
Abstract
The discovery that heterozygous disruption of Becn1 in mice leads to spontaneous tumor development was the first direct link arguing for a predominantly tumor-suppressor function of autophagy. However, the mechanisms by which BECN1 restrains tumorigenesis and whether autophagy-independent functions of BECN1 contribute to its tumor-restraining potential remained unexplored. We recently described a novel function of BECN1—regulation of oncogene MCL1 protein levels. Our results show that BECN1 regulates MCL1 levels in an inverse-reciprocal manner, whereby changes in the levels of one of the 2 proteins inversely affects the proteasomal degradation of the other. Importantly, this mechanism is independent of autophagy and of the physical interaction between BECN1 and MCL1. In vitro and in vivo analysis using several models, including patient-derived melanoma cells and tissue samples from patients with melanocytic lesions at different stages, showed that the identified mechanism of inverse coregulation between BECN1 and MCL1 significantly contributes to their opposing roles in tumorigenesis.
Keywords:
Whether autophagy acts to promote or suppress tumorigenesis has been the subject of a long debate with evidence supporting both sides of the argument. The first direct link between autophagy and tumorigeneisis was the correlation between Becn1/BECN1 monoallelic deletion and development of tumors in mice and several types of human cancer. Due to the essential role of BECN1 in autophagy, these findings were presented, and remain so far, as one of the strongest lines of evidence arguing for a mainly tumor-suppressive role of autophagy. However, whether autophagy solely accounts for BECN1 tumor suppression capacity remained unexplored. Comparative analysis of mouse models disrupted for either Becn1 or other autophagy regulators (e.g. Atg5, Atg7) led to the speculation that BECN1 plays unique roles in autophagy-independent processes that may contribute to its tumor-restraining potential. However, very little is known about such functions of BECN1 and their relevance to tumor suppression. Moreover, while the mechanistic functions of BECN1 in the process of autophagy are well studied, less is known about the mechanisms that fine-tune the levels of BECN1 itself.
The discovery that BECN1 binds BCL2, BCL2L1/Bcl-xL, and MCL1, was a landmark finding that linked the fields of apoptosis and autophagy. Ever since, subsequent studies focused on the implications of disrupting these interactions on the induction of autophagy in different contexts and by different stimuli such as activation of certain oncogenes (e.g., HRAS) or treatment with some drugs. Ectopic overexpression of anti-apoptotic members of the BCL2 family inhibits starvation-induced, BECN1-mediated autophagy. In our study, we initially assessed the contribution of the endogenous levels of those proteins to the regulation of basal, housekeeping autophagy. Depletion of MCL1 is sufficient on its own to induce basal autophagy in cells cultured under nutrient-rich conditions, and this phenomenon is associated with an increase in BECN1 levels. Ablation of either BCL2 or BCL2L1, however, does not exert similar effects. Upregulation of BECN1 is frequently observed in response to several other autophagy-inducing stimuli. The specific link between the levels of MCL1 and BECN1 was further confirmed by stabilizing MCL1 through depletion of the E3 ligase HUWE1/Mule or PMAIP1/Noxa, both involved in MCL1 degradation. In each case, varying magnitudes of MCL1 stabilization result in a tightly corresponding decrease in BECN1 levels. Finally, this tight inverse correlation became evident by observing that ablation of BECN1, but not ATG5 or ATG7, leads to a dramatic increase in MCL1 levels.
Our dissection of the molecular mechanisms underlying this coregulation ruled out transcriptional or translational regulation. As we observed that the 2 proteins are degraded mainly through the proteasome, we focused on the possibility of mutual modulation of ubiqutination and proteasomal degradation. As neither MCL1 nor BECN1 are known to exhibit direct ubiquitin ligase or deubiquitinase activity, we explored the possibility that they may indirectly regulate the ubiquitination of each other through modulating the interaction or the function of other ubiquitination regulators. Mass spectrometric analysis of entire MCL1 and BECN1 interactomes revealed that the deubiquitinase USP9X interacts with both MCL1 and BECN1. Interestingly, the interaction of either MCL1 or BECN1 with USP9X and the inverse reciprocal regulation is independent of MCL1-BECN1 physical interaction as disruption of MCL1-BECN1 binding does not affect their individual interaction with USP9X. Further analysis showed that both proteins compete for binding the same region on USP9X and thereby binding of one displaces the other with subsequent increased ubiquitination and proteasomal degradation. This mechanism of regulation is thus distinguished from the already established link between BECN1 and BCL2 proteins.
Elevation of MCL1 levels contributes to chemoresistance and relapse in several types of tumors. Due to its key anti-apoptotic properties, tumor-promoting functions of MCL1 have been largely attributed to evading apoptosis. However, emerging reports now suggest apoptosis-independent roles for MCL1 in the regulation of cellular metabolism, which may contribute to its tumorigenic capacity. Melanoma is an example of tumors in which MCL1 plays crucial tumor-promoting roles.
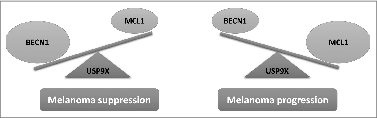
Our in vitro and in vivo experiments using several models, including patient-derived melanoma cells and analysis of tissue samples from benign nevi, primary, and metastatic melanomas show a strong negative correlation between MCL1 and BECN1 levels and demonstrate that the MCL1-BECN1 axis is specifically modulated during melanoma progression. As tumors progress to a more malignant phenotype, the levels of BECN1 decrease and MCL1 subsequently increase in a significant interdependent manner. This inverse correlation was further evident by examining normal and malignant tissues from the same melanoma patients, which revealed that the MCL1-BECN1 axis is tipped toward an increase in BECN1 in normal tissues and in MCL1 in tumors ().
Figure 1. Schematic presentation of the reciprocal regulation and functional counteraction of the MCL1-BECN1 axis in melanoma. High levels of the tumor suppressor BECN1 maintain the levels of the oncogene MCL1 in check. Imbalance toward a decrease in BECN1 and subsequent increase in MCL1 levels is associated with melanoma progression.
In conclusion, our work unravels unexplored areas of the contribution of BECN1 and MCL1 to tumorigenesis that is far from the traditional view of these 2 proteins. Mediating the proteasomal degradation of MCL1 according to the mechanism described is thus a novel autophagy-independent function of BECN1 that contributes to its tumor-suppressive functions. From another angle, mutations that lead to increased MCL1 levels may contribute to tumorigenesis by destabilizing BECN1 and thus evading its tumor-suppressive functions.