significant correlation provides no definitive answer (does not exclude that correlation exists) due to the small sample size, and it needs to be further confirmed by increasing the number of cases. Research has confirmed [Citation14] that FCD IIIc is observed in SWS with meningeal hemangiomatosis, and abnormal cortical stratification characterized by L2 and L3 or L4-L6 fusion has been identified.
Conclusion
In summary, most epileptic foci of SWS were associated with FCD IIIc. The areas of leptomeningeal angiomatosis, the degree of cortical calcification, and the degree of cerebral atrophy in SWS were not associated with seizures. There was a significant positive correlation between the leptomeningeal angiomatosis area and the degree of brain atrophy in infants with SWS. The more extensive the cerebral lobes involved in leptomeningeal angiomatosis, the more obvious the cerebral atrophy in infants. For children with SWS with refractory epilepsy symptoms, complete resection of the epileptic foci should be performed as soon as possible. Children with SWS have leptomeningeal angiomatosis on the surface of the cerebral cortex, and post-operative pathological analysis could be standardized to determine whether there is FCD IIIc.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Silverstein M, Salvin J. Ocular manifestations of Sturge–Weber syndrome. Curr Opin Ophthalmol. 2019;30(5):301–5. doi:10.1097/ICU.0000000000000597.
- Anbuselvan S, Venkatachalam P. Sturge–Weber syndrome and glaucoma. J Pharm Bioallied Sci. 2021;13(Suppl 2):S1765–S1768. doi:10.4103/jpbs.jpbs_354_21.
- Wang DD, Blümcke I, Coras R, Zhou WJ, Lu DH, Gui QP, Hu JX, Zuo HC, Chen SY, Piao YS. Sturge–Weber syndrome is associated with cortical dysplasia ILAE type IIIc and excessive hypertrophic pyramidal neurons in brain resections for intractable epilepsy. Brain Pathol. 2015;25(3):248–55. doi:10.1111/bpa.12172.
- Higueros E, Roe E, Granell E, Baselga E. Sturge–Weber syndrome: a review. Actas Dermosifiliogr. 2017;108(5):407–17. doi:10.1016/j.ad.2016.09.022.
- Bianchi F, Auricchio AM, Battaglia DI, Chieffo DRP, Massimi L. Sturge–Weber syndrome: an update on the relevant issues for neurosurgeons. Childs Nerv Syst. 2020;36(10):2553–70. doi:10.1007/s00381-020-04695-3.
- Pathak BD, Sharma S, Adhikari A, Simkhada N, Ghimire B, Aryal N. Sturge–Weber syndrome with bilateral Port-Wine stain. Case Rep Pediatr. 2022; 2022(4):2191465. doi:10.1155/2022/2191465.
- Arinzechi EO, Ogunrin OA, Nwosu CM, Nwani PO, Enwereji KO, Asomugha LA, Dimkpa U. Seizure frequency and risk of cognitive impairment in people living with epilepsy in a sub-urban community in South Eastern Nigeria. J Clin Neurosci. 2019;59(1):98–105. doi:10.1016/j.jocn.2018.10.120.
- Pedraza MI, de Lera M, Bos D, Calleja AI, Cortijo E, Gómez-Vicente B, Reyes J, Coco-Martín MB, Calonge T, Agulla J, et al. Brain Atrophy and the risk of futile endovascular reperfusion in acute ischemic stroke. Stroke. 2020;51(5):1514–21. doi:10.1161/STROKEAHA.119.028511.
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. doi:10.1111/epi.13709.
- Swarup O, Waxmann A, Chu J, Vogrin S, Lai A, Laing J, Barker J, Seiderer L, Ignatiadis S, Plummer C, et al. Long-term mood, quality of life, and seizure freedom in intracranial EEG epilepsy surgery. Epilepsy Behav. 2021;123(10):108241. doi:10.1016/j.yebeh.2021.108241.
- Fjaer R, Marciniak K, Sundnes O, Hjorthaug H, Sheng Y, Hammarström C, Sitek JC, Vigeland MD, Backe PH, Øye AM, et al. A novel somatic mutation in GNB2 provides new insights to the pathogenesis of Sturge–Weber syndrome. Hum Mol Genet. 2021;30(21):1919–31. doi:10.1093/hmg/ddab144.
- Comi AM, Sahin M, Hammill A, Kaplan EH, Juhász C, North P, Ball KL, Levin AV, Cohen B, Morris J, 2015 Sturge-Weber Syndrome Research Workshop, et al. Leveraging a Sturge–Weber gene discovery: an agenda for future research. Pediatr Neurol. 2016;58(5):12–24. doi:10.1016/j.pediatrneurol.2015.11.009.
- Dompmartin A, Vander Vleuten CJM, Dekeuleneer V, Duprez T, Revencu N, Désir J, Te Loo DMWM, Flucke U, Eijkelenboom A, Schultze Kool L, et al. GNA11-mutated Sturge–Weber Syndrome has distinct neurologic and dermatologic features. Eur J Neurol. 2022; 6(6):1–8. doi:10.1111/ene.15452.
- Powell S, Fosi T, Sloneem J, Hawkins C, Richardson H, Aylett S. Neurological presentations and cognitive outcome in Sturge–Weber syndrome. Eur J Paediatr Neurol. 2021;34(9):21–32. doi:10.1016/j.ejpn.2021.07.005.
- Miyata H, Kuwashige H, Hori T, Kubota Y, Pieper T, Coras R, Blümcke I, Yoshida Y. Variable histopathology features of neuronal dyslamination in the cerebral neocortex adjacent to epilepsy-associated vascular malformations suggest complex pathogenesis of focal cortical dysplasia ILAE type IIIc. Brain Pathol. 2022; 1(1):e13052. doi:10.1111/bpa.13052.