
Abstract
Disruption of the complex of BECN1 with BCL2 or BCL2L1/BCL-XL is an essential switch that turns on cellular autophagy in response to environmental stress or treatment with BH3 peptidomimetics. Recently, it has been proposed that BCL2 and BCL2L1/BCL-XL may inhibit autophagy indirectly through a mechanism dependent on the proapoptotic BCL2 family members, BAX and BAK1. Here we report that the BH3 mimetic, ABT-737, induces autophagy in parallel with disruption of BCL2-BECN1 binding in 2 different apoptosis-deficient cell types lacking BAX and BAK1, namely in mouse embryonic fibroblasts cells and in human colon cancer HCT116 cells. We conclude that the BH3 mimetic ABT-737 induces autophagy through a BAX and BAK1-independent mechanism that likely involves disruption of BECN1 binding to antiapoptotic BCL2 family members.
Abbreviations:
- ACTB, actin, β
- Baf A1, bafilomycin A1
- BAK1, BCL2-antagonist/killer 1
- BAX, BCL2-associated X protein
- BCL2, B-cell CLL/lymphoma 2
- BECN1, Beclin 1, autophagy-related
- DKO, double-knockout
- FBS, fetal bovine serum
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- HBSS, Hanks’ balanced salt solution
- HRP, horseradish peroxidase
- MAP1LC3/LC3, microtubule-associated protein 1 light chain 3
- MCL1, myeloid cell leukemia 1
- MEFs, mouse embryonic fibroblasts
- MTOR, mechanistic target of rapamycin
- PBS, phosphate-buffered saline
- SQSTM1, sequestosome 1
- STS, staurosporine
- WT, wild type
BECN1/Beclin 1 is a central regulator of autophagy, a lysosomal degradation pathway crucial in diverse cellular processes, including protein and organelle quality control, nutrient and energy generation, life and death decisions, and immune regulation.Citation1-3 The autophagy function of BECN1 requires its interaction with the class III phosphatidylinositol 3-kinase (PtdIns3K, whose catalytic subunit is PIK3C3/VPS34), and the autophagy protein ATG14, and is regulated by a variety of nonexclusive, overlapping mechanisms including (1) its subcellular localization; (2) its dimerization state; (3) its interactions with other core members of the class III PtdIns3K complex (i.e., PIK3C3, ATG14); (4) its interactions with positive and negative regulatory proteins; and (5) its post-translational modifications (e.g., ubiquitination, phosphorylation, proteolysis).Citation3-11 These multiple mechanisms of regulation serve to integrate diverse cellular signals with autophagic vesicle nucleation and can bypass upstream signals that are autophagy inhibitory or stimulatory such as MTOR activation or MTOR repression, respectively.Citation8,12
The binding of BCL2 or BCL2L1/BCL-XL is the best-studied negative regulatory mechanism of the autophagy function of BECN1.Citation5,13,14 Both BCL2 and BCL2L1 (as well as viral BCL2 orthologs) bind BECN1 directly via its BH3 domain, and the dissociation of BCL2 or BCL2L1 complexes with BECN1 is essential for autophagy upregulation in response to stress, both in cultured cells and in vivo. This binding may be inhibited by competitive disruption by BH3-only proteins or BH3 peptidomimetic drugs,Citation15,16 post-translational modifications of BECN1 that promote its dissociation from BCL2 or BCL2L1,Citation17,18 or posttranslational modifications of BCL2 that promote its dissociation from BECN1. For example, MAPK8/JNK1-mediated multisite phosphorylation of BCL2 is essential for starvation-induced disruption of BCL2-BECN1 binding and starvation-induced autophagy in vitro,Citation19 and mice lacking these BCL2 phosphorylation sites are deficient in starvation-induced and exercise-induced autophagy in vivo.Citation20 Furthermore, mutations in BCL2, BCL2L1 or viral BCL2s that block interaction with BECN1 fail to inhibit autophagy,Citation21,22 and mutations in BECN1 that block its interaction with BCL2 family members confer resistance to their antiautophagy effects.Citation21,23 The crystal structures of BCL2 family members, including BCL2L1 and viral BCL2, bound to the BH3 domain of BECN1 have been solvedCitation22,24-26 and predict that mutations that block the antiautophagy activity of BCL2 family members are crucial for physical interaction with BECN1.
Taken together, these observations provide strong support for the prevailing paradigm that BCL2, BCL2L1, and viral BCL2-like proteins inhibit autophagy through a direct interaction with BECN1. Recently, Lindqvist et al. have proposed that prosurvival BCL2 family members do not inhibit components of the autophagy pathway directly, but rather, inhibit autophagy indirectly as a consequence of inhibition of proapoptotic BCL2 family members, BAX and BAK1.Citation27,28 This conclusion is based in part on the failure to observe induction of autophagy in MEFs lacking BAX and BAK1 after 4-h treatment with ABT-737, a BH3 peptidomimetic agent that has previously been shown to disrupt BCL2L1-BECN1 binding and induce autophagy.Citation15 In the present study, we sought to examine whether a longer ABT-737 treatment period could induce autophagy in MEFs lacking BAX and BAK1 and whether ABT-737 could induce autophagy in a second cell type, HCT116 cells, lacking BAX and BAK1.
Our results indicate that short-term treatment (12 h) of both wild-type HCT116 or BAX BAK1 double-knockout (DKO) HCT116 cells leads to autophagy and disruption of the BCL2-BECN1 complex. However, 12-h treatment with ABT-737 fails to induce autophagy or disruption of BCL2-BECN1 binding in either wild-type or in Bax Bak1 DKO MEFs, but a longer duration treatment period (48 h) induces comparable levels of autophagy and disruption of BCL2-BECN1 binding in both wild-type and in Bax Bak1 DKO MEFs. These findings demonstrate that different cell types require different durations of ABT-737 treatment to observe autophagy induction; yet, the ability of ABT-737 to induce autophagy uniformly correlates with its ability to disrupt BCL2-BECN1 binding but fails to correlate with the presence or absence of the BAX and BAK1 proapoptotic machinery. Thus, BAX and BAK1 are dispensable for ABT-737-mediated autophagy, suggesting that BCL2 family members do not activate autophagy by indirectly inhibiting activation of BAX and BAK1. Rather, BCL2 family members likely inhibit autophagy through direct interactions with the autophagy protein BECN1.
Results
BAX and BAK1 are dispensable for ABT-737-induced autophagy in HCT116 cells
Lindqvist et al.Citation24,25 report that MEFs lacking the antiapoptotic protein, MCL1, but not MEFs lacking MCL1 and the proapoptotic molecules, BAX and BAK1, undergo autophagy, as measured by MAP1LC3B (LC3B-I) to LC3B-II conversion, after 4-h treatment with 1 μM ABT-737. To evaluate whether ABT-737-induced autophagy is universally independent of BAX and BAK1, we used another cell line, namely HCT116 colon carcinoma cells, which we genetically engineered to lack BAX and BAK1 expression ().
Figure 1. ABT-737 induces autophagy and dissociation of the BECN1-BCL2 complex in HCT116 cells lacking BAX and BAK1. (A) Western blot detection of BAX and BAK1 in parental HCT116 cells (WT) and in HCT116 cells lacking either BAX (BAX−/−), BAK1 (BAK1−/−), or both BAX and BAK1 (DKO). (B) Cell death (as measured by flow cytometric staining of propidium iodide (PI) uptake) of WT or BAX BAK1 DKO HCT116 cells following treatment with the indicated dose of staurosporine (STS) treatment for the indicated duration. Results represent mean ± s.d. of triplicate samples. (C) Western blot detection of LC3B conversion and SQSTM1 degradation in the indicated cell types treated with rapamycin (1 μM, 4 h) in the presence or absence of 50 nM Baf A1. GAPDH was used as a loading control. (D) Representative photomicrographs of WT HCT116 cells or BAX BAK1 DKO HCT116 cells stably expressing GFP-LC3B and control-treated or treated with rapamycin (1 μM, 4 h). Hoechst (blue) represents nuclear staining. GFP-LC3B puncta (green) correspond to autophagosomes. Scale bars: 10 μm. (E) Quantification of the number of GFP-LC3B puncta per cell in cells treated as in (D) in the absence or presence of 50 nM Baf A1. Results represent mean ± s.d. of triplicate samples (500 cells analyzed per sample). (F) Western blot detection of LC3B conversion and SQSTM1 degradation in the indicated cell type treated with the indicated dose of ABT-737 for 12 h. GAPDH was used as a loading control. (G) Representative photomicrographs of WT HCT116 cells and HCT116 BAX BAK1 DKO cells stably expressing GFP-LC3B and control-treated or treated with ABT-737 (10 μM, 12 h). Hoechst (blue) represents nuclear staining. GFP-LC3B puncta (green) correspond to autophagosomes. Scale bars: 10 μm. (H) Quantification of the number of GFP-LC3B puncta per cell in cells treated as in (G) in the absence or presence of 50 nM Baf A1. Results represent mean ± s.d. of triplicate samples (500 cells analyzed per sample). (I) Immunoprecipitation of endogenous BECN1 with endogenous BCL2 in WT or in BAX BAK1 DKO HCT116 cells following the indicated treatment. Cells were subjected to rapamycin treatment (1 μM, 4 h), starvation (HBSS, 4 h), or the indicated dose of ABT-737 for 12 h. ACTB is shown as a loading control. For (B), (E) and (H), NS = not significant, and *** = P < 0.001 for the indicated comparison between genotypes in each condition; Student t test.
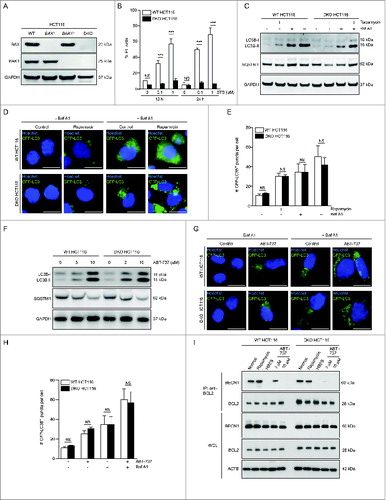
First, we confirmed that HCT116 BAX BAK1 DKO cells are resistant to cell death by measuring the percentage of propidium iodide-positive cells 12 h and 24 h after treatment with increasing concentrations of the apoptosis-inducing kinase inhibitor, staurosporine. The majority of wild-type HCT116 cells were dead within 12 h of 1 μM staurosporine treatment, whereas no cell death was observed in HCT116 BAX BAK1 DKO cells after 12 h or 24 h of 1 μM staurosporine treatment (). Next, we evaluated whether HCT116 BAX BAK1 DKO cells underwent autophagy in response to treatment with the MTOR inhibitor rapamycin. Rapamycin increased autophagic flux to similar levels in wild-type HCT116 cells and HCT116 BAX BAK1 DKO cells, as measured by increased LC3B-I to LC3B-II conversion (with a further increase in LC3B-II after treatment with the lysosomal inhibitor bafilomycin A1/Baf A1), increased degradation of the autophagy receptor SQSTM1/p62 (which was blocked by Baf A1), and increased numbers of GFP-LC3B puncta (which further increased in the presence of Baf A1) ().
The ability of HCT116 BAX BAK1 DKO cells to undergo autophagy but to resist cell death induction demonstrated that they are a useful reagent for testing the hypothesis that ABT-737-induced autophagy requires the BAX and BAK1 apoptotic machinery. In contradiction to this hypothesis, we found that 12 h of ABT-737 treatment resulted in similar levels of autophagy induction in both wild-type HCT116 and HCT116 BAX BAK1 DKO cells. There was a dose-dependent increase in LC3B-II conversion and SQSTM1 degradation in both cell types (), and treatment with 10 μM ABT-737 resulted in similar numbers of increased GFP-LC3B puncta in both cell types (which were further increased by Baf A1, indicating a complete autophagic response) ().
Next, we investigated the effects of ABT-737 on the autophagy-inhibitory BECN1-BCL2 complex by examining the amount of endogenous BECN1 that coimmunoprecipitates with endogenous BCL2. The dose-dependent increase in ABT-737-induced autophagy was paralleled by a dose-dependent disruption of BECN1 binding to BCL2 in wild-type and HCT116 DKO cells; 3 μM of ABT-737 treatment partially disrupted BECN1-BCL2 binding, whereas the BECN1-BCL2 complex was no longer detectable by immunoprecipitation after 10 μM ABT-737 treatment (). The disruption of BECN1-BCL2 binding by 12-h treatment with 10 μM ABT-737 was similar to that observed after 4 h of amino acid starvation, whereas rapamycin had no effect on BECN1-BCL2 binding. Taken together, our results in HCT116 BAX BAK1 DKO cells indicate that BAX and BAK1 are dispensable for both rapamycin and ABT-737-induced autophagy. However, ABT-737-induced autophagy, but not rapamycin-induced autophagy, is accompanied by disruption of BECN1-BCL2 binding.
BAX and BAK1 are dispensable for ABT-737-induced autophagy in MEFs
Given our findings in HCT116 BAX BAK1 DKO cells indicating that BAX and BAK1 are dispensable for ABT-737-induced autophagy, we used identical conditions (10 μM ABT-737 for 12 h) to examine the effects of ABT-737 on autophagy in previously characterizedCitation29 wild-type and Bax Bak1 DKO MEFs (). Similar to HCT116 cells lacking BAX and BAK1, we confirmed that MEFs lacking BAX and BAK1 are resistant to staurosporine-induced cell death (), but sensitive to rapamycin-induced autophagy (). No differences were observed between wild-type or Bax Bak1 DKO MEFs with respect to rapamycin-induced LC3B-II conversion, SQSTM1 degradation, or numbers of GFP-LC3B puncta in the presence or absence of Baf A1. However, in contrast to HCT116 cells, 10 μM treatment of ABT-737 for 12 h failed to induce autophagy (as measured by LC3B-II conversion, SQSTM1 degradation, and numbers of GFP-LC3B puncta) in either wild-type MEFs or Bax Bak1 DKO MEFs (). Consistent with the failure of ABT-737 to induce autophagy, there was no disruption of BECN1-BCL2 binding after 12 h of ABT-737 treatment in either wild-type MEFs or Bax Bak1 DKO MEFs ().
Figure 2. ABT-737 induces autophagy and dissociation of the BECN1-BCL2 complex after prolonged, but not shorter duration treatment, in wild-type or in Bax and Bak1 KO MEFs. (A) Western blot detection of BAX and BAK1 in wild-type (WT) MEFS and in MEFS lacking either BAX (Bax−/−), BAK1 (Bak1−/−), or both BAX and BAK1 (DKO). (B) Cell death (as measured by flow cytometric staining of propidium iodide (PI) uptake) of WT or Bax Bak1 DKO MEFs following treatment with the indicated dose of staurosporine (STS) treatment for the indicated duration. Results represent mean ± s.d. of triplicate samples. (C) Western blot detection of LC3B conversion and SQSTM1 degradation in the indicated cell types treated with rapamycin (1 μM, 4 h) in the presence or absence of 50 nM Baf A1. GAPDH was used as a loading control. (D) Representative photomicrographs of WT MEFs or Bax Bak1 DKO MEFs stably expressing GFP-LC3B and control-treated or treated with rapamycin (1 μM, 4 h). Hoechst (blue) represents nuclear staining. GFP-LC3B puncta (green) correspond to autophagosomes. Scale bars: 10 μm. (E) Quantification of the number of GFP-LC3B puncta per cell in cells treated as in (D) in the absence or presence of 50 nM Baf A1. Results represent mean ± s.d. of triplicate samples (500 cells analyzed per sample). (F) Western blot detection of LC3B conversion and SQSTM1 degradation in the indicated cell type treated with the indicated dose of ABT-737 for 12 h. GAPDH was used as a loading control. (G) Representative photomicrographs of WT MEFs or Bax Bak1 DKO MEFs stably expressing GFP-LC3B and control-treated or treated with ABT-737 (10 μM, 12 h). Hoechst (blue) represents nuclear staining. GFP-LC3B puncta (green) correspond to autophagosomes. Scale bars: 10 μm. (H) Quantification of the number of GFP-LC3B puncta per cell in cells treated as in (G) in the absence or presence of 50 nM Baf A1. Results represent mean ± s.d. of triplicate samples (500 cells analyzed per sample). (I) Immunoprecipitation of endogenous BECN1 with endogenous BCL2 in WT or Bax Bak1 DKO MEF cells following the indicated treatment. Cells were subjected to starvation (HBSS, 4 h), or ABT-737 for 12 h or 48 h. Western blot detection of LC3B-I to LC3B-II conversion and levels of SQSTM1 were performed to assess autophagy in the same samples used for immunoprecipitation of BECN1-BCL2 complexes. ACTB is shown as a loading control. For (B), (E) and (H), NS = not significant, and *** = P < 0.001 for the indicated comparison between genotypes in each condition; Student t test.
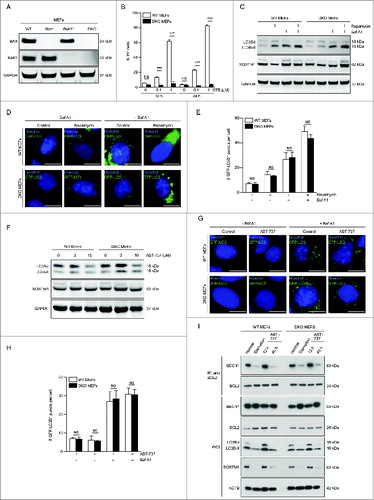
These observations suggested that MEFs may display a cell type-specific relative resistance to the effects of ABT-737 that are independent of BAX and BAK1. To examine this hypothesis, we treated MEFs for a longer duration with ABT-737, and found that after 48 h treatment, there was disruption of BECN1-BCL2 binding, marked conversion of LC3B-I to LC3B-II, and marked SQSTM1 degradation in both wild-type or Bax Bak1 DKO MEFs (). The magnitude of these effects was similar to those observed with amino acid starvation, a well-known inducer of autophagy that requires disruption of BECN1-BCL2 binding for its actions. Thus, although MEFs (independently of their BAX and BAK1 status) fail to undergo ABT-737-induced autophagy in similar conditions that induce autophagy in HCT116 cells, both wild-type and Bax Bak1 DKO cells undergo ABT-737-induced autophagy and disruption of BECN1-BCL2 binding with a longer duration of exposure to ABT-737.
Discussion
Our results, using 2 distinct apoptosis-deficient cell types genetically engineered to lack BAX and BAK1, indicate that BAX and BAK1 are dispensable for ABT-737-induced autophagy. The BH3 peptidomimetic induces autophagy in both HCT116 and MEF cells lacking BAX and BAK1, but the duration of treatment required for these effects is longer in MEFs cells (independently of their genotype) than in HCT116 cells. In HCT116 cells, we show a correlation between dose-dependent induction of autophagy and disruption of BECN1-BCL2 binding (i.e., stronger autophagy induction and more complete disruption of the BECN1-BCL2 complex with 10 μM vs. 3 μM treatment) and in MEFs, we show a correlation between treatment duration-dependent induction of autophagy and disruption of BECN1-BCL2 binding (i.e., autophagy induction and decreased BECN1-BCL2 binding with 48 h but not 12 h of exposure to ABT-737).
These findings are inconsistent with the model proposed by Lindqvist et al. in which BCL2 family members regulate autophagy only indirectly by activating BAX and BAK1.Citation27,28 In our studies, the BH3 mimetic, ABT-737, which can competitively disrupt the binding of both proapoptotic proteins and the BECN1 autophagy protein with antiapoptotic BCL2 family members (BCL2, BCL2L2, and BCL2L1), induces autophagy in cells lacking BAX and BAK1. Therefore, this autophagy induction cannot be an indirect consequence of BAX and BAK1 and/or BAX and BAK1-dependent apoptotic processes. Rather, the strong association between the ability of ABT-737 to induce autophagy in a given experimental condition (even in cells lacking BAX and BAK1) and its ability to disrupt binding between BECN1 and BCL2 favors the classic model discussed by Lindqvist et al. in which BCL2 family members inhibit autophagy by binding to BECN1 via the BH3 binding groove.Citation28 This model is consistent with our previous results regarding the role of ABT-737 and BH3-only proteins in promoting autophagy through competitive disruption of the binding of BCL2 or BCL2L1 to BECN1,Citation15 as well as multiple other lines of evidence that BCL2 family members regulate autophagy through interaction with BECN1, including the demonstrations that (1) post-translational modifications of BCL2 or BECN1 that regulate their interaction with each other also regulate autophagy;Citation17-19,30 and (2) substitution mutations in BCL2 family members at amino acid residues that interface in crystal structures with the BH3 domain of BECN1 block their antiautophagy activity.Citation22,25
We note that there are several factors that may explain the apparently discrepant conclusions between the current study (and preceding papers demonstrating that BCL2 family members function as antiautophagy proteins through their interaction with the BH3 domain of BECN1) and the study by Lindqvist et al.Citation27 First, in the ABT-737 experiments, Lindqvist et al. used only a short duration (4 h) of ABT-737 treatment in Bax, Bak1, and Mcl1 KO MEFs, and compared Bax, Bak1, and Mcl1 KO MEFs to Mcl1 KO MEFs. We directly compared wild-type MEFs with Bax Bak1 DKO MEFs, and it is possible that concurrent Mcl1 deletion somehow affects ABT-737 sensitivity. Moreover, we examined a longer duration of ABT-737 treatment (48 h), and found that longer (but not shorter, i.e., 12 h) duration of exposure to ABT-737 treatment induced autophagy in wild-type and in Bax Bak1 DKO MEFs. Thus, the time course used by Lindqvist et al. may have missed the ability of ABT-737 to induce autophagy in MEFs in a BAX and BAK1-independent manner. Second, the dose of ABT-737 used by Lindqvist et al. (1 μM) is significantly lower than the doses which we found were required to disrupt BECN1-BCL2 binding in either HCT116 cells or MEFs (data not shown). As Lindqvist et al. do not directly examine the effects of ABT-737 on the interaction between BECN1 and either BCL2 or BCL2L1, it is unknown whether the regimen of ABT-737 that failed to induce autophagy had any effects on BECN1 binding to antiapoptotic BCL2 family members.
In addition to the ABT-737 experiments, the model proposed by Lindqvist et al. that BCL2 regulates autophagy indirectly through a BAX and BAK1-dependent mechanism is based on the observation that overexpression of antiapoptotic BCL2 family members (BCL2, BCL2L1, or MCL1) in BAX and BAK1-deficient MEFs fails to alter autophagy, as assessed by levels of LC3B lipidation.Citation27 However, the authors do not examine whether overexpression of these proteins alters LC3B lipidation (or other readouts of autophagy induction) in wild-type MEFs. They also do not determine whether overexpression of these proteins affected levels of BCL2 family member-BECN1 interactions. As several different post-translational events regulate BCL2-binding to BECN1, the overexpression of BCL2 family members per se may not be sufficient to lead to increased BECN1 binding and increased autophagy inhibition.
In conclusion, our results demonstrate that the ability of ABT-737 to induce autophagy is independent of BAX and BAK1, but is strongly associated with disruption of BECN1-BCL2 binding. These findings are inconsistent with the possibility that competitive BH3 mimetics induce autophagy through the indirect activation of BAX and BAK1-dependent apoptosis and are consistent with a model in which such agents induce autophagy through competitive disruption of binding between BCL2 family members and the BECN1 autophagy protein.
Materials and Methods
Cell lines and cell culture conditions
Previously described transformed wild-type, Bax KO, Bak1 KO, or Bax Bak1 DKO MEFsCitation29 were cultured in Dulbecco's modified essential medium (Gibco, 11965-092 or 11995-065) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids, 100 mM sodium pyruvate, 10 mM HEPES buffer, 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. For GFP-LC3B autophagy assays, GFP-LC3B stable cell lines were generated by transfecting WT or Bax Bak1 DKO MEFs with prepackaged viral particles expressing recombinant GFP-LC3B (LentiBrite GFP-LC3B Lentiviral Biosensor; Millipore, 17-10193), according to the manufacturer's instructions. Briefly, cells were plated in a chamber slide and transduced with lentiviral particles at a multiplicity of infection of 20 for 24 h. Then, the medium was replaced and cells were visualized after 48 h to monitor transduction efficiency. HCT116 colon carcinoma cell lines lacking BAX, BAK1, or both BAX and BAK1 were generated using homologous recombination (for BAXCitation31) or zinc finger nuclease technology (for BAK1). Wild-type or BAX BAK1 DKO HCT116 cells expressing GFP-LC3B were generated by transduction with LentiBrite GFP-LC3B. HCT116 cell lines were cultured in McCoy medium (Gibco-Invitrogen, 26600-023) supplemented with 10% FBS, 10 mM HEPES buffer, 100 units/mL penicillin G sodium and 100 μg/mL streptomycin sulfate. All cells were cultured at 37°C and 5% CO2. For starvation experiments, cells were cultured in Hanks’ balanced salt solution (HBSS; Sigma-Aldrich, 55021C).
Chemicals and antibodies
Baf A1 and staurosporine were purchased from Sigma-Aldrich (B1793 and S4400 respectively); rapamycin from either Tocris Bioscience or Sigma-Aldrich (1292/1 and R0395, respectively), and ABT-737 from Selleck Chemicals (S1002).
The following antibodies were used for western blot experiments: anti-BAK1 (Cell Signaling Technology, 3814; 1:1,000 dilution), anti-BAX (Cell Signaling Technology, 2772; 1:1,000 dilution); anti-GAPDH Millipore, ab2303; 1:1,000 dilution), anti-LC3B (Cell Signaling Technology, 2775; 1:1000 dilution), and anti-SQSTM1 (Abnova, H00008878-M01;1:5,000 dilution). For immunoprecipitation of BCL2, a monoclonal anti-BCL2 antibody was used (Santa Cruz Biotechnology, sc-7382AC; 1:50 dilution), and immunoprecipitates were subjected to western blot analysis with anti-BCL2 (Santa Cruz Biotechnology, sc-7382HRP; 1:250 dilution) or anti-BECN1 (Santa Cruz Biotechnology, sc-11427; 1:250 dilution). Western blot analyses of whole cell lysates in samples used for BCL2-BECN1 coimmunoprecipation were performed using the following antibodies: anti-BECN1 (sc-11427; 1:1000 dilution), anti-BCL2 (sc-7382HRP; 1:100 dilution), anti-SQSTM1 (H00008878-M01; 1:3,500 dilution), anti-LC3B (Novus Biologicals, NB100-2220; 1:1,000 dilution), and anti-ACTB/β-actin HRP (Santa Cruz Biotechnology, sc-8432HRP; 1:2,000 dilution).
Western blot analysis and coimmunoprecipitation experiments
For western blot analyses, approximately 5 × 105 cells were washed with cold phosphate-buffered saline (PBS; Gibco-Invitrogen, 10010023) and lysed using standard procedures. Twenty-five μg of proteins were separated according to molecular weight on NuPAGE Novex Bis-Tris 4–12% precast gels (Invitrogen, NW04120BOX and NP0321BOX) and electrotransferred to ImmobilonTM PVDF membranes (Bio-Rad, 162-0176 and 162-0177). Nonspecific binding sites were blocked with 5% nonfat powdered milk (w:v) plus 0.05% Tween 20 (v:v; Sigma, P2287 or P1379) in Tris-Buffered Saline (50 mM Tris-Cl, pH 7.6; 150 mM NaCl) for 1 h, followed by overnight incubation at 4°C with primary antibodies. Bands were detected using appropriate horseradish peroxidase (HRP)-labeled secondary antibodies (Southern Biotech, 1030-05 and 4030-05) coupled with the SuperSignal West Pico chemoluminiscent substrate (Thermo Scientific-Pierce, 34077). Either GAPDH or ACTB antibodies were used to monitor equal loading of lanes.
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldeyde (w:v in PBS) for 15 min at room temperature. Nuclear staining was performed using Hoechst 33342 (Molecular Probes-Invitrogen, H1399). Nonautomated fluorescence microscopy assessments were performed on an IRE2 microscope (Leica Microsystems, Wetzlar, Germany) equipped with a DC300F camera. For automated immunofluorescence microscopy, images from plates processed as described above were acquired using a BD pathway 855 automated microscope (BD Imaging Systems, San Jose, CA, USA) equipped with a 40× objective (Olympus, Center Valley, CA, USA) and coupled to a robotized Twister II plate handler (Caliper Life Sciences, Hopkinton, MA, USA). Images were then analyzed either for the presence of GFP-LC3B+ (green) puncta in the cytoplasm and for nuclear staining by means of the BD Attovision software (BD Imaging Systems). The surface of each cell was segmented and subdivided into a cytoplasmic and a nuclear region according to the manufacturer's standard protocol. The RB 2 × 2 and Marr-Hildreth algorithms were used to quantify the number of cytoplasmic GFP-LC3B+ puncta per cell.
Flow cytometry
Cell viability was determined using propidium iodide (Molecular Probes, P3566; 1 mg/ml). Cells were trypsinized, labeled with the fluorochrome at 37°C, and analyzed by flow cytometry using a fluorescence-activated cell sorter (FACS-Scan, Becton Dickinson, La Jolla, CA, USA).
Statistical analyses
Data are reported as mean ± s.d. of triplicate determinations and all experiments were repeated at least 2 times, with each independent experiment yielding similar results. Data were analyzed using Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) or Excel (Microsoft Co., Redmond, WA, USA) and statistical significance was assessed using a 2-tailed Student t test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Douglas Green (St. Jude Hospital, Memphis) and Bert Vogelstein (John Hopkins Hospital, Baltimore) for providing critical reagents, and Haley Harrington for assistance with manuscript preparation.
Funding
This work was supported by CPRIT grant RP120718-P1 (B.L.), NIH grants R01CA109618 and U19AI109725 (B.L.), and GK is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008; 15:344-57; PMID:18804433; http://dx.doi.org/10.1016/j.devcel.2008.08.012
- Fu LL, Cheng Y, Liu B. Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol 2013; 45:921-4; PMID:23420005; http://dx.doi.org/10.1016/j.biocel.2013.02.007
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355-62; PMID:20356743; http://dx.doi.org/10.1016/j.tcb.2010.03.002
- Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008; 27 Suppl 1:S137-48; PMID:19641499; http://dx.doi.org/10.1038/onc.2009.51
- He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 2010; 22:140-9; PMID:20097051; http://dx.doi.org/10.1016/j.ceb.2010.01.001
- Abrahamsen H, Stenmark H, Platta HW. Ubiquitination and phosphorylation of Beclin 1 and its binding partners: Tuning class III phosphatidylinositol 3-kinase activity and tumor suppression. FEBS Lett 2012; 586:1584-91; PMID:22673570; http://dx.doi.org/10.1016/j.febslet.2012.04.046
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/10.1016/j.cell.2013.08.015
- Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013; 494:201-6; PMID:23364696; http://dx.doi.org/10.1038/nature11866
- Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J 2010; 29:515-6; PMID:20125189; http://dx.doi.org/10.1038/emboj.2009.377
- Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 2010; 29:1717-9; PMID:20101204; http://dx.doi.org/10.1038/onc.2009.519
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/10.1126/science.1225967
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4:600-6; PMID:18497563; http://dx.doi.org/10.4161/auto.6260
- Decuypere JP, Parys JB, Bultynck G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012; 1:284-312; PMID:24710477; http://dx.doi.org/10.3390/cells1030284
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/10.1038/sj.emboj.7601689
- Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Marino G, BenYounes A, Bénit P, Rustin P, Maiuri MC, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011; 30:3918-29; PMID:21460857; http://dx.doi.org/10.1038/onc.2011.104
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/10.1038/embor.2008.246
- Gurkar AU, Chu K, Raj L, Bouley R, Lee SH, Kim YB, Dunn SE, Mandinova A, Lee SW. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun 2013; 4:2189; PMID:23877263; http://dx.doi.org/10.1038/ncomms3189
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/10.1016/j.molcel.2008.06.001
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511-5; PMID:22258505; http://dx.doi.org/10.1038/nature10758
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008; 4:989-97; PMID:18797192; http://dx.doi.org/10.4161/auto.6803
- Su M, Mei Y, Sanishvili R, Levine B, Colbert CL, Sinha S. Targeting gamma-herpesvirus 68 Bcl-2-mediated down-regulation of autophagy. J Biol Chem 2014; 289:8029-40; PMID:24443581; http://dx.doi.org/10.1074/jbc.M113.515361
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 2007; 282:13123-32; PMID:17337444; http://dx.doi.org/10.1074/jbc.M700492200
- Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL's target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol 2007; 372:223-35; PMID:17659302; http://dx.doi.org/10.1016/j.jmb.2007.06.069
- Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS, Jung JU, Oh BH. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS pathogens 2008; 4:e25; PMID:18248095; http://dx.doi.org/10.1371/journal.ppat.0040025
- Lindqvist LM, Heinlein M, Huang DC, Vaux DL. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci U S A 2014; 111:8512-7; PMID:24912196; http://dx.doi.org/10.1073/pnas.1406425111
- Lindqvist LM, Vaux DL. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy 2014; 10:1474-5; PMID:24991825; http://dx.doi.org/10.4161/auto.29639
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind to prosurvival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001; 15:1481-1486; PMID:11410528; http://dx.doi.org/10.1101/gad.897601
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 2013; 19:1478-88; PMID:24141421; http://dx.doi.org/10.1038/nm.3322
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science 2000; 290:989-92; PMID:11062132; http://dx.doi.org/10.1126/science.290.5493.989