
Abstract
The autophagy-lysosomal pathway (ALP) regulates cell homeostasis and plays a crucial role in human diseases, such as lysosomal storage disorders (LSDs) and common neurodegenerative diseases. Therefore, the identification of DNA sequence variations in genes involved in this pathway and their association with human diseases would have a significant impact on health. To this aim, we developed Lysoplex, a targeted next-generation sequencing (NGS) approach, which allowed us to obtain a uniform and accurate coding sequence coverage of a comprehensive set of 891 genes involved in lysosomal, endocytic, and autophagic pathways. Lysoplex was successfully validated on 14 different types of LSDs and then used to analyze 48 mutation-unknown patients with a clinical phenotype of neuronal ceroid lipofuscinosis (NCL), a genetically heterogeneous subtype of LSD. Lysoplex allowed us to identify pathogenic mutations in 67% of patients, most of whom had been unsuccessfully analyzed by several sequencing approaches. In addition, in 3 patients, we found potential disease-causing variants in novel NCL candidate genes. We then compared the variant detection power of Lysoplex with data derived from public whole exome sequencing (WES) efforts. On average, a 50% higher number of validated amino acid changes and truncating variations per gene were identified. Overall, we identified 61 truncating sequence variations and 488 missense variations with a high probability to cause loss of function in a total of 316 genes. Interestingly, some loss-of-function variations of genes involved in the ALP pathway were found in homozygosity in the normal population, suggesting that their role is not essential. Thus, Lysoplex provided a comprehensive catalog of sequence variants in ALP genes and allows the assessment of their relevance in cell biology as well as their contribution to human disease.
Introduction
The lysosome plays a central role in cellular clearance and energy metabolism by ensuring degradation and recycling of a variety of substrates. While extracellular substrates reach the lysosome via the endocytic and phagocytic pathways, intracellular material follows the autophagic pathway, mainly through the fusion of autophagosomes with lysosomes.Citation1-4 In addition, recent studies have revealed that the lysosome is also involved in signaling pathways that play a crucial role in cell homeostasis and growth.Citation1
The physiological importance of lysosome-mediated pathways is underlined by their involvement in human diseases. Lysosomal storage diseases are due to mutations in more than 50 genes encoding lysosomal soluble acidic hydrolases, integral membrane proteins, activators, transporters, or nonlysosomal proteins that are necessary for lysosomal function.Citation5–7 The precise molecular diagnosis is very important as new therapies are becoming available for specific types of LSDs.Citation8 Unfortunately, for the majority of these disorders, the diagnosis is difficult due to considerable clinical overlap and clinical variability. This is particularly true for neuronal ceroid lipofuscinosis, a genetically heterogeneous subtype of LSDs with a devastating phenotype that can be caused by mutations of at least 13 different genes.Citation9 Mutations in the autophagy-lysosomal (ALP) pathway can also cause common neurodegenerative disorders, such as Parkinson and Alzheimer diseases.Citation10-15 It is likely that mutations in the ALP pathway will be identified in an increasing number of diseases.
Next-generation sequencing is revolutionizing our view of life sciences by allowing the investigation of variations in DNA/RNA sequences at a genome scale.Citation16 In the last few years, whole exome sequencingCitation17 or whole genome sequencing (WGS)Citation18 approaches have received unlimited consideration as universal tests for most Mendelian conditions, with the exceptions of those caused by complex structural variations. As a cheaper and more focused alternative to WGS, WES has been demonstrated to be an excellent and cost-effective solution with a consistent number of advantages and, above all, a higher throughput on coding sequences that favors the identification of novel disease genes. However, accumulating data indicate that WES may not be a totally reliable tool to assess sequence variations in a given gene. Efficiency of WES is undermined by nonuniform targeting and coverage that, for a significant fraction of the exome, is too low to allow thorough variant detection.Citation19 In addition, false positive calls are frequently observed in segmental duplications and processed pseudogenes. This reduces WES sensitivity as well as specificity, leading to labor-intensive validation steps usually based on conventional PCR reactions and Sanger sequencing. For practical reasons, the validation steps are usually restricted to specific genes, missing the remaining variability of the exome. Finally, there is an increasing need to properly manage incidental findings detected by WES/WGS.Citation20
NGS-based targeted approaches, i.e. the sequencing of selected genes and genomic regions of interest, have been developed as an alternative to WES/WGS.Citation19 Using targeted approaches the ethical issues related to incidental findings are very limited. Our aim was to develop a robust sequencing platform to identify the sequence variations in the ALP pathway. A similar approach was used to detect sequence variations in mitochondrial pathways.Citation21,22
A potential obstacle to building a panel of bona fide ALP genes is represented by the fact that the identity of many key players is still unknown. Therefore, we used multiple bioinformatic tools to identify a comprehensive collection of ALP genes. The final, nonredundant, ALP list contains 891 genes, including 106 autophagy genes, 194 lysosomal genes, and 627 genes with a role in the endocytic pathway ().
Figure 1. Subsets of genes that are part of the ALP gene list. Schematic diagram showing the composition of the Lysoplex gene list (n = 891 genes). Please note that some of the selected genes belong to more than one category.
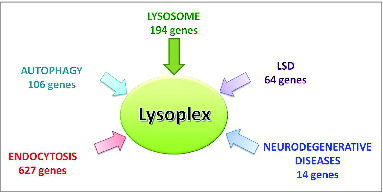
Approximately 140 out of the total 891 genes, are mutated in human Mendelian diseases. We used the Haloplex enrichment protocol, which permitted accurate sequencing and high sequence coverage.Citation23 This workflow was named Lysoplex.
Results
Generation of the ALP gene list
We selected the following sets of genes to generate the Lysoplex list: 1) a subset of genes encoding proteins with either an established role in autophagy or predicted by at least 2 of the following bioinformatics tools: AmiGO,Citation24 Netview,Citation25 UniProt,Citation26 and MSigDBCitation27 (see also Materials and Methods and Table S1); 2) a set of genes encoding proteins with lysosomal localizationCitation28-31 selected using a similar procedure involving both literature searchCitation32-34 and bioinformatic predictions; 3) all genes that are mutated in LSDs, including those coding for nonlysosomal proteinsCitation35,36 and the transcription factor TFEB, a master regulator of lysosomal biogenesis and autophagy,Citation1,28,37-40 as well as 3 closely related paralogs (Mit/TFE members);Citation41 4) genes with a known/predicted role in the endocytic pathway;Citation42 5) genes involved in familial forms of ParkinsonCitation43 and Alzheimer diseases.Citation44 As a result, the ALP list includes 891 genes (Table S1), for which all predicted exons and at least 10 nucleotides of flanking intronic sequences were included. The target region collectively spans 2.054 megabases of human DNA coding sequence ().
Lysoplex design and validation with mutation-known samples
To obtain a homogeneous and accurate coverage of the 891 ALP genes selected as indicated above, we designed capture oligonucleotide probes using the HaloPlex™ Target Enrichment System, which represents an excellent technology for complex targeting with high sensitivity and specificity.Citation23 This procedure is based on restriction enzyme digestion of genomic DNA with multiple endonucleases and hybridization capture with probes that also work in the subsequent amplification step. We used this platform to sequence ∼100 DNA samples. On average, sequence reads that mapped to the ALP gene target represented more than 92% of total reads (), showing efficient and specific probe capture. This minimal “off-targeting” reduces the sequencing demand in comparison with other enrichment protocolsCitation45 and increases on-target coverage. Using a high-throughput setting of 32 libraries/lane, the average coverage depth was 40x for 95% and 100x for 80% of the ALP-target, which is significantly higher than the average depth we reached on the same genes with WES (). This setting led to a high specificity in variant calls, as demonstrated by the validation of 128/128 well covered (n > 20) variants tested by Sanger sequencing (100%).
Figure 2. Performance of the Lysoplex toolkit. (A) Average yield of the Lysoplex capture. More than 92% of sequence reads truly map on ALP targets, indicating a very limited off-targeting in comparison with other methods. (B) Comparison between Lysoplex and WES. Comparison of the coverage distribution on all the 891 ALP genes between whole exome sequencing (blue boxes) and Lysoplex (red boxes). (C, D) Comparison of sequence coverage between WES and Lysoplex in 2 disease genes, namely CLN3 (C) and MTMR2 (D). Red boxes, Lysoplex; blue boxes, WES. Bold lines represent the median of all samples tested, the boxes include 50% of the values and the whiskers represent minimum and maximum values, excluding the outliers that are plotted individually.
To test the sensitivity of Lysoplex, we utilized a training set of 16 genomic DNAs from patients affected by 14 different forms of LSDs, whose molecular diagnosis was already known except for 2 cases (Table S2). These patients were affected by the following types of LSDs: Mucopolysaccharidoses (MPS) types I, II, IIIB and VI, Fabry disease, Niemann-Pick disease type C1 (NPC1), mucolipidosis type III (MLIII), late infantile neuronal ceroid lipofuscinosis 5 (CLN5), Gaucher disease, multiple sulfatase deficiency (MSD), metachromatic leukodystrophy (MLD), Pompe disease, and Danon disease.
The application of the Lysoplex procedure to the training set of 16 LSD patients allowed us to detect all of the previously known mutations and to identify the disease-causing mutations in the 2 patients in whom molecular diagnosis had not been previously made (Table S2).
Lysoplex application to molecular diagnosis of NCLs
To test the efficacy of Lysoplex in the analysis of patients with unknown/elusive molecular defects, we applied it to the search for disease-causing mutations in patients with clinically diagnosed NCLs, which are caused by mutations in at least 13 different genes.Citation9,46 We carried out Lysoplex on 48 NCL patients, whose gene defect had not been previously detected (Table S3).
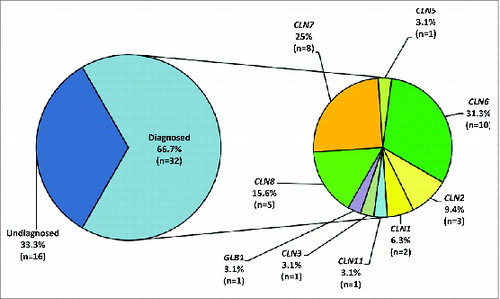
describes in “exploded pies” the success rate of Lysoplex to identify causative variants in the NCL patients. We detected both pathogenic variants (homozygous or compound heterozygous) in 29/48 cases. In 3 additional cases (; L33, L57 and L58), we only found a single heterozygous mutation in known NCL genes (CLN3, CLN6), which were compatible with the observed phenotype. We postulate that these patients may harbor a second mutation in the same gene that is not detectable, e.g., a deep intronic mutation or a large copy number variation.
Figure 3. Success rate of Lysoplex in NCL patients. Schematic diagram summarizing the results of the Lysoplex procedure in the NCL patients analyzed. An overall detection rate of 67% was observed. In particular, mutations were identified in 8 NCL known genes. In one case, we found pathogenic mutations in the GLB1 gene (see text for details).
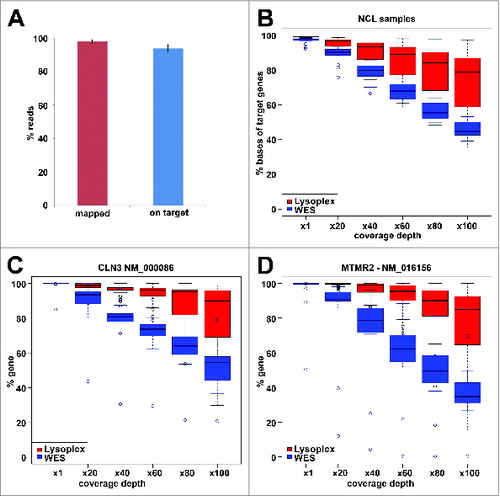
Table 1. Summary of mutations identified in NCL patients analyzed
In total, pathogenic mutations were identified in 32 NCL patients in the following disease genes: CLN1 (n = 2), CLN2 (n = 3), CLN3 (n = 1), CLN5 (n = 1), CLN6 (n = 10), CLN7 (n = 8), CLN8 (n = 5), CLN11 (n = 1) and GLB1 (n = 1). Patient L44 turned out to be compound heterozygous for 2 mutations in the GLB1 gene, encoding β-galactosidase. Interestingly, mutations in GLB1 are responsible for GM1-gangliosidosis,Citation47 a neurodegenerative type of LSD whose phenotype partially overlaps with that of CLN1. Overall, in all patients analyzed, the molecular diagnosis was in agreement with the clinical diagnosis, taking into account the phenotypic variability observed in NCLs. This is further exemplified by the fact that mutations in the CLN6 gene can cause both late infantile as well as adult NCL phenotypes ().
Of note, patient L42 had been previously proposed to represent a novel CLN locus, namely CLN9,Citation48 based on a distinctive fibroblast phenotype, gene expression pattern, ultrastructural analysis of storage material, enzymatic activities, and sequencing of NCL genes. However, we found that this patient carries a homozygous CLN2 mutation.
Taken as a whole, the use of Lysoplex allowed us to identify causative mutations in 67% of the NCL patients () Eighteen patients had never received any test (“naïve” group), whereas the remaining 30 individuals had been tested for some NCL genes (“pre-screened”) but without positive findings. Interestingly, one pre-screened family had previously been studied by both linkage analysis and WES (; patients L37 and L38). There was, however, no substantial difference in the success rate between the pre-tested and the naïve group.
Interestingly, in addition to the causative mutations in a given disease gene, we also identified in some patients additional mutations in genes involved in other diseases (Table S4). This may add further complexity to the genotype/phenotype correlation.
We then looked for recessive mutations in novel NCL candidate genes, which could exert a pathogenic role in the 16 patients in whom Lysoplex could not identify any mutations in known disease genes. We focused our attention on loss-of-function variants such as frame-shift, nonsense, splice defects, and potentially damaging nonsynonymous amino acid substitutions in nondisease genes. We considered as novel candidate NCL genes those that showed these variants on both alleles in at least one NCL patient or those mutated in 2 unrelated patients. Following these selection criteria, we identified 3 potential candidate genes: STAB2 (stabilin 2), AGAP1 (ArfGAP with GTPase domain, ankyrin repeat and PH domain 1) and PLCG2 (phospholipase C, gamma 2 [phosphatidylinositol-specific]) (Table S5). However, functional analyses and in vivo studies in animal models are needed before we can conclude that these are causative mutations. Overall, we identified 37 different pathogenic mutations, including 21 novel mutations. They include 25 amino acid changes, 5 indels (insertions or deletions) including an insertion of 17 nucleotides, 5 nonsense mutations, and 2 splice site variants ().
Sequence variation in ALP genes
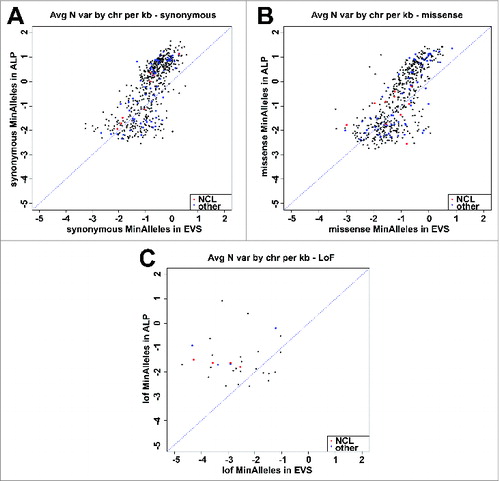
To assess the efficiency of Lysoplex in detecting sequence variations in ALP genes, we compared its performance to pre-existing public resources, primarily the Exome Variant Server (EVS). To avoid any bias due to sample selection, we first counted the number of synonymous variations/allele/kb in our patient cohort as compared with EVS data. We plotted each gene as a dot in a logarithmic scale where the X-axis represents the number of sequence variations/allele/kb in EVS and the Y-axis represents the same value obtained in the Lysoplex panel (). This analysis revealed that the vast majority of genes show a much higher number of variations in the Lysoplex samples vs. EVS. These data were also confirmed for all other categories of sequence substitutions, including missense and loss-of-function mutations (). This demonstrates that Lysoplex is much more sensitive than EVS in detecting sequence variability in ALP genes. We did not consider small insertions or deletions that grossly deviate from Hardy-Weinberg equilibrium, since it is well known that a high number of false positive variants are erroneously annotated in EVS and in all other databases.
Figure 4. Comparison between the number of sequence variations found by Lysoplex and WES. Plotted data indicate the log of the number of variations/kb found in each gene by Lysoplex (Y-axis) and by public database (X-axis). If the sensitivity was identical, dots should tend to align. Dots above the line indicate a superior sensitivity of Lysoplex. Red dots indicate NCL genes and blue dots other disease genes. (A) synonymous variations; (B) missense variations; (C) loss-of-function variations.
Overall, we found 61 clear loss-of-function mutations (i.e., nonsense, indels leading to frameshifts and splice variants) and 488 missense variations with a high probability to exert a damaging effect on the corresponding protein product. The above mutations were found in a total of 316 different genes (Table S6). Interestingly, some of the loss-of-function variations were also found in the normal population at a frequency higher than 1% (). Some of the genes (n = 6) carrying common loss-of-function variations in the normal population are involved in autophagy (ATG3), endocytosis (ATP6V1G3, CBLC, SEC14L4) and membrane trafficking (INPP5D). These findings suggest that total loss-of-function in these genes does not appear to have major effects.
Table 2. List of common loss-of-function sequence variations (frequency > 1%) in ALP genes
Discussion
In the past few years, remarkable progress has been made in the characterization of the autophagic lysosomal pathway, discovering wide-ranging pathogenic roles in addition to vital homeostatic functions.Citation1,49-51 Autophagy is involved in various conditions such as genetic diseases,Citation52 metabolic disorders,Citation53,54 neurodegenerative diseases,Citation55 infections,Citation56 and cancer.Citation57,58
While great attention has been given to the study of the mechanisms underlying the regulation and execution of autophagy in health and disease, the intrinsic genetic variation of the system in the human population has so far been ignored. Lysoplex was developed to map such genetic variation. When compared with public genome variant databases, the Lysoplex panel provided a significantly more accurate list of genetic variants that may affect the ALP pathways, in normal individuals and in patients.
When applied to the molecular diagnosis of LSDs, Lysoplex revealed a number of significant advantages over WES or WGS approaches. For example, the data load was approximately 1/10 and 1/100 compared to WES and WGS, respectively. This reduces costs and efforts, making Lysoplex applicable to a large number of samples. In addition, Lysoplex proved to be much more robust in terms of target reproducibility, specificity, and sensitivity, and its depth was such that there was no need for Sanger validation steps (i.e. to confirm variations or distinguish between homozygous and heterozygous status). The extent of off-targeting was minimal (8%) although slightly higher than that obtained with Motorplex (2%).Citation23 However, Motorplex covers a target region that is 4-fold smaller than Lysoplex. It is also important to underline that Lysoplex is a very versatile tool as the list of genes to be analyzed can be easily updated as new relevant genes are identified.
In our study we detected all disease-causing mutations in a set of LSD patients. Lysoplex was successful in identifying the molecular defect in 67% of the NCL patients analyzed, which is a significant result when considering both the locus and allele heterogeneity of this class of diseases. Remarkably, patient L42 belongs to one of the 2 pairs of NCL siblings of German and Serbian descent, respectively, who were previously proposed to be affected by mutations in an undefined CLN9 locus.Citation48 Lysoplex analysis allowed us to reclassify L42 as CLN2. Since the other sib pair (of Serbian origin) was reclassified earlier after homozygosity mapping as CLN5,Citation59 no further evidence for an elusive CLN9 gene exists. Two others of the pre-tested patients, L37 and L38 (), had been previously analyzed by WES, but the pathogenic mutations were originally missed due to unclear family history (presence of one additional sibling with an uncertain diagnosis). Furthermore, in one of the patients originally diagnosed as affected by NCL, we identified missense mutations in both alleles of the GLB1 gene, resulting in the molecular diagnosis of GM1 gangliosidosis. This disease is associated with accumulation of saposin and subunit c of the V-ATPase, and is characterized by common manifestation of ER and oxidative stressCitation60–62 with a resulting phenotype that overlaps that observed in NCL. This suggests that some cases with a clinical diagnosis of NCL may indeed have GM1-gangliosidosis. There is another report in which a patient diagnosed with adult NCL was found to carry heterozygous mutations in SGSH defective in MPS IIIA (Sanfilippo syndrome A) and associated with lysosomal accumulation of heparin sulfate.Citation63 Thus, Lysoplex appears to be instrumental for the differential diagnosis and correct classification between different types of LSDs. This result emphasizes the value of testing multiple genes for the same broad class of genetic disease (e.g., LSDs) with partially overlapping phenotypes.
Considering that ALP genes may be candidates for other genetic conditions, and that approximately 85% of them are not yet assigned to any disease, Lysoplex can effectively be used for the discovery of novel disease genes. Lysoplex application to a relatively small set of patients resulted in the identification of several homozygous or compound heterozygous mutations in putative novel disease genes for NCLs. These are: STAB2, that encodes a cell receptor that enables the scavenging and clearance of multiple ligands from the circulation salvaging their building block monomers by lysosomal degradation, as well as MAPK1/ERK2-MAPK3/ERK1 activated signaling,Citation64,65 AGAP1 that encodes an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein,Citation66 and PLCG2 that encodes a transmembrane signaling enzyme whose gain-of-function mutations have been associated with autoimmune and inflammatory diseases mediated by B cells.Citation67 More data, such as the identification of additional families and/or functional assays, are needed to definitely prove the involvement of the above candidate genes in NCL pathogenesis.
Another application of Lysoplex may be in the field of modifier genes, complex disorders, and polygenic inheritance. It is well known that patients that share the same mutations may have different phenotypic spectrum. Thus, the effect of the primary molecular defects may be modified by the presence of additional and variable elements located in other genes that encode proteins involved in the same pathways. Remarkably, we have identified additional mutations in other disease genes in patients harboring pathogenic mutations in NCL genes (Table S4). This may represent disease modifiers that explain intra-familial phenotypic variability. However, to test their role as modifier genes, genotype-phenotype correlation studies are needed on a much larger number of cases.
There is increasing evidence that mutations in genes involved in the ALP pathway may predispose to common neurodegenerative diseases.Citation1 Cohorts of patients affected by common neurodegenerative diseases, such as Parkinson and Alzheimer diseases, may be screened using Lysoplex to identify additional genes predisposing to such diseases. It may also be interesting to use Lysoplex for the screening of somatic mutations in ALP genes in tumor samples due to the known involvement of autophagy in cancer.Citation57
Finally, Lysoplex is an exceptionally efficient tool to map amino acid sequence variations in proteins encoded by ALP genes. Such amino acid sequence variations may have functional relevance. Indeed, we have observed a high frequency of rare, nonconserved, amino acid changes as well as truncating variants. The observation of such protein variation may not be a total surprise, considering the predicted human genome variability, but it is remarkable considering the crucial roles of ALP genes in cell homeostasis. Much of this variability is overlooked when performing global WES or WGS approaches. In practice, WES or WGS data are usually filtered and the discordant variability is not dissected gene by gene according to function. Furthermore, when the coverage is low, the variability is always submerged by numerous sequencing errors and gaps that, in contrast, do not affect Lysoplex.
This catalog of functional variants may serve 2 main purposes. It provides a useful tool for basic researchers to study the biological relevance of protein variants in the ALP pathway. At the same time, clinical researchers can use this catalog to identify candidate causative or modifier genes for human diseases.
Materials and Methods
Patients
The training set of 16 genomic DNA samples belonged to patients affected by 14 different LSDs (Table S2). In all cases, the enzyme activities were measured in cultured fibroblasts, using artificial fluorogenic substrates, according to standard procedures. NCL patients received a diagnosis based on clinical evaluation, biochemical assays, and electron microscopy analysis. Some of the patients had already undergone some molecular tests (). All procedures on patients were approved by the Ethics Boards of the participating institutions and adhered to the tenets of the Declaration of Helsinki.
Selection of Lysoplex genes
The complete gene list includes 891 members (Table S1): 98 previously described lysosomal genes;Citation29 9 genes colocalizing with the lysosomal marker LAMP2;Citation68 the 4 MIT family members;Citation41 all the known lysosomal and nonlysosomal protein-coding genes with a role in the different LSDsCitation36,69 selected by using the Online Mendelian Inheritance in Man (OMIM).Citation70,71
Lysoplex also includes 10 genes involved in hereditary forms of Parkinson disease and 4 involved in hereditary forms of Alzheimer disease. Moreover, bioinformatics prediction tools, including AmiGO,Citation24 Netview,Citation25 and UniProtCitation26 were used. For lysosomal genes, in each database the complete human gene list was downloaded restricting the output to the “lysosome” term: the AmiGO list included 306 genes, the Netview list included 484 genes and the UniProt list included 305 genes. The 3 lists were compared in pairs: 83 genes predicted to be lysosomal by at least 2 out of 3 tools were considered good candidate genes and thus added to the final list. In Table S1 this subset of genes is listed and associated with the specific bioinformatics tools' pair: 16 genes were shared by AmiGO and Netview lists, 63 genes were shared by AmiGO and UniProt lists, one gene was shared by Netview and UniProt lists and 3 genes were shared by all 3 lists.
As for the lysosomal genes, the selection of the 106 genes mainly playing a role in the autophagic pathway was performed by both literature analysisCitation33,34 and bioinformatics tools. In detail, 73 genes were mapped to 12 autophagic subcategories on the GO hierarchy rooted in the “Autophagy”(GO:0006914) accession term; 2 more genes (SNX3 and CHPT1) were introduced because they were predicted as “autophagic vacuole” genes by Netview.
For the selection of genes with a known role in the endocytic pathway and trafficking, both literature analysisCitation42,72–76 (n = 325) and the Molecular Signatures Database (MSigDB)Citation27,77 were used. MSigDB, which is one of the most widely used repositories of well-annotated gene sets, was used to dissect lists of genes included in different “endocytosis” gene sets: we thus found 53 genes annotated by the AmiGO term (GO:0006897) “endocytosis”; 14 genes annotated by the GO term (GO:0030139) “endocytic vescicle”; 23 genes collected in the GO term (GO:0006898) “receptor mediated endocytosis”; 11 genes annotated by the GO term (GO:0030100) “regulation of endocytosis”; 178 genes included in the “endocytosis” term in KEGGCitation78,79 and finally 23 genes included in the ReactomeCitation80 gene set term “transferrin endocytosis and recycling.”
Finally, the information about the specific cellular compartment, biological function and biological process associated with each Lysosplex gene was obtained by EntrezGene.Citation81
Targeted sequencing analysis
The libraries were sequenced using the HiSeq1000 system (Illumina inc., San Diego, CA, USA). The generated sequences were analyzed using an in-house pipeline designed to automate the analysis workflow and composed of modules performing every step using the appropriate tools available to the scientific community or developed in-house.Citation82 Paired sequencing reads were aligned to the reference genome (UCSC, hg19 build) using BWACitation83 and sorted with SAMtoolsCitation84 and Picard (http://picard.sourceforge.net). Genome Analysis Toolkit (GATK)Citation85 with parameters adapted to the Haloplex-generated sequences was then used to perform local realignment around insertions-deletions (indel) and single nucleotide variants (SNV) and small indel calling. The called SNV and indel variants produced with both platforms were annotated using ANNOVARCitation86 with: the relative position in genes using the RefSeqCitation87 gene model, amino acid change, presence in dbSNP v137,Citation88 frequency in NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS), 1,000 genomes project,Citation89 multiple cross-species conservationCitation90,91 and prediction scores of damaging on protein activity.Citation92–96 The annotated variants were then imported in the internal variation database, which stores all the variations found in internal sequencing projects. The database is then queried to generate the filtered list of variations and the internal database frequency in samples with unrelated phenotype is used as further annotation and filtering criterion. The alignments at candidate positions were visually inspected using the Integrative Genomics Viewer (IGV).Citation97
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1043077_supplemental_files.zip
Download Zip (135.9 KB)Acknowledgments
We are grateful to Manuela Dionisi and Annalaura Torella and the TIGEM NGS Core for the NGS data generation and the TIGEM Bioinformatics core for support in sequencing data analysis. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We also thank Nicola Brunetti-Pierri and Antonella de Matteis for help in ALP gene selection. We thank all patients and families as well as referring physicians for providing patient samples and data.
Funding
The research leading to these results has received funding from the European Research Council under the European Research Council (Grant Agreement n. 250154 to AB), from the European Union Seventh Framework Program (Grant agreement n° 281234 to AS and TB), from Fondazione Telethon (grant n. TGM11CB to AB and TGM11Z06 to V.N.), from Pennsylvania University (grant n. MPS I-13-01 to AB), from Team Sanfilippo Foundation and Sanfilippo Children's Research Foundation (to AB) and from Beyond Batten Disease Foundation (to AB).
References
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; PMID:23609508; http://dx.doi.org/10.1038/nrm3565
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Kaushik S, Kiffin R, Cuervo AM. Chaperone-mediated autophagy and aging: a novel regulatory role of lipids revealed. Autophagy 2007; 3:387-9; PMID:17438364; http://dx.doi.org/10.4161/auto.4246
- Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 2011; 7:673-82; PMID:21646866; http://dx.doi.org/10.4161/auto.7.7.14733
- Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 2009; 1793:684-96; PMID:19111581; http://dx.doi.org/10.1016/j.bbamcr.2008.12.001
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 2004; 5:554-65; PMID:15232573; http://dx.doi.org/10.1038/nrm1423
- Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends Neurosci 2011; 34:401-10; PMID:21723623; http://dx.doi.org/10.1016/j.tins.2011.05.006
- Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Ann Rev Med 2015; 66:471-86; PMID:25587658; http://dx.doi.org/10.1146/annurev-med-122313-085916
- Schulz A, Kohlschutter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim Biophys Acta 2013; 1832:1801-6; PMID:23602993; http://dx.doi.org/10.1016/j.bbadis.2013.04.008
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004; 351:1972-7; PMID:15525722; http://dx.doi.org/10.1056/NEJMoa033277
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. New Engl J Med 2009; 361:1651-61; PMID:19846850; http://dx.doi.org/10.1056/NEJMoa0901281
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006; 38:1184-91; PMID:16964263; http://dx.doi.org/10.1038/ng1884
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/10.1126/science.1096284
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011; 89:168-75; PMID:21763483; http://dx.doi.org/10.1016/j.ajhg.2011.06.008
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146-58; PMID:20541250; http://dx.doi.org/10.1016/j.cell.2010.05.008
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2009; 11:31-46; PMID:19997069; http://dx.doi.org/10.1038/nrg2626
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010; 42:30-5; PMID:19915526; http://dx.doi.org/10.1038/ng.499
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008; 456:53-9; PMID:18987734; http://dx.doi.org/10.1038/nature07517
- Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet 2013; 14:295-300; PMID:23478348; http://dx.doi.org/10.1038/nrg3463
- Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, Mullikin JC, Biesecker LG. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet 2012; 91:97-108; PMID:22703879; http://dx.doi.org/10.1016/j.ajhg.2012.05.021
- Dames S, Chou LS, Xiao Y, Wayman T, Stocks J, Singleton M, Eilbeck K, Mao R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J Mol Diagn 2013; 15:526-34; PMID:23665194; http://dx.doi.org/10.1016/j.jmoldx.2013.03.005
- Wong LJ. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion 2013; 13:379-87; PMID:23473862; http://dx.doi.org/10.1016/j.mito.2013.02.001
- Savarese M, Di Fruscio G, Mutarelli M, Torella A, Magri F, Santorelli F, Comi GP, Bruno C, Nigro V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun 2014; 2:100; PMID:25214167; http://dx.doi.org/10.1186/s40478-014-0100-3
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics 2009; 25:288-9; PMID:19033274; http://dx.doi.org/10.1093/bioinformatics/btn615
- Belcastro V, Siciliano V, Gregoretti F, Mithbaokar P, Dharmalingam G, Berlingieri S, Iorio F, Oliva G, Polishchuck R, Brunetti-Pierri N, et al. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res 2011; 39:8677-88; PMID:21785136; http://dx.doi.org/10.1093/nar/gkr593
- Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011; 2011:bar009; PMID:21447597; http://dx.doi.org/10.1093/database/bar009
- Liberzon A. A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol Biol 2014; 1150:153-60; PMID:24743996; http://dx.doi.org/10.1007/978-1-4939-0512-6_9
- Schwake M, Schroder B, Saftig P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013; 14:739-48; PMID:23387372; http://dx.doi.org/10.1111/tra.12056
- Luzio JP, Hackmann Y, Dieckmann NM, Griffiths GM. The Biogenesis of Lysosomes and Lysosome-Related Organelles. Cold Spring Harb Perspect Biol 2014; 6:a016840; PMID:25183830; http://dx.doi.org/10.1101/cshperspect.a016840
- Schroder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics 2010; 10:4053-76; PMID:20957757; http://dx.doi.org/10.1002/pmic.201000196
- Hu ZZ, Valencia JC, Huang H, Chi A, Shabanowitz J, Hearing VJ, Appella E, Wu C. Comparative bioinformatics analyses and profiling of lysosome-related organelle proteomes. Int J Mass Spectrom 2007; 259:147-60; PMID:17375895; http://dx.doi.org/10.1016/j.ijms.2006.09.024
- Tanida I. Autophagy basics. Microbiol Immunol 2011; 55:1-11; PMID:21175768; http://dx.doi.org/10.1111/j.1348-0421.2010.00271.x
- Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy 2011; 7:477-89; PMID:21293178; http://dx.doi.org/10.4161/auto.7.5.14811
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Lubke T, Lobel P, Sleat DE. Proteomics of the lysosome. Biochim Biophys Acta 2009; 1793:625-35; PMID:18977398; http://dx.doi.org/10.1016/j.bbamcr.2008.09.018
- Segatori L. Impairment of homeostasis in lysosomal storage disorders. IUBMB Life 2014; 66:472-7; PMID:25044960; http://dx.doi.org/10.1002/iub.1288
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463; http://dx.doi.org/10.1126/science.1174447
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/10.1038/emboj.2012.32
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013; 15(6):647-58; PMID:23604321; http://dx.doi.org/10.1038/ncb2718
- Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci 2014; 71:2483-97; PMID:24477476; http://dx.doi.org/10.1007/s00018-014-1565-8
- Di Fiore PP, von Zastrow M. Endocytosis, Signaling, and Beyond. Cold Spring Harb Perspect Biol 2014; 6(8); PMID:25085911; http://dx.doi.org/10.1101/cshperspect.a016865
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 2014; 111:2626-31; PMID:24510904; http://dx.doi.org/10.1073/pnas.1318306111
- Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol 2013; 12:92-104; PMID:23237904; http://dx.doi.org/10.1016/S1474-4422(12)70259-4
- Bodi K, Perera AG, Adams PS, Bintzler D, Dewar K, Grove DS, Kieleczawa J, Lyons RH, Neubert TA, Noll AC, et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J Biomole Tech 2013; 24:73-86; PMID:23814499
- Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta 2013; 1832:1866-81; PMID:23402926; http://dx.doi.org/10.1016/j.bbadis.2013.01.019
- Caciotti A, Donati MA, Procopio E, Filocamo M, Kleijer W, Wuyts W, Blaumeiser B, d'Azzo A, Simi L, Orlando C, et al. GM1 gangliosidosis: molecular analysis of nine patients and development of an RT-PCR assay for GLB1 gene expression profiling. Hum Mutat 2007; 28:204; PMID:17221873; http://dx.doi.org/10.1002/humu.9475
- Schulz A, Dhar S, Rylova S, Dbaibo G, Alroy J, Hagel C, Artacho I, Kohlschütter A, Lin S, Boustany RM. Impaired cell adhesion and apoptosis in a novel CLN9 Batten disease variant. Ann Neurol 2004; 56:342-50; PMID:15349861; http://dx.doi.org/10.1002/ana.20187
- Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Klionsky DJ. The autophagy connection. Dev Cell 2010; 19:11-2; PMID:20643346; http://dx.doi.org/10.1016/j.devcel.2010.07.005
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Cox TM, Cachon-Gonzalez MB. The cellular pathology of lysosomal diseases. J Pathol 2012; 226:241-54; PMID:21990005; http://dx.doi.org/10.1002/path.3021
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/10.1038/nature07976
- Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol 2014; 10:322-37; PMID:24663220; http://dx.doi.org/10.1038/nrendo.2014.35
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 2008; 131:1969-78; PMID:18187492; http://dx.doi.org/10.1093/brain/awm318
- Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 2009; 16:57-69; PMID:18772897; http://dx.doi.org/10.1038/cdd.2008.130
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Thera 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047
- Cicchini M, Karantza V, Xia B. Molecular pathways: autophagy in cancer-a matter of timing and context. Clin Cancer Res 2015; 21:498-504; PMID:25165101; http://dx.doi.org/10.1158/1078-0432.CCR-13-2438
- Haddad SE, Khoury M, Daoud M, Kantar R, Harati H, Mousallem T, Alzate O, Meyer B, Boustany RM. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis 2012; 33:3798-809; PMID:23160995; http://dx.doi.org/10.1002/elps.201200472
- Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet 2008; 17:469-77; PMID:17989065; http://dx.doi.org/10.1093/hmg/ddm324
- Elleder M, Sokolova J, Hrebicek M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol 1997; 93:379-90; PMID:9113203; http://dx.doi.org/10.1007/s004010050629
- Inui K, Wenger DA. Concentrations of an activator protein for sphingolipid hydrolysis in liver and brain samples from patients with lysosomal storage diseases. J Clin Invest 1983; 72:1622-8; PMID:6415115; http://dx.doi.org/10.1172/JCI111121
- Sleat DE, Ding L, Wang S, Zhao C, Wang Y, Xin W, Zheng H, Moore DF, Sims KB, Lobel P. Mass spectrometry-based protein profiling to determine the cause of lysosomal storage diseases of unknown etiology. Mol Cell Proteomics 2009; 8:1708-18; PMID:19383612; http://dx.doi.org/10.1074/mcp.M900122-MCP200
- Pandey MS, Weigel PH. A hyaluronan receptor for endocytosis (HARE) link domain N-glycan is required for extracellular signal-regulated kinase (ERK) and nuclear factor-kappaB (NF-kappaB) signaling in response to the uptake of hyaluronan but not heparin, dermatan sulfate, or acetylated low density lipoprotein (LDL). J Biol Chem 2014; 289:21807-17; PMID:24942734; http://dx.doi.org/10.1074/jbc.M114.565846
- Laurent TC, Fraser JR. Hyaluronan. FASEB J 1992; 6:2397-404; PMID:1563592
- Nie Z, Stanley KT, Stauffer S, Jacques KM, Hirsch DS, Takei J, Randazzo PA. AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J Biol Chem 2002; 277:48965-75; PMID:12388557; http://dx.doi.org/10.1074/jbc.M202969200
- Yu P, Constien R, Dear N, Katan M, Hanke P, Bunney TD, Kunder S, Quintanilla-Martinez L, Huffstadt U, Schröder A, et al. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity 2005; 22:451-65; PMID:15845450
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66; PMID:21752829; http://dx.doi.org/10.1093/hmg/ddr306
- Chapel A, Kieffer-Jaquinod S, Sagné C, Verdon Q, Ivaldi C, Mellal M, Thirion J, Jadot M, Bruley C, Garin J, Gasnier B, Journet A. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 2013; 12(6):1572-88.
- Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 2009; 37:D793-6; PMID:18842627; http://dx.doi.org/10.1093/nar/gkn665
- McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 2007; 80:588-604; PMID:17357067; http://dx.doi.org/10.1086/514346
- Braun V, Deschamps C, Raposo G, Benaroch P, Benmerah A, Chavrier P, Niedergang F. AP-1 and ARF1 control endosomal dynamics at sites of FcR mediated phagocytosis. Mol Biol Cell 2007; 18:4921-31; PMID:17914058; http://dx.doi.org/10.1091/mbc.E07-04-0392
- D'Angelo G, Vicinanza M, De Matteis MA. Lipid-transfer proteins in biosynthetic pathways. Curr Opin Cell Biol 2008; 20:360-70; PMID:18490149; http://dx.doi.org/10.1016/j.ceb.2008.03.013
- Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA. Function and dysfunction of the PI system in membrane trafficking. EMBO J 2008; 27:2457-70; PMID:18784754; http://dx.doi.org/10.1038/emboj.2008.169
- Wigge P, McMahon HT. The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci 1998; 21:339-44; PMID:9720601; http://dx.doi.org/10.1016/S0166-2236(98)01264-8
- Weber-Boyvat M, Zhong W, Yan D, Olkkonen VM. Oxysterol-binding proteins: functions in cell regulation beyond lipid metabolism. Biochem Pharmacol 2013; 86:89-95; PMID:23428468; http://dx.doi.org/10.1016/j.bcp.2013.02.016
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27:1739-40; PMID:21546393; http://dx.doi.org/10.1093/bioinformatics/btr260
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28:27-30; PMID:10592173; http://dx.doi.org/10.1093/nar/28.1.27
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999; 27:29-34; PMID:9847135; http://dx.doi.org/10.1093/nar/27.1.29
- Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 2011; 39:D691-7; PMID:21067998; http://dx.doi.org/10.1093/nar/gkq1018
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 2011; 39:D52-7; PMID:21115458; http://dx.doi.org/10.1093/nar/gkq1237
- Mutarelli M, Marwah V, Rispoli R, Carrella D, Dharmalingam G, Oliva G, di Bernardo D. A community-based resource for automatic exome variant-calling and annotation in Mendelian disorders. BMC Genomics 2014; 15(Suppl 3):S5; PMID:25078076; http://dx.doi.org/10.1186/1471-2164-15-S3-S5
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/10.1093/bioinformatics/btp324
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; http://dx.doi.org/10.1093/bioinformatics/btp352
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43:491-8; PMID:21478889; http://dx.doi.org/10.1038/ng.806
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38:e164; PMID:20601685; http://dx.doi.org/10.1093/nar/gkq603
- Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 2012; 40:D130-5; PMID:22121212; http://dx.doi.org/10.1093/nar/gkr1079
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29:308-11; PMID:11125122; http://dx.doi.org/10.1093/nar/29.1.308
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061-73; PMID:20981092; http://dx.doi.org/10.1038/nature09534
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010; 20:110-21; PMID:19858363; http://dx.doi.org/10.1101/gr.097857.109
- Goode DL, Cooper GM, Schmutz J, Dickson M, Gonzales E, Tsai M, Karra K, Davydov E, Batzoglou S, Myers RM, et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res 2010; 20:301-10; PMID:20067941; http://dx.doi.org/10.1101/gr.102210.109
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 2011; 32:894-9; PMID:21520341; http://dx.doi.org/10.1002/humu.21517
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7:575-6; PMID:20676075; http://dx.doi.org/10.1038/nmeth0810-575
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics 2013; 76:7.20.1-41.
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073-81; PMID:19561590; http://dx.doi.org/10.1038/nprot.2009.86
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46:310-5; PMID:24487276; http://dx.doi.org/10.1038/ng.2892
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2012; 14:178-92; PMID:22517427; http://dx.doi.org/10.1093/bib/bbs017