
Abstract
Levels of autophagy markers rise upon treatment of cells with antidepressants. However, it was not known whether this phenomenon might be linked to other antidepressant pathways or to any physiological effect. In this punctum, we summarize and discuss our recent findings that provide evidence for a role of the cochaperone FKBP5/FKBP51 (FK506 binding protein 5) in autophagy as a prerequisite for antidepressant action in cells, mice, and humans. FKBP5 associates with BECN1, changes its phosphorylation and protein levels and enhances markers of autophagy and autophagic flux. The effects of antidepressants on autophagy as well as their physiological effects in mice and human depend on FKBP5.
Depression is a devastating mental disorder affecting millions of people worldwide. Overall, currently available antidepressants are effective, but less than half of the patients reach sustained remission. The development of new antidepressants is at least partially hampered by the fact that the mechanism of action of the current drugs is still incompletely understood. Autophagy is one of the molecular mechanisms that have been linked to antidepressants. Genetic studies in humans provided evidence that the HSP90 cochaperone FKBP5 is linked to antidepressant treatment response. FKBP5 is known as a regulator of NR3C1/glucocorticoid receptor and thus of the physiological stress response. More recently, additional molecular functions of FKBP5 have been reported, among them the inhibition of AKT1 through recruitment of the phosphatase PHLPP (PH domain and leucine rich repeat protein phosphatase). AKT1, in turn, controls the activity of several targets, including BECN1, an established regulator of autophagy.
We have thus embarked on this study to evaluate a potential role of FKBP5 in autophagy and to test whether this might underlie the link of FKBP5 to antidepressant action. Our goal was a translational study combining molecular and cellular analyses with animal and human studies.
Using co-immunoprecipitation in extracts from cells and from mouse brain tissue we revealed that FKBP5 associates with BECN1, in addition to the previously described interactors AKT1 and PHLPP. FKBP5 also promotes association of AKT1 with BECN1 and at the same time dephosphorylation of BECN1. Mechanistically, this can be explained by the inactivation of AKT1 by FKBP5 through dephosphorylation of AKT1 by corecruitment of PHLPP. Furthermore, FKBP5 increases the levels of BECN1, LC3B-II/-I, and ATG12, and enhances the autophagic flux.
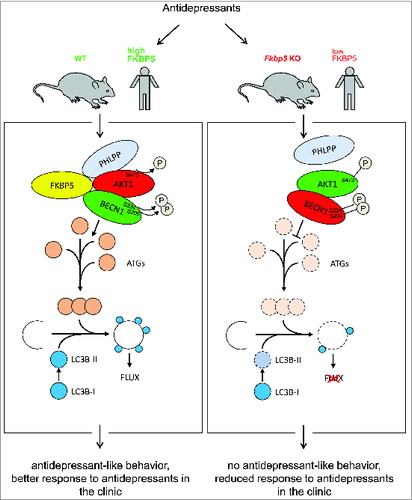
In cells, the antidepressants paroxetine and amitriptyline also increase the levels of BECN1, LC3B-II/-I, and ATG12, decrease the phosphorylation of AKT1 and enhance the autophagic flux. Ectopic overexpression of FKBP5 further stimulates these antidepressant effects, while deletion of FKBP5 largely abolishes them. To assess a possible synergism between FKBP5 and antidepressants on autophagy in mice, we made use of Fkbp5 knockout mice. Treatment with antidepressants leads to decreased phosphorylation of AKT1 and to higher levels of the autophagic markers BECN1, LC3B-II/-I and ATG12 in the brains of wild-type mice, but not in mice without FKBP5. Intriguingly, in parallel to the loss of the effect of antidepressants on markers of autophagy, there is no effect on depression-like behavior in mice without FKBP5, but only in wild-type mice.
To test whether these findings can be translated to humans, we first determined the levels of FKBP5 and markers of autophagy in peripheral blood mononuclear cells (PBMCs) from a group of healthy subjects. The levels of FKBP5 positively correlate with the abundance of the autophagy markers BECN1, LC3B-II/-I, and ATG12, and show a negative correlation with the phosphorylation of AKT1, consistent with a role of FKBP5 as an autophagy inducer. Next, we treated the PBMCs ex vivo with antidepressants; we found that the extent of the response of the autophagy markers and phosphorylation of AKT1 to this treatment correlates with the protein levels of FKBP5 in these PBMCs. When we used PBMCs from depressed patients for the same type of experiments, not only are the same correlations manifested, but there is also a strong correlation between the cellular ex vivo responsiveness of autophagic markers to antidepressants with the clinical treatment success of the respective patients. In other words, the inducibility of autophagic markers is predictive for the clinical outcome.
Taken together, our findings shed light on a novel mechanism of FKBP5 in priming autophagic pathways which appears to set the stage for antidepressant action (see for a graphical summary). Physiological and clinical relevance is indicated by the animal experiments and by the results in a human sample of depressed patients pointing to the ex vivo antidepressant responsiveness of autophagy proteins as predictive markers for clinical treatment outcome.
Figure 1. Model of FKBP5′s role in directing antidepressant effects to autophagy pathways. FKBP5 forms a complex with BECN1, AKT/PKB, and the PHLPP phosphatase. PHLPP dephosphorylates AKT1 at serine 473 and thereby reduces its kinase activity. Inactive AKT1 is recruited to BECN1, resulting in less phosphorylation at serine 234 and serine 295, which triggers downstream signaling of ATG genes. Antidepressants affect the same pathways depending on the expression level of FKBP5. This mechanism could explain the FKBP5 dependency of antidepressants’ effect in cells, animals, and humans.
Another physiological aspect of our results emerges from the fact that FKBP5 is not only a regulator of NR3C1, but also a target of this transcription factor. Thus, we speculate that FKBP5 may mediate the effects of stress on autophagy. In our study, we provide experimental evidence for this by showing that the synthetic glucocorticoid dexamethasone enhances markers of autophagy in wild-type cells, but not in cells devoid of FKBP5.
Several questions and research directions arise from our findings: On the molecular and mechanistic level, it would be interesting to learn which domains of FKBP5 are involved. Does it act as a cochaperone of HSP90? Is FKBP5 involved in both macroautophagy and chaperone-mediated autophagy? Is its close homolog FKBP4/FKBP52 also able to elicit autophagic processes? Moreover, how does FKBP5 increase the levels of BECN1? Elucidation of this mechanism might yield a novel way of inducing autophagy. It will also be of interest to determine which antidepressants elicit autophagy and which use other mechanisms. Further, while our data provide strong evidence that autophagy is required for the action of at least some antidepressants, we still cannot exclude the possibility that autophagy is a mere epiphenomenon. Therefore, it will be important to assess antidepressant-like properties of autophagy inducers. Finally we would like to mention the possibility that it might not be the classical effect of autophagy—providing energy, maintaining protein and organelle homeostasis—that converges with antidepressant actions, but rather the morphological/mechanistic aspects of autophagy resulting in membrane reorganization and thus altered electrophysiological processes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Gassen NC, Hartmann J, Zschocke J, Stepan J, Hafner K, Zellner A, Kirmeier T, Kollmannsberger L, Wagner KV, Dedic N, et al. Association of FKBP51 with Priming of Autophagy Pathways and Mediation of Antidepressant Treatment Response: Evidence in Cells, Mice, and Humans. Nestler E, ed. Plos Medicine 2014; 11(11):e1001755; http://dx.doi.org/10.1371/journal.pmed.1001755