
Abstract
Telomere dysfunction plays a complex role in tumorigenesis. While dysfunctional telomeres can block the proliferation of incipient cancer clones by inducing replicative senescence, fusion of dysfunctional telomeres can drive genome instability and oncogenic genomic rearrangements. Therefore, it is important to define the regulatory pathways that guide these opposing effects. Recent work has shown that the autophagy pathway regulates both senescence and genome instability in various contexts. Here, we apply models of acute telomere dysfunction to determine whether autophagy modulates the resulting genome instability and senescence responses. While telomere dysfunction rapidly induces autophagic flux in human fibroblast cell lines, inhibition of the autophagy pathway does not have a significant impact upon the transition to senescence, in contrast to what has previously been reported for oncogene-induced senescence. Our results suggest that this difference may be explained by disparities in the development of the senescence-associated secretory phenotype. We also show that chromosome fusions induced by telomere dysfunction are comparable in autophagy-proficient and autophagy-deficient cells. Altogether, our results highlight the complexity of the senescence-autophagy interface and indicate that autophagy induction is unlikely to play a significant role in telomere dysfunction-driven senescence and chromosome fusions.
Abbreviations:
- ACD/Tpp1, adrenocortical dysplasia homolog (mouse)
- ATG5, autophagy-related 5, ATG7, autophagy-related 7
- B2M, β-2-microglobulin
- HBSS, Hank's buffered salt solution
- HMECs, human mammary epithelial cells
- MEFs, mouse embryonic fibroblasts
- MT-HsTER, mutant template-Homo sapiens template-containing RNA
- MT-MmTER, mutant template-Mus musculus template-containing RNA
- OIS, oncogene-induced senescence
- RBBP8/CtIP, retinoblastoma binding protein 8
- SA-β-Gal, senescence-associated β-galactosidase
- SASP, senescence associated secretory phenotype
- TDIS, telomere dysfunction-induced senescence
- TERT, telomerase reverse transcriptase
- TIFs, telomere dysfunction-induced foci
Introduction
Telomeres are composed of tandem TTAGGG DNA repeats that maintain genome stability by recruiting protein complexes called shelterins, which protect chromosome ends from recognition as double-strand DNA breaks. Upon extensive loss of telomeric sequence from replicative shortening,Citation1 the resulting dysfunctional telomeres activate a canonical DNA damage responseCitation2 that promotes senescence and/or genome instability depending upon the cellular context. Telomere dysfunction-induced senescence (TDIS) is an important barrier to tumor development because it can block the proliferation of incipient cancer cells.Citation3,4 However, telomere dysfunction can also induce genome-destabilizing chromosome fusions that promote tumor initiation, progression, and evolution.Citation5-7
In addition to telomere dysfunction, cellular senescence can be triggered by constitutive activation of oncogenes such as HRASG12V,Citation8 or loss of tumor suppressors including PTEN.Citation9 Similar to TDIS, oncogene-induced senescence (OIS) is proposed to be an important barrier to tumor progression and to be caused, at least in part, by a DNA damage response secondary to replication stress.Citation4,10-13 Nonetheless, there are fundamental differences between these 2 types of senescence.Citation10,13 First, OIS can occur independently of a DNA damage response.Citation14 Second, OIS and TDIS exhibit qualitatively and quantitatively distinct gene expression profiles, with differences in expression of Wnt target genes and chromatin regulators.Citation15 Finally, while both OIS and TDIS induce secretion of myriad cytokines and other factors as part of the senescence-associated secretory phenotype (SASP), the SASP differs both in magnitude and composition between these 2 modes of senescence.Citation16,17
Recently, macroautophagy (hereafter referred to as autophagy) has been uncovered as an important regulator of OIS because of its role in supporting the production of a robust SASP that promotes the senescence transition in response to oncogenic HRASG12V or Kaposi sarcoma-associated herpesvirus cyclin (v-cyclin).Citation17-20 However, other studies suggest that autophagy can block senescence in certain circumstances, highlighting the complex relationship between autophagy and senescence.Citation21-23 Although increased autophagy has been observed in response to dysfunctional telomeres, the role of autophagy in TDIS remains unclear.Citation24-26
In addition, autophagy has been implicated in the maintenance of genome stability. Initial studies showed that autophagy promotes genome stability by maintaining cellular metabolic homeostasis,Citation27 and more recent work in yeast and mammalian cells suggests that autophagy directly influences DNA damage repair by modulating levels of the key repair protein RBBP8/CtIP.Citation28,29 Moreover, autophagy guards against aneuploidy by regulating cell cycle progression and cytokinesis.Citation30,31 In light of these emerging roles for autophagy in senescence and genome instability, 2 critical sequelae of telomere dysfunction, we sought to delineate the potential functions of autophagy in the cellular response to acute telomere dysfunction. Our results reveal the autophagy-independent nature of senescence and genome instability driven by targeted telomere dysfunction.
Results
Telomere dysfunction induces autophagic flux in human fibroblasts
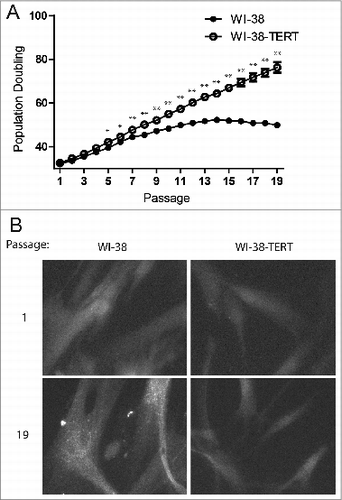
To determine whether autophagy is involved in the cellular response to telomere dysfunction, we first examined autophagy status in the context of replicative senescence in WI-38 human fibroblasts. Because WI-38 cells lack telomerase activity, their telomeres shorten during each S phase until reaching a critically short length that induces a DNA damage response and replicative senescence.Citation1,2 As a control, we generated WI-38 cells stably expressing TERT (telomerase reverse transcriptase; WI-38-TERT cells), which confers robust telomerase activity and blocks replicative telomere shortening.Citation32 As expected, the primary WI-38 fibroblasts lacking TERT entered replicative senescence over a period of extended culture, while the WI-38-TERT cells proliferated indefinitely (). To monitor autophagosome formation during senescence, we used cells that stably express GFP fused to MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β; GFP-MAP1LC3B/LC3B) and observed that the senescent WI-38 fibroblasts showed an increase in GFP-tagged LC3B puncta while the WI-38-TERT cells did not (). Similar results were observed in human mammary epithelial cells (HMECs) upon transition to agonescence, a senescence-associated growth plateau resulting from telomere dysfunction-induced growth arrest (Fig. S1).Citation33
Figure 1. Punctate GFP-LC3B in replicative senescent human fibroblasts. (A) Growth curves of primary WI-38 and WI-38-TERT fibroblasts in extended culture. Error bars represent standard deviation among 3 wells grown in parallel. (B) Representative images of GFP-LC3B puncta in WI-38 or WI-38-TERT cells at passage 1 and 19 after initiation of culture. A 40X objective was used for imaging. * and ** indicate significance at P < 0.05 and P < 0.01 respectively.
In contrast to OIS, replicative senescence appears gradually as the onset of telomere dysfunction occurs asynchronously within a proliferating cell population,Citation34,35 thereby complicating the rigorous quantification of autophagic flux as well as mechanistic studies of autophagy in this context. Accordingly, to more effectively scrutinize the relationship between telomeric dysfunction and autophagy, we employed a well-defined acute model of TDIS. Telomerase is minimally composed of a TERT protein and a template-containing RNA (TER). We utilized a mutant form of human TER containing base changes in the template region, hereafter referred to as MT-HsTER for mutant template Homo sapiens TER (or in the case of mice, MT-MmTER for mutant template Mus musculus TER).Citation36,37 When MT-HsTER is overexpressed, it joins with endogenous TERT to form mutant telomerase enzyme that adds variant TTTGGG telomeric repeats to chromosome ends (), thereby disrupting shelterin binding and inducing acute telomere dysfunction.Citation36,37 MT-HsTER overexpression in WI-38-TERT cells caused rapid telomere dysfunction as demonstrated by the induction of DNA damage foci at telomeres (telomere dysfunction-induced foci, TIFs; ). Furthermore, MT-HsTER induced senescence rapidly and relatively uniformly over a period of 7 to 10 d, as demonstrated by dramatically reduced cell proliferation and the appearance of enlarged cells positive for senescence-associated β-galactosidase (SA-β-gal; ).
Figure 2. For figure legend, see page 531.
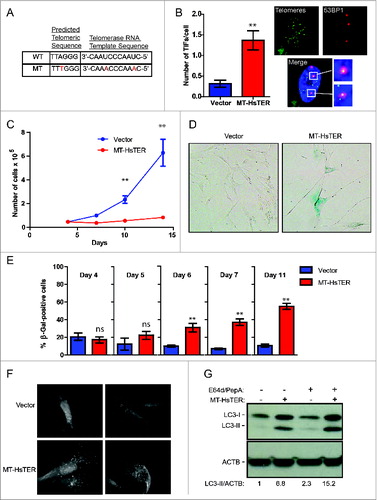
We next asked whether autophagy is induced during MT-HsTER-driven senescence. Indeed, an accumulation of GFP-LC3B puncta was observed in MT-HsTER-expressing cells, but not in vector controls (). In addition, immunoblotting for LC3B confirmed an increase in the lipidated form of LC3B (LC3-II), which associates with the autophagosome membrane, in MT-HsTER-expressing cells (). Autophagic flux was also increased in MT-HsTER cells compared to vector and WT-HsTER controls when cells were treated with the lysosomal protease inhibitors E64d and pepstatin A to block LC3-II turnover (, Fig. S2). Overall, these results indicate that MT-HsTER-induced telomeric dysfunction is sufficient to induce both autophagy and senescence.
Autophagy does not regulate the transition to TDIS
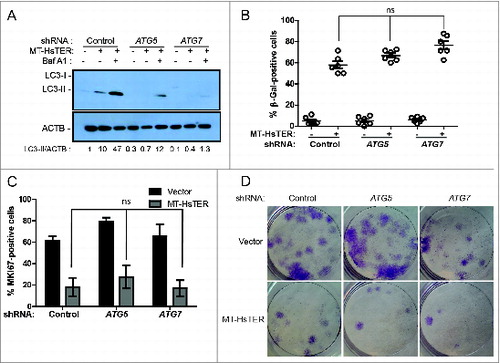
Prior work implicates autophagy in promoting the transition to OIS,Citation18,19 suggesting that the induction of autophagy we observed in response to telomeric dysfunction may similarly contribute to TDIS. To test this possibility, we used shRNAs targeting ATG5 and ATG7, 2 proteins essential for the early steps of autophagosome formation.Citation38 In WI-38-TERT cells, expression of these shRNAs reduced target mRNA and protein levels (Fig. S3). Knockdown of ATG5 or ATG7 also resulted in a reduction in LC3-II formation following MT-HsTER treatment in the presence or absence of the lysosomal v-ATPase inhibitor bafilomycin A1 (BafA1) (), indicating that these shRNAs effectively block autophagy flux. In contrast to previous studies of OIS,Citation18,19 MT-HsTER-induced TDIS was largely unaffected by inhibition of autophagy. The accumulation of SA-β-gal-positive cells after MT-HsTER treatment was not significantly affected by depletion of ATG5 or ATG7 (, Fig. S3). Additionally, MKI67/Ki67 staining to identify cells actively engaged in the cell cycle did not show significant differences between autophagy-proficient and autophagy-deficient cells expressing MT-HsTER (). Lastly, autophagy inhibition did not affect the ability of WI-38 fibroblasts expressing MT-HsTER to grow in a colony-forming assay (). In fact, we observed fewer and smaller colonies of shATG7-treated cells in a colony-forming assay regardless of the addition of MT-HsTER. This data is consistent with previous results that autophagy inhibition alone may impair cell growth depending on the cell type and level of autophagy inhibition.Citation22
Figure 3. Autophagy inhibition does not affect the senescence transition following MT-HsTER treatment of WI-38 fibroblasts. (A) LC3B immunoblotting in WI-38-TERT fibroblasts transduced with a control, ATG5, or ATG7 shRNA and treated with 25 nM BafA1, 7 d after MT-HsTER addition. Relative amounts of LC3-II are indicated below the blots. (B) SA-β-gal staining 7 d after MT-HsTER addition. SA-β-gal staining showed a significant increase between vector and MT-HsTER cells (P < 0.05) but no significant differences were observed between the MT-HsTER-treated samples. Error bars represent standard error. (C) MKI67 staining in WI-38-TERT fibroblasts expressing a control, ATG5, or ATG7 shRNA, 7 d after MT-HsTER transduction. MT-HsTER-treated samples showed a significant depletion of MKI67-positive cells compared to controls (P < 0.05). Error bars represent standard deviation. (D) Crystal violet-stained colonies showing colony forming efficiency upon platting 3 × 103 cells 8 d after MT-HsTER addition. Results are representative of at least 3 independent experiments.
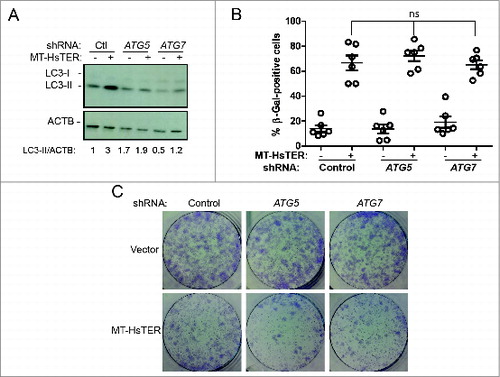
Previous studies of autophagy in OIS have predominantly utilized BJ fibroblasts.Citation13,16,18,19 In contrast to WI-38 fibroblasts, senescence in BJ fibroblasts can be reversed upon inhibition of TP53/p53, likely due to differences in CDKN2A/p16 expression between these 2 cell types.Citation39 We therefore tested whether autophagy inhibition alters the progression of TDIS in BJ-TERT fibroblasts. Despite robust depletion of ATG5 and ATG7 mRNA and protein as well as effective inhibition of induced autophagic flux (, Fig. S4), the accumulation of SA-β-gal-positive cells following MT-HsTER treatment in BJ fibroblasts was unaffected (). Similarly, autophagy inhibition did not affect the ability of MT-HsTER-treated BJ fibroblasts to grow in a colony-forming assay (). Together, these data argue that autophagy does not play a significant role in MT-HsTER-induced senescence in human fibroblast cell lines.
Figure 4. Autophagy inhibition does not affect the senescence transition following MT-HsTER treatment of BJ fibroblasts. (A) LC3B immunoblotting in BJ-TERT fibroblasts transduced with a control, ATG5, or ATG7 shRNA 7 d after MT-HsTER addition. Relative amounts of LC3-II are indicated below the blots. (B) SA-β-gal staining 7 d after MT-HsTER addition. SA-β-gal staining showed a significant increase between vector and MT-HsTER cells (P < 0.05) but no significant differences were observed between the MT-HsTER-treated samples. Error bars represent standard error. (C) Crystal violet-stained colonies showing colony forming efficiency upon plating 3 × 103 cells at day 8 after MT-HsTER addition. Data is representative of at least 3 independent experiments.
The transition to TDIS occurs in the absence of a robust SASP
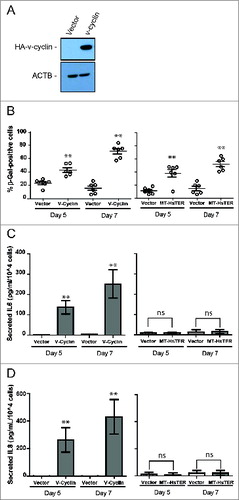
Previous work suggests that autophagy supports the SASP during the transition to OIS, and autophagy inhibition slows the transition to OIS by delaying SASP induction.Citation18-20 Because the SASP reinforces senescence in an autocrine and paracrine fashion,Citation16,17,40 we reasoned that MT-HsTER-induced TDIS may be less dependent on autophagy induction than OIS due to a difference in the SASP between the 2 types of senescence. Notably, previous work has shown that the SASP from replicative senescent cells is reduced in overall magnitude compared to that produced from OIS.Citation16 To test this possibility in our system, we further scrutinized how MT-HsTER-induced SASP differs from that induced by the v-cyclin oncogene. We first confirmed that v-cyclin expression () elicited high levels of senescence in BJ fibroblasts, as shown by the progressive accumulation of SA-β-gal-positive cells (). Importantly, similar numbers of senescent cells were observed in response to MT-HsTER and v-cyclin expression during the transition to senescence (). Next, we monitored the secretion of SASP factors during the transition to senescence. Since autophagy has been linked to the efficient synthesis and secretion of IL6 and IL8 during OIS, we focused on these 2 SASP cytokines in our studies.Citation18–20 In agreement with previous findings, v-cyclin overexpression induced rapid and robust secretion of both IL6 and IL8 during the transition to senescence ().Citation19 In contrast, the secretion of IL6 and IL8 was not significantly increased during this timeframe in the MT-HsTER-treated cells (). Thus, both IL6 and IL8 production are much more limited in our model of TDIS compared to v-cyclin-induced OIS during the period of senescence transition. Despite these profound differences in the elaboration of these key SASP factors, BJ fibroblasts exhibit comparable levels of senescence in response to either telomeric dysfunction or v-cyclin expression during the period of senescence transition ().
Figure 5. Senescence transition in the absence of a robust SASP in MT-HsTER-expressing BJ fibroblasts. (A) Immunoblot for HA to detect overexpressed HA-v-cyclin protein in BJ fibroblasts 7 d after transduction. (B) SA-β-gal staining during the period of senescence transition—5 and 7 d after MT-HsTER or HA-v-cyclin addition—in BJ-TERT fibroblasts. Error bars represent standard error. (C) IL6 and (D) IL8 Elisa from conditioned media harvested from BJ-TERT fibroblasts at various time points after MT-HsTER or HA-v-cyclin addition. Error bars represent standard deviation. Elisa data includes 2 independent experiments. SA-β-Gal data is representative of at least 3 independent experiments. ** indicates significance at P < 0.01 when compared to the vector control.
Autophagy does not influence telomere dysfunction-induced chromosome fusions
In addition to inducing senescence, telomere dysfunction causes genomic instability primarily through the production of chromosome fusions, which occur upon recognition of the de-protected chromosome ends by the DNA damage repair machinery.Citation41 In an attempt to “repair” the exposed chromosome ends, the DNA repair machinery fuses chromosomes together through classical or alternative nonhomologous end joining (C-NHEJ and A-NHEJ, respectively).Citation42 Telomere dysfunction-induced fusions can then drive further instability through iterative breakage-fusion-bridge cycles during subsequent mitoses.Citation43
Given the ability of autophagy to modulate DNA repair,Citation28,29 we sought to determine if autophagy influences the rate or pattern of telomere dysfunction-induced chromosome fusions. Such fusions are challenging to study in primary human cells with intact checkpoints because telomere dysfunction induces rapid cell cycle arrest.Citation44 In contrast, because immortalized mouse embryonic fibroblasts (MEFs) do not exhibit cell cycle arrest in response to telomeric dysfunction, they are a well-established and highly useful model for the study of telomere dysfunction-induced genomic instability, including chromosome fusions.Citation41,42 Therefore, to examine the role of the autophagy pathway in telomere dysfunction-induced chromosome fusions, we used mouse TERT-expressing immortalized MEFs from littermate Atg5+/+ or atg5−/- mice and littermate Atg7+/+ or atg7−/- mice. We first induced telomere dysfunction by expressing MT-MmTER in autophagy-proficient or autophagy-deficient immortalized MEFs. Autophagy was enhanced in response to MT-MmTER in autophagy-proficient MEFs following nutrient starvation with Hank's buffered salt solution (HBSS) and treatment with BafA1 (). In addition, MT-MmTER treatment significantly induced chromosome fusions as monitored by metaphase analysis (). Importantly, the number of fusions did not significantly differ in the autophagy-proficient and autophagy-deficient cells, suggesting that autophagy did not influence the fusion process ().
Figure 6. For figure legend, see page 535.
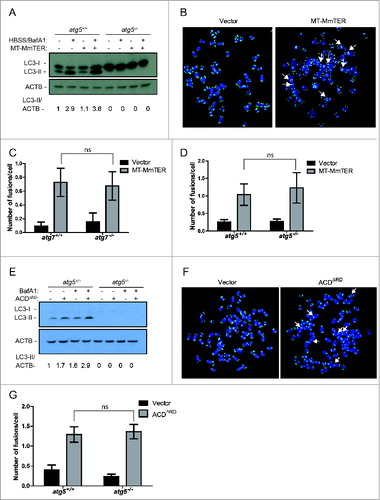
Chromosome fusions induced by MT-MmTER are thought to arise predominantly through ATM-dependent C-NHEJ.Citation45,46 In contrast, fusions induced by overexpression of ACDΔRD/Tpp1ΔRD, a dominant-negative form of the telomeric shelterin component ACD/Tpp1, occur through A-NHEJ, a fundamentally different DNA repair mechanism.Citation42 While C-NHEJ is ligase IV-dependent and usually occurs through ligation of blunt DNA ends, A-NHEJ typically requires 3’ end processing and generates microhomology at the fusion point.Citation47 Of note, the RBBP8 protein, which is reportedly regulated by autophagy, has been implicated in A-NHEJ and chromosome fusions induced by ACDΔRD.Citation28,42 We therefore asked whether ACDΔRD- induced chromosome fusions differ in autophagy-competent and autophagy-deficient immortalized MEFs. ACDΔRD induced autophagic flux () and chromosome fusions (). However, comparable numbers of fusions were observed in response to ACDΔRD expression independently of the autophagy status in Atg5+/+ and atg5−/- as well as in Atg7+/+ and atg7−/- MEFs (, data not shown). Based on these cumulative results, we conclude that the autophagy pathway does not have an impact on the development of chromosome fusions induced by MT-MmTER and ACDΔRD.
Discussion
Cellular senescence and chromosome fusions are 2 key outcomes of telomere dysfunction that critically affect tumor initiation and progression.Citation4,5,7 Previous work in other systems has shown that telomere dysfunction induces autophagic flux,Citation24-26 and that autophagy regulates senescence and genome instability in some contexts.Citation18,19,27,28 Here, we have directly tested whether autophagy guides the response to acute telomere dysfunction. We find that while autophagic flux is indeed induced by several distinct types of telomere dysfunction, the resulting senescence and chromosome fusions are essentially unaffected by autophagy status.
The role of autophagy in the senescence response has been studied predominantly in the context of oncogene activation. A previous study has uncovered that inhibition of autophagy delays the transition to senescence following overexpression of oncogenic HRASG12V,Citation18 suggesting that autophagy induction promotes the senescent phenotype. A role for autophagy activation in promoting oncogene-induced senescence was subsequently identified in other systems.Citation19,23 However, independent studies have found that the autophagy pathway can instead inhibit senescence in a variety of contexts.Citation21,22 For example, autophagy inhibition in human fibroblasts triggers cellular senescence under certain conditions,Citation22 and knockdown of ATG5 in mouse fibroblasts promotes Ras-induced senescence.Citation21 These conflicting results could be due to a number of experimental differences, including the level of autophagy inhibition, the type of cells used, and the type of senescence trigger under study. Overall, the results from these studies reinforce that the connections between autophagy and cellular senescence remain complex and poorly defined.Citation23
Our results indicate that autophagy activation is not required for the transition to MT-HsTER-induced TDIS, thus providing additional evidence that senescence and autophagy are not always tightly linked. Moreover, previous studies of both HRASG12V and v-cyclin-induced senescence have uncovered important roles for autophagy in the production of IL6 and IL8, 2 key SASP cytokines.Citation18,19 In contrast to these models of OIS, we do not observe the robust elaboration of either cytokine in autophagy-competent or autophagy-deficient cells undergoing MT-HsTER-induced TDIS. Our results suggest that the SASP is more limited in the transition to MT-HsTER-induced TDIS than OIS, which may partly explain the different role of the autophagy pathway in these 2 types of senescence. Important questions for future study include determining whether autophagy serves critical functions in other types of senescence, such as that induced by CDKN2A overexpression,Citation48 and whether the impact of autophagy on different kinds of senescence parallels the level of key SASP mediators involved in each. In addition, it will be important to establish whether the role of autophagy in senescence differs between the mesenchymal cells used in this study and various epithelial cell types that also undergo telomere dysfunction-induced cell cycle arrest, such as HMECs.Citation33
Cells experiencing telomere dysfunction accumulate chromosome fusions, resulting in significant genome instability.Citation41,43 Initial studies examining the role of autophagy in genome stability conclude that autophagy suppresses chromosome instability by maintaining cellular metabolic homeostasis.Citation27 Subsequent work has suggested that autophagy also modulates genome stability by controlling DNA damage repair mechanisms and cell cycle progression. In yeast, autophagy targets the DNA damage repair protein Sae2 (orthologous to human RBBP8) for autophagic degradation upon its acetylation,Citation28 and autophagy has also been implicated in regulating levels of RBBP8 in human colon cancer cells.Citation29 These data suggest that autophagy may control DNA damage repair in mammalian cells through the regulation of RBBP8, a key protein mediating the choice between nonhomologous and homologous repair pathways.Citation49 Autophagy has also been shown to influence cell cycle progression. For example, studies in yeast have shown that modulating autophagy upon nutrient starvation causes a delay in cell cycle progression and defects in nuclear division, leading to aneuploidy.Citation30 In mammals, autophagy regulates protein levels of active RHOA in a SQSTM1/p62-dependent manner. When autophagy is disrupted, active RHOA accumulates in areas surrounding the midbody and causes cytokinesis failure, multinucleation, and aneuploidy.Citation31 While changes in DNA repair mechanisms and cell cycle progression could potentially influence the extent and pattern of telomere fusions, we find no change in these readouts despite the complete loss of autophagic activity in immortalized atg5 or atg7 knockout MEFs. Thus, autophagy does not influence telomere dysfunction-induced genome instability in this cellular context. Nonetheless, our results do not rule out a role for autophagy in repair of other DNA lesions at telomeres or throughout the rest of the genome, including those that rely on homologous recombination for resolution.
As autophagy inhibitors reach the clinic for the treatment of a variety of cancers, it is critical to understand how these therapeutic agents affect basic cellular processes that influence cancer initiation and progression, including the dysfunctional telomere response. While autophagy inhibition for the purposes of anticancer therapy may impinge on a variety of cellular pathways, potentially eliciting unwanted side effects, our results suggest that impaired autophagy is unlikely to alter the acute response to telomere dysfunction.
Materials and Methods
Cell culture
Human BJ and WI-38 fibroblasts were obtained from ATCC and immortalized by retroviral expression of TERT. Immortalized Atg5+/+ and atg5−/- as well as Atg7+/+ and atg7−/- MEFs were obtained from Noburu Mizushima (University of Tokyo) and Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) respectively. HMECs were grown in mammary epithelial growth medium (MEGM) supplemented with hEGF, hydrocortisone, and insulin (Lonza, CC-2551, CC-3151, CC-3150). All other cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, 11965) supplemented with 10% FBS. All cells were grown at 37°C in 5% CO2. For viral transduction, cells were infected for 24 h with lentivirus or 48 h with retrovirus, and antibiotic selection was started 24 h after virus removal at the following concentrations: puromycin (Life Technologies, A11138–02) at 1.5 ug/mL for 2 to 3 d, or blasticidin (Life Technologies, A11139–03) at 8 ug/mL for 3 d. For the pSMB retroviruses expressing control, ATG5, or ATG7 shRNAs, BJ and WI-38 fibroblasts were infected for 8 h followed by a second 24-h infection.
Plasmids and viruses
Retroviral ACDΔRD/Tpp1ΔRD plasmid was kindly provided by Sandy Chang (Yale University). The TER-expressing lentivector included a wild-type or mutant TER gene driven by the IU1 promoter and a puromycin resistance gene driven by the CMV promoter.Citation50 Lentiviral vectors expressing MT-MmTER and a retroviral vector expressing mouse TERT were kindly provided by Amir Goldkorn (University of Southern California). The pSMB retroviral vectors expressing shRNAs against ATG5 or ATG7, as well as the retroviral HA-v-cyclin pBMN plasmid, were a gift from Andrew Leidal (Dalhousie University). The shRNA targeting sequences were as follows: ATCTCGCTTGGGCGAGAGTAAG (nontargeted control); CTTTGATAATGAACAGTGAGA (ATG5); GGAGTCACAGCTCTTCCTTAC (ATG7). The retroviral pBABE GFP-LC3B plasmid has been previously described and is available through AddGene (22405).Citation51 Lentiviruses and retroviruses were produced as previously described.Citation45
Cell proliferation, colony formation, SA- β-gal assays
Growth curves were carried out by plating 4.5 × 104 cells per well of a 6-well plate and counting cell number by hemocytometer every 3 d, which is defined as one passage. SA-β-gal was measured according to manufacturer's instructions (Sigma-Aldrich, G5635). Six fields from 3 separate wells were analyzed for each of 3 independent experiments. Flow cytometry detection of SA- β-gal was performed as previously described.Citation52 Briefly, cells were treated for 1 h with 100 nM BafA1 (Sigma-Aldrich, B1793) followed by addition of 33 uM C12FDG (Life Technologies, D2893) for 1.5 h and analysis using a BD FACS Calibur instrument (Mississauga, ON, Canada). To examine colony formation, 3 × 103 cells were plated in each well of a 6-well plate in triplicate 7 or 8 d after infection with MT-HsTER-expressing lentivirus. Colonies were fixed and stained with 0.05% crystal violet (Sigma, C3886) 9 d (BJ fibroblasts) or 14 to 18 d (WI-38 fibroblasts) after plating. Washed and dried plates representative of 3 independent experiments were imaged.
Immunofluorescence and fluorescent in situ hybridization (FISH)
Cells were seeded on coverslips and fixed with 4% paraformaldehyde (Electron Microscopy Services, 15710) for 10 min. After permeabilization with 0.5% NP-40 (Sigma, 18896), cells were blocked in PBG (0.2% [w/v] gelatin from cold water fish skin [Sigma, G7765], 0.5% [w/v] bovine serum albumin [Sigma, A2153] in 1X phosphate-buffered saline) and incubated overnight with a 1:100 dilution of MKI67/Ki67 antibody (Millipore, AB9260). To obtain metaphases, MEFs were treated with 0.1 μg/ml colcemid (Invitrogen, 15212) for 2 to 3 h. FISH was performed as previously described.Citation53 FISH-IF for TIFs was performed on coverslips with a 53BP1 antibody as previously described and validated (Novus Biologicals, NB100–304).Citation54 Telomeric DNA was detected with a PNA FISH probe (TMR-OO-5′-[CCCTAA]3–3′, Panagene) and all images were obtained using a Metasystems Metafer slide scanning platform (70 Bridge Street Suite 100, Newton, MA 02458) in conjunction with a Zeiss Axio Imager Z2 upright microscope (Dublin, CA USA) using a Zeiss 63X 1.4 NA Plan APOCHROMAT lens (Dublin, CA USA) and Metafer software. Fusions were scored in a blinded fashion.
Gene expression
For quantitative RT-PCR, RNA was harvested using an RNeasy extraction kit (Qiagen, 74104). 50 ng of RNA was used in a quantitative RT-PCR reaction using the Brilliant II SYBR Green One-Step QRT-PCR kit (Agilent Technologies, 600825). mRNA expression relative to B2M was determined by calculating 2^(- ΔΔCT). Primers used for amplification are listed in .
Table 1. Primers used for quantitative RT-PCR analysis
IL6 and IL8 ELISA
The concentration of secreted IL6 and IL8 was determined from 24-h conditioned full media harvested at the indicated time points using the R&D Quantikine Elisa kit (R&D Systems, D6050, D8000C). The data was normalized to cell number at the time of conditioned media harvest and reported as ng per 104 cells over the 24-h period. The ELISA data for IL6 and IL8 derive from 2 independent experiments, each of which included triplicate measurements.
Autophagy assays
To examine autophagy flux, cells were treated with a single dose of BafA1 (25 nM) (Sigma-Aldrich, B1793) for 1 h, E64d (10 ug/mL) and pepstatin A (10 ug/mL) for 5 h, or a vehicle control (DMSO or 100% ethanol) (Sigma-Aldrich, P5318, 11707; Enzo, PI107) as previously described.Citation55,56 For starvation assays, cells were washed 3 times with phosphate-buffered saline (PBS; Life Technologies, 14190) followed by HBSS (Invitrogen, 14170) treatment for 1 h.
Immunoblotting
Cells were lysed in RIPA buffer supplemented with a protease and phosphatase-inhibitor cocktail (Thermo Scientific, 78440). Protein concentrations were determined using a BCA protein assay kit (Thermo Scientific 23225). Samples were run on 4–20% Tris-glycine gels after dilution in 2X Laemmli sample buffer supplemented with β-mercaptoethanol. Immunoblotting for LC3-II was performed with a 1:1000 dilution of LC3B antibody that has been previously described and is now commercially available (EMD Millipore, ABC232).Citation51 Immunoblotting for ATG5 was performed using a 1:500 dilution of ATG5 antibody (Cell Signaling Technology, 2630). Immunoblotting for ATG7 was performed using a 1:100 dilution of ATG7 antibody (Santa Cruz Biotechnology, sc-8668). Immunoblotting for HA-v-cyclin was performed using a 1:1000 dilution of HA antibody (Covance, MMS-101P). Immunoblotting for ACTB/β-actin was performed using a 1:30,000 dilution of ACTB/β-actin-HRP antibody (Abcam, ab20272). To quantify bands, Fiji software was used to measure band intensity of the protein of interest divided by the band intensity of ACTB.
Statistics
Data for chromosome fusions was analyzed using a nonparametric Kruskal-Wallis test, followed by a pairwise Wilcoxon signed-rank test with a Holm-Sidak correction for multiple comparisons. Data for ELISAs, growth curves, SA- β-gal analysis, and MKI67 staining was performed using a One-Way ANOVA, followed by pairwise Student t tests with a Holm-Sidak correction for multiple comparisons. All statistical tests were done using R. Data was plotted using GraphPad Prism.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
2014AUTO0528R1_supplemental_files.pdf
Download PDF (470.5 KB)Acknowledgments
We thank Ajay Ravindranathan, Andrew Leidal, and Morgan Diolaiti for review of the manuscript and insightful feedback. We would like to thank Drs. Amir Goldkorn, Sandy Chang, Andrew Leidal, Noboru Mizushima, and Masaaki Komatsu for providing research materials.
Funding
Grant support includes the NIH (K08 CA134552 to BAS, R01 CA188404 to JD) and American Cancer Society IRG-97–150–14 to BAS. This material is based upon work supported by the National Science Foundation under Grant No. 1144247 to FAM.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:36; PMID:13905658
- Fabrizio d'Adda di F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003; 426:4; PMID:14608368
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO 2012; 31:22; PMID:22569128; http://dx.doi.org/10.1038/emboj.2012.132
- Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 2005; 26:7; PMID:15471900
- Bailey SM, Murnane JP. Telomeres, chromosome instability and cancer. Nucleic Acids Res 2006; 34:9; PMID:16682448; http://dx.doi.org/10.1093/nar/gnj009
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007; 338:3; PMID:17687332
- Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES, Hu J, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012; 148:11; PMID:22341455; http://dx.doi.org/10.1016/j.cell.2012.01.039
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:9; PMID:9019410; http://dx.doi.org/10.1016/S0092-8674(00)81852-8
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:5; PMID:16079851
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al.. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:4; PMID:17136093; http://dx.doi.org/10.1038/nature05268
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA, et al.. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436:5; PMID:16079837; http://dx.doi.org/10.1038/nature03841
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS.. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436:4; PMID:16079850; http://dx.doi.org/10.1038/436004a
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre' M, Nuciforo PG, Bensimon A, et al.. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:4; PMID:17136094; http://dx.doi.org/10.1038/44404a
- Haferkamp S, Tran SL, Becker TM, Scurr LL, Kefford RF, Rizos H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging 2009; 1:14; PMID:20157537
- Nelson DM, McBryan T, Jeyapalan JC, Sedivy JM, Adams PD. A comparison of oncogene-induced senescence and replicative senescence: implications for tumor suppression and aging. Age 2014; 36(3):9637; PMID:24647599; http://dx.doi.org/10.1007/s11357-014-9637-0
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor PLOS Biol 2008; 6:15; PMID:19053174; http://dx.doi.org/10.1371/journal.pbio.0060301
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133:12; PMID:18555778; http://dx.doi.org/10.1016/j.cell.2008.03.039
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Gene Dev 2009; 23:5; PMID:19279323; http://dx.doi.org/10.1101/gad.519709
- Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012; 11:13; PMID:22341465; http://dx.doi.org/10.1016/j.chom.2012.01.005
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011; 332:4; PMID:21512002; http://dx.doi.org/10.1126/science.1205407
- Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A 2012; 109:5; PMID:22198765; http://dx.doi.org/10.1073/pnas.1118531109
- Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PoS One 2011; 6:e23367; PMID:21858089; http://dx.doi.org/10.1371/journal.pone.0023367
- Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy 2013; 9:4; PMID:23422284; http://dx.doi.org/10.4161/auto.23922
- Zhou WJ, Deng R, Zhang XY, Feng GK, Gu LQ, Zhu XF. G-quadruplex ligand SYUIQ-5 induces autophagy by telomere damage and TRF2 delocalization in cancer cells. Mol Cancer Ther 2009; 8:10; PMID:19996277; http://dx.doi.org/10.1158/1535-7163.MCT-09-0244
- Orlotti NI, Cimino-Reale G, Borghini E, Pennati M, Sissi C, Perrone F, Palumbo M, Daidone MG, Folini M, Zaffaroni N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 2012; 8:9; PMID:22627293; http://dx.doi.org/10.4161/auto.20519
- Aoki H, Iwado E, Eller MS, Kondo Y, Fujiwara K, Li GZ, Hess KR, Siwak DR, Sawaya R, Mills GB, et al. Telomere 3′ overhang-specific DNA oligonucleotides induce autophagy in malignant glioma cells. FASEB J 2007; 21:22; PMID:17449721; http://dx.doi.org/10.1096/fj.06-6941com
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Gene Dev 2007; 21:14; PMID:17510285; http://dx.doi.org/10.1101/gad.1545107
- Robert T VF, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471:74-9; PMID:21368826; http://dx.doi.org/10.1038/nature09803
- Rajendran P, Kidane AI, Yu TW, Dashwood WM, Bisson WH, Löhr CV, Ho E, Williams DE, Dashwood RH. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013; 8:11; PMID:23770684; http://dx.doi.org/10.4161/epi.24710
- Matsui A, Kamada Y, Matsuura A. The Role of autophagy in genome stability through suppression of abnormal mitosis under starvation. PLoS Genet 2013; 9:e1003245; PMID:23382696; http://dx.doi.org/10.1371/journal.pgen.1003245
- Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, Ilie M, Rubera I, Tauc M, Barale S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res 2013; 73:11; PMID:23704209; http://dx.doi.org/10.1158/0008-5472.CAN-12-4142
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science 1998; 279:3; PMID:9454332
- Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol 2002; 34:8; PMID:12200033
- Kill IR, Faragher RG, Lawrence K, Shall S. The expression of proliferation-dependent antigens during the lifespan of normal and progeroid human fibroblasts in culture. J Cell Sci 1994; 2:8; PMID:7911472
- Martin-Ruiz C, Saretzki G, Petrie J, Ladhoff J, Jeyapalan J, Wei W, Sedivy J, von Zglinicki T. Stochastic variation in telomere shortening rate causes heterogeneity of human fibroblast replicative life span. J Biol Chem 2004; 279:7; PMID:14963037
- Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, Blackburn EH. A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci U S A 2001; 98:5; PMID:11438744; http://dx.doi.org/10.1073/pnas.241497098
- Marusíc L, Anton M, Tidy A, Wang P, Villeponteau B, Bacchetti S. Reprogramming of telomerase by expression of mutant telomerase RNA template in human cells leads to altered telomeres that correlate with reduced cell viability. Mol Cell Biol 1997; 17:7; PMID:9343401
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:13; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2009.12.034
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:10; PMID:12912919
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15:12; PMID:23770676; http://dx.doi.org/10.1038/ncb2784
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42:333; PMID:18680434; http://dx.doi.org/10.1146/annurev.genet.41.110306.130350
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 2010; 29:12; PMID:20588252; http://dx.doi.org/10.1038/emboj.2010.142
- Murnane JP. Telomere dysfunction and chromosome instability. Mutation Res 2012; 730:8; PMID:21575645; http://dx.doi.org/10.1016/j.mrfmmm.2011.04.008
- Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science 2002; 295:3; PMID:11923537
- Stohr BA, Xu L, Blackburn EH. The terminal telomeric DNA sequence determines the mechanism of dysfunctional telomere fusion. Mol Cell 2010; 39:7; PMID:20670897; http://dx.doi.org/10.1016/j.molcel.2010.06.020
- Stohr BA, Blackburn EH. ATM mediates cytotoxicity of a mutant telomerase RNA in human cancer cells. Cancer Res 2008; 68:8; PMID:18593932; http://dx.doi.org/10.1158/0008-5472.CAN-08-0504
- Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet 2013; 47:22; PMID:24050180; http://dx.doi.org/10.1146/annurev-genet-110711-155540
- Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 2011; 286:7; PMID:21880712; http://dx.doi.org/10.1074/jbc.M111.257071
- Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009; 459:3; PMID:19357644; http://dx.doi.org/10.1038/nature07955
- Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, Blackburn EH. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res 2004; 64:7; PMID:15256453; http://dx.doi.org/10.1158/0008-5472.CAN-03-2623
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:9; PMID:18094039
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:8; PMID:20010931; http://dx.doi.org/10.1038/nprot.2009
- Diolaiti ME, Cimini BA, Kageyama R, Charles FA, Stohr BA. In situ visualization of telomere elongation patterns in human cells. Nuclei Acids Res 2013; 41:9; PMID:23963699; http://dx.doi.org/10.1093/nar/gkt689
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol 2003; 13:7; PMID:12956959; http://dx.doi.org/10.1016/S0960-9822(02)01378-7
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:13; PMID:21119005; http://dx.doi.org/10.1091/mbc.E10-06-0500
- N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009; 10:6; PMID:19148225; http://dx.doi.org/10.1038/embor.2008.238