
Abstract
Mutations in the PINK1 and PARK2/PARKIN genes are associated with hereditary early onset Parkinson disease (PD), and in cell lines the corresponding gene products play a critical role in mitophagic clearance of damaged mitochondria. In neurons, however, where the extraordinary cellular shapes pose particular challenges for maintaining healthy mitochondria, the pathways of mitophagy are less well understood. Both the location at which mitophagy occurs and the involvement of PINK1 and PARK2 have been controversial. Here we review our recent study where we found that selective damage to a subset of axonal mitochondria causes them to be engulfed within autophagosomes and cleared locally within the axon without the need for transport back to the soma. We also found this process to be completely dependent on neuronal PINK1 and PARK2.
In cell lines, mitochondrial damage, such as loss of mitochondrial membrane potential, leads to the accumulation of PINK1, a Ser/Thr kinase, on the outer membrane which in turn recruits PARK2, an E3 ubiquitin ligase, to activate the autophagic machinery and cause the engulfment of dysfunctional mitochondria. Parkinson disease (PD) is characterized by neuronal degeneration and thus the role of these proteins in neurons is of particular importance. Moreover, the vast majority of neuronal mitochondria are not in the soma, but in the axonal and dendritic processes. It is these distal processes, far from the soma where the lysosomal machinery was thought to be located, that are likely to degenerate first in PD.
It was suggested that mitochondria with reduced membrane potential are transported back to the soma to be degraded. In contrast, we had previously demonstrated that PINK1 and PARK2 promote proteosomal degradation of the mitochondrial motor adaptor protein RHOT/Miro, leading to the motility arrest of axonal mitochondria (). PINK1 and PARK2 also inhibit fusion of damaged mitochondria with healthy ones by triggering the degradation of the fusion proteins MFN1 (mitofusin 1) and MFN2. Loss of RHOT and MFN1/2 effectively quarantines damaged mitochondria and further suggests that subsequent degradation of the mitochondrion should occur locally. We set out therefore to define the pathway of mitochondrial clearance from axons: Is mitophagy initiated locally? Do autophagosomes containing defective mitochondria move in a retrograde manner to the soma? Are PINK1 and PARK2 involved?
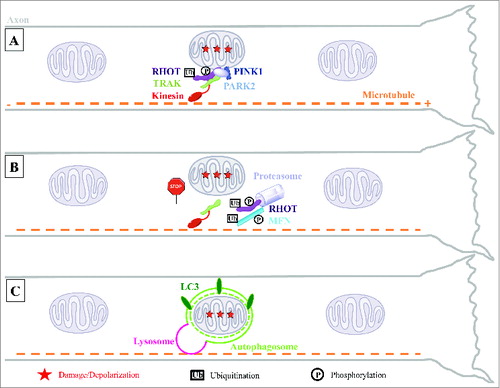
Figure 1. Model for local mitophagy of damaged mitochondria in neuronal axons. (A) Damage to axonal mitochondria leads to the accumulation of PINK1 and PARK2 on the mitochondrial outer surface where they interact with the motor complex composed of the RHOT/Miro and TRAK/Milton adaptor proteins and the kinesin motor protein. (B) PINK1- and PARK2-mediated proteasomal degradation of RHOT arrests both anterograde and retrograde motility of these organelles. This, and the parallel degradation of MFN1/2, are an early action of the PINK1 and PARK2 pathway prior to mitophagy and may help quarantine damaged mitochondria by preventing their fusion with healthy neighbors. (C) Accumulation of PARK2 on the stationary damaged mitochondria promotes their local engulfment within LC3-positive autophagosomes. Mitophagosomes fuse with axonal lysosomes to form autolysosomes and their mitochondrial content is degraded locally within the axon.
One limitation of the previous studies exploring neuronal mitophagy has been the nonphysiological level of mitochondrial damage induced with chemical uncouplers, which are toxic to neurons due to their strict dependence on mitochondrial ATP production. For this reason, we mimicked more physiological levels of mitochondrial dysfunction with 2 approaches that damaged only a subpopulation of axonal mitochondria in cultured hippocampal neurons. First, mito-KillerRed, a genetically encoded photosensitizer targeted to mitochondria, was activated in a few axonal mitochondria to cause ROS-mediated damage, while preserving the integrity of mitochondria outside the small irradiated region. As a second approach, antimycin A, a specific inhibitor of complex III, was applied to only a segment of axons through the use of microfluidic devices. We found that axonal mitochondria depolarized with either method become colocalized with stationary GFP-LC3-positive autophagosomes. We concluded that autophagosome formation does in fact occur along the axon. In several instances, the mitochondrial marker inside an autophagosome disappeared over time, suggesting the acidification or lysosomal degradation of the autophagosomal contents. Contrary to the prevalent notion that lysosomes are primarily located in the soma, we found a significant population of lysosomes in hippocampal axons. In response to mitochondrial damage, a fraction of axonal lysosomes are recruited to these stationary mitochondria-containing autophagosomes (mitophagosomes) to facilitate their degradation (). Unlike the retrograde transport model, local mitophagy would obviate the need for damaged mitochondria to be transported along the axon and would prevent further spread of reactive oxygen species. We do not rule out the possibility that the autolysosomes may move to the soma to recycle their contents at later time points than we examined. In addition, the depolarization-induced mitophagosomes we observed may be quite distinct from those that form in the absence of mitochondrial damage which, as observed by Maday and Holzbaur, are motile, and arise primarily near the tips of axons.
What are the requirements for axonal mitophagy? In non-neuronal cells both PINK1 and PARK2 are required. In neurons, however, PARK2 recruitment is only reported in the soma by some studies but not others. Induction of localized mitochondrial damage allowed us to demonstrate the accumulation of an expressed YFP-tagged PARK2 on damaged axonal mitochondria, consistent with our previous finding that the arrest of mitochondrial motility in response to damage is PARK2-dependent. Most importantly, mitophagy is completely abolished in either pink1-/- or park2-/- hippocampal axons indicating that the presence of endogenous PINK1 and PARK2 are both essential for the axonal pathway of damage-induced mitophagy. The involvement of PINK1 and PARK2 in axonal mitophagy implies that axonal degeneration in PD may in fact be due to the accumulation of dysfunctional mitochondria. We speculate that defects in PINK1-PARK2-mediated mitophagy predisposes neurons to death under conditions of high stress, which may arise in old age in humans due to the accumulation of mitochondrial damage over time.
One additional feature of axonal mitophagy was revealed by our live-cell imaging: the process can occur quite rapidly in axons. Both PARK2 and LC3 are recruited to the mitochondria in less than 30 min, and the subsequent loss of mitochondrial markers can occur in less than 1 h. Mitophagy is transient and asynchronous and therefore only 20–40% of the depolarized mitochondria might be captured in a PARK2- or LC3-positive state at a given time, unless lysosomes are inhibited. Nonetheless, we never saw clearance of 100% of the depolarized mitochondria; depolarization alone may not be sufficient to trigger mitophagy and one or more of the mitophagy effectors may be in limiting supply. However, it is clear that the fundamental PINK1-PARK2 pathway that has been so clearly elucidated in cell lines is also active in neurons and indeed acts both rapidly and locally in neuronal processes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.