
Abstract
ATG101 is an essential component of the ULK complex responsible for initiating cellular autophagy in mammalian cells; its 3-dimensional structure and molecular function, however, are currently unclear. Here we present the X-ray structure of human ATG101. The protein displays an open HORMA domain fold. Both structural properties and biophysical evidence indicate that ATG101 is locked in this conformation, in contrast to the prototypical HORMA domain protein MAD2. Moreover, we discuss a potential mode of dimerization with ATG13 as a fundamental aspect of ATG101 function.
Abbreviations
| Atg | = | autophagy related |
| AUC | = | analytical ultracentrifugation |
| BEST | = | band-selective excitation short-transient |
| βME | = | β-mercaptoethanol |
| DLS | = | dynamic light scattering |
| FOM | = | figure of merit |
| HORMA | = | Hop1p/Rev7p/MAD2 |
| MAD2L1 | = | mitotic arrest deficient 2-like protein 1 |
| NMR | = | nuclear magnetic resonance |
| RB1CC1 | = | RB1-inducible coiled-coil 1 |
| SAD | = | single-wavelength anomalous diffraction |
| SEC | = | size exclusion chromatography |
| TROSY | = | transverse relaxation-optimized spectroscopy |
| ULK | = | unc-51 like autophagy activating kinase. |
Introduction
Eukaryotic cells have evolved several mechanisms enabling them to degrade cytoplasmic constituents: while the proteasome plays a major role in the breakdown of relatively short-lived proteins, autophagy is the only route to degradation of larger structures including entire organelles. Today, the term autophagy is used to denote several distinct pathways converging at the lysosomal compartment; the most prominent of these is macroautophagy (subsequently referred to as autophagy), which critically contributes both to basal turnover of biopolymers and to maintenance of homeostasis under conditions of stress, such as nutrient or oxygen deprivation. Indeed, dysregulation of autophagy has been implicated in a number of human diseases, such as neurodegenerative disorders and cancer.Citation1 Pioneering studies in the yeast Saccharomyces cerevisiae have led to the identification of a core machinery of autophagy-related (Atg) proteins, many of which are conserved throughout eukaryotic evolution.Citation2 The Atg1 complex, for instance, which plays a crucial role during the initiation stage of autophagy, is composed of the serine/threonine kinase Atg1, the regulatory component Atg13, and a scaffold containing Atg11 and/or Atg17-Atg31-Atg29. In mammalian cells, ULK (unc-51 like autophagy activating kinase) 1 and ULK2 have been recognized as counterparts of yeast Atg1, and an Atg13 ortholog has been identified as well.Citation3 In addition, the ULK complex contains a scaffold protein called RB1CC1 (RB1-inducible coiled-coil 1), and a protein of hitherto unknown function, ATG101. While RB1CC1 has recently been proposed to be distantly related to yeast Atg11,Citation4 ATG101 orthologs are found in lower metazoans, but are absent in S. cerevisiae.
Human ATG101 is a cytosolic polypeptide composed of 218 amino acid residues, without obvious sequence similarity to any other protein. It has been discovered independently by 2 groups,Citation5,6 who consistently showed that ATG101 is essential for autophagy in human cells and is incorporated into the ULK complex via interaction with ATG13. Similar results have recently been obtained for Caenorhabditis elegansCitation7 and Drosophila melanogaster.Citation8 Intriguingly, ATG101 has been reported to increase the lifetime of its binding partner ATG13 by protecting it from proteasomal degradation,Citation5 and to stabilize basal phosphorylation of ULK1 and ATG13.Citation6 While bioinformatics analyses suggest ATG101 to be distantly related to HORMA (Hop1p/Rev7p/MAD2) domain-containing proteins,Citation8 its precise 3-dimensional structure and its mode of action have remained elusive.
Results
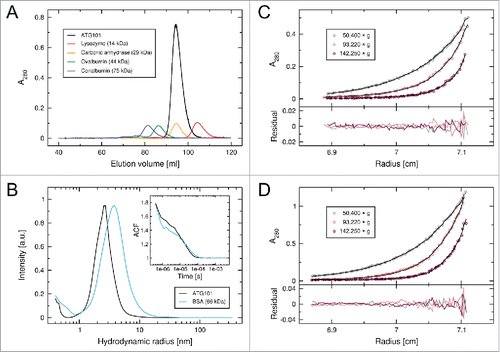
Following heterologous expression in Escherichia coli, full-length human ATG101 was purified from the soluble fraction of bacterial lysates or, alternatively, from inclusion bodies, which resulted in significantly higher yields. In order to exclude artifacts related to the refolding procedure, we recorded 2-dimensional nuclear magnetic resonance (NMR) data for representative preparations of 15N-labeled ATG101 (Fig. S1). The spectra of refolded and nonrefolded protein are virtually indistinguishable, implying an identical environment of amide groups and thus an unaltered structure of the refolded material. ATG101 elutes from a size exclusion chromatography (SEC) column with a retention volume indicative of a monomeric state, even when injected at very high concentrations (); this observation is consistent with results reported by Hosokawa et al., who have investigated the size distribution of ATG101-containing complexes in cytosol preparations from HeLa cells. In addition to larger assemblies containing additional components of the ULK complex, they detect a significant fraction of apparently monomeric endogenous ATG101.Citation6 In order to further investigate potential oligomer formation by ATG101, we performed dynamic light scattering (DLS) and analytical ultracentrifugation (AUC) experiments. The hydrodynamic radius distribution, as determined from the DLS autocorrelation function (), displays a single peak at 2.48 ± 0.20 nm, corresponding to a diffusion coefficient of 8.5 (7.9–9.3) × 10−11 m2/s. This value is in good agreement with the theoretical diffusion coefficient (8.0 × 10−11 m2/s) calculated from our ATG101 crystal structure (see below). In contrast, dimeric assemblies inferred from prominent lattice contacts would be expected to show significantly slower diffusion, with coefficients on the order of 6.0 × 10−11 m2/s. AUC sedimentation equilibria were established with 2 different starting concentrations of ATG101 (). Monte Carlo analysis of concentration profiles yielded a single species with a molecular mass of 24.1 ± 0.3 kDa, which is very close to the theoretical mass of human ATG101 (25.0 kDa). Taken together these data confirm that purified ATG101 is a monomer in solution.
Figure 1. Human ATG101 is a monomer in solution. (A) SEC elution profile of ATG101 (black), together with a set of reference proteins (see legend for details). ATG101 was injected at a concentration of 400 µM (10 mg/mL). (B) Hydrodynamic radius distribution of ATG101 (56 µM, black), as determined by DLS (a.u., arbitrary units). Data for bovine serum albumin (BSA, blue) are included for comparison. The inset shows the autocorrelation function of scattered light intensity for both samples. (C, D) Sedimentation equilibrium profiles of ATG101 (circles) obtained by AUC. Experiments were performed with protein solutions containing 10 µM (C) and 20 µM (D) ATG101, using 3 different rotational speeds. Black lines indicate the respective exponential fits; residual differences are shown below. Since the model matches the data over the entire radial distribution, ATG101 oligomerization can be effectively excluded at concentrations twice the starting values.
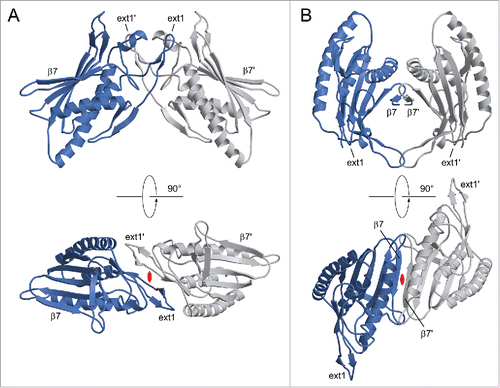
ATG101 was crystallized in vapor diffusion experiments, using PEG3350 as a precipitating agent. Crystals belonged to space group P2, with one molecule per asymmetric unit, and generally displayed significant diffraction anisotropy. Phases were determined via single-wavelength anomalous diffraction (SAD) using a dataset collected from a selenomethionine-containing ATG101 crystal. After an initial model had been established, the structure was refined against a native data set extending to resolutions between 2.6 Å (along a*) and 1.9 Å (along b* and c*, Fig. S2). The C-terminal 8 amino acid residues could not be traced in the electron density and are therefore likely to be disordered. ATG101 belongs to the α+β class of proteins and displays a layered structure in which a 5-stranded β-sheet is attached to a central scaffold of 3 α-helices (). A search for similar folds in the Protein Data Bank revealed human MAD2L1 (mitotic arrest deficient 2-like protein 1, shortly MAD2), a protein comprising a single HORMA domain, as the closest match (). MAD2, which does not share significant sequence similarity with ATG101, is a component of the mitotic spindle assembly checkpoint and has been found to occur in 2 different conformations.Citation9 While newly expressed MAD2 preferentially folds into the open state (O), a closed state (C) can be attained by relocation of 2 segments located at the termini, resulting in binding partners being trapped by a loop dubbed the safety belt. This conversion is thought to be promoted autocatalytically by pre-existing C-MAD2, involving an O-MAD2-C-MAD2 asymmetric dimer, which has been characterized by X-ray crystallography.Citation10 Overall, the conformation of ATG101 corresponds to the open state of MAD2, as evidenced by the N-terminal segment being inserted as strand β1 between β5 and αC and the C-terminal residues constituting the opposite edge of the β-sheet (). Nevertheless, several differences are noteworthy. First of all, β1 is much longer in ATG101 (10 residues) than in O-MAD2 (4 or 5 residues), with a proportional disparity in the number of hydrogen bonds. As a result, significantly more energy would be required to mobilize this portion of the molecule (). Second, the C terminus differs topologically in that it forms a single antiparallel β-strand (β7) in ATG101, whereas in O-MAD2 it comprises 2 strands (β7, β8) forming a type-I Ψ-loop together with β6 (). Even if the 8 terminal residues not traceable in the electron density are taken into account, the ATG101 C terminus is too short to traverse the β-sheet and insert as a hairpin in place of β1, as observed in the closed conformation of MAD2. Together, these considerations strongly suggest that ATG101 will not undergo a conformational transition, but is locked in the open HORMA domain fold, analogous to the effect of C-terminal truncation in MAD2.Citation10 Further support for this conclusion is provided by NMR spectroscopy. In the case of MAD2, the open state is well established to be stable at 4°C, but to undergo conformational equilibration with the closed state upon overnight incubation at 30°C. Since the 2D NMR spectra of the 2 states differ significantly, formation of the closed state of MAD2 is reflected by the appearance of a second set of resonances.Citation11 In contrast, we found that 15N-labeled ATG101 did not show significant spectral changes after incubation at 30°C for almost 5 d (). We note that the number of resonances recorded in our BEST-TROSY spectra exceeded the number inferred from the amino acid sequence by about 10%. While this observation does suggest a limited amount of conformational exchange in ATG101 (possibly related to transient interactions of protein molecules in a highly concentrated solution), it is not obviously affected by prolonged incubation at 30°C. Moreover, its extent is incompatible with an intramolecular relocation of secondary structure elements as observed for MAD2, which would affect a much larger number of resonances. An additional striking feature of ATG101 is the presence of 3 large insertions relative to MAD2, which are all located at one pole of the molecule (). The first 2 result in formation of an extended finger-like structure containing 2 short β-strands (β2′ and β3′) between αA and β2 (ext1) and elongation of the β4-β5 hairpin involving extra strand segments β4′ and β5′ (ext2), respectively. The third insertion (ext3) located between αC and β6 contains a short irregular helix and a loop, herein referred to as the capping loop (CL), that appears to shield the hydrophobic core of ATG101, substituting for the β7-β8 loop present in MAD2 (). While defining the precise significance of these structural elements requires further investigation, they are likely to reflect an adaptation of the canonical HORMA fold to a novel function, such as binding of a yet-to-be-identified protein involved in regulation or execution of the autophagy process. It is interesting to note that ext1 is involved in an important lattice contact of the ATG101 crystal structure (). Quantitative analysis with QtPISACitation12 suggests that the respective dimeric assembly may be stable in solution; a second contact (), which is chiefly established by strands β7 of adjacent molecules, is classified as metastable. While the results outlined in demonstrate that purified ATG101—under the conditions used in our studies—is a monomer in solution, association tendencies revealed by crystal packing might be relevant for homo- or heteromeric interactions in specialized microenvironments, such as the multiprotein ULK complex. Here, additional components may stabilize ATG101 oligomers or, alternatively, bridge non-interacting molecules. Note that the coprecipitation results reported by Hegedűs et al., suggesting association of ATG101 with ATG101 in Drosophila cell lysates,Citation8 are compatible with either scenario.
Figure 2. Structure of human ATG101. (A) Ribbon diagram illustrating the open HORMA domain fold of the protein. Helices are labeled by capital letters, β-strands are numbered sequentially. Note the extensions relative to MAD2 (dotted box), which are located in close proximity in the 3-dimensional structure. A loop segment (Ser108 to Ser113) exhibiting weak electron density (indicative of enhanced flexibility) is drawn in dark gray and marked by a tilde. CL, capping loop. (B) The hydrogen bonding network anchoring strand β1 (dark gray) to β5 (light gray) and the neighboring αC helix. Most side chains are omitted for visual clarity. (C) Topology diagram of ATG101, highlighting the extensions indicated above.
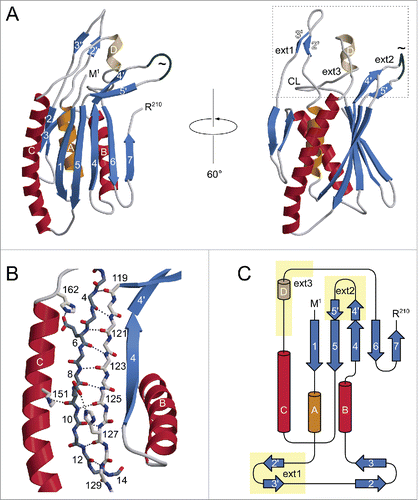
Figure 3. Comparison of ATG101 with MAD2. (A) Stereo view of ATG101 (blue, this study) superimposed on O-MAD2 (yellow, PDB 2V64, chain H). The root-mean-square distance between the 2 structures is 2.04 Å, considering 140 pairs of Cα atoms. For visual clarity, backbone traces are shown in a splined fashion, thus reducing clutter particularly in the helical regions. (B) While O-MAD2 (yellow) contains a ψ-loop in its C-terminal segment, the corresponding portion of ATG101 (blue) forms a simple β-hairpin. The capping loop (CL) occupies the position of the β7-β8 region missing in ATG101, covering part of the hydrophobic core. Both traces are shown in the context of the O-MAD2 structure (surface representation including residues 9 to 89 and 95 to 144). Numbers in brackets indicate additional residues not resolved in the 2 crystal structures.
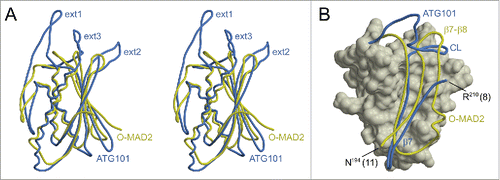
Figure 4. BEST-TROSY NMR data of 15N-labeled ATG101 recorded before (red) and after (blue) incubation at 30°C for 115 h (contoured differently for visual clarity). The spectrum is mostly unchanged, with small deviations likely due to aggregation occurring at the protein concentration employed (600 µM). Equilibration with an alternative fold, as observed for MAD2, would be reflected by the appearance of a separate set of resonances for the majority of amide groups.
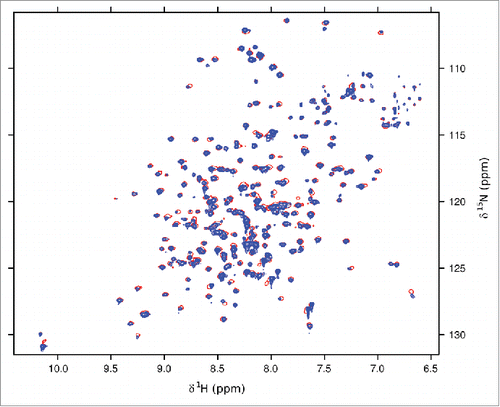
Figure 5. Lattice contacts of the ATG101 crystal structure with possible significance for oligomeric assemblies. Interface #1 (A) involves one of the insertions to the MAD2 fold (ext1) and has a pronounced hydrophobic character, whereas #2 (B) is dominated by hydrogen bonds mostly formed by the C-terminal strand β7. Interacting molecules are related by crystallographic 2-fold axes (indicated in red).
In the context of the ULK complex, ATG101 is thought to directly interact with ATG13. Several lines of evidence support this notion. First of all, Hosokawa et al. have detected ATG101 in a cytosolic SEC fraction which contains ATG13 but none of the other established components of the ULK complex.Citation6 Using immunoprecipitation experiments and fluorescence imaging, Mercer et al. demonstrate that recruitment of ATG101 into the ULK complex depends on the presence of ATG13.Citation5 Finally, Liang et al. report yeast 2-hybrid screens with C. elegans proteins, revealing a dominant interaction of EPG-1/ATG13 and EPG-9/ATG101. Binding of EPG-1 and EPG-9 has been subsequently verified by in vitro affinity isolation assays.Citation7
Human ATG13 is a protein of 517 amino acid residues (isoform 1), which has been predicted to contain a compactly folded domain in its N-terminal half, and a poorly ordered C-terminal segment. Importantly, affinity isolation experimentsCitation5 and immunoprecipitation studiesCitation8 have indicated ATG101 binding to depend on the N-terminal domain of ATG13. The crystal structure of Atg13 (residues 1 to 260) from the yeast Lachancea thermotolerans has been determined recently.Citation13 Whereas this protein was found to also adopt a HORMA domain fold, its structure reveals characteristics of the closed MAD2 conformation. In fact, its N-terminal segment provides insufficient length to form a β-strand between β5 and αC, a requirement for a closed-to-open transition. We therefore propose that the ATG13 HORMA domain (ATG13H) is locked in the closed conformation, analogous to the effect of N-terminal truncation in MAD2.Citation10
Together with published results these considerations strongly suggest that HORMA domains representing the 2 conformers previously established for the prototypical HORMA protein MAD2 mediate the ATG101-ATG13 interaction. We therefore hypothesize this complex to assemble along the lines of the O-MAD2-C-MAD2 “conforma-tional” dimer.Citation10 In order to explore this possibility, we have built a homology model of the human ATG101-ATG13H complex (Supplementary information, Fig. S3, S4), which is based on the L. thermotolerans Atg13 structure (PDB 4J2G) and the ATG101 structure determined in this study, with relative orientations guided by the O-MAD2-C-MAD2 dimer (PDB 2V64). The interface involves surfaces of complementary shape and charge, and quantitative analysis reveals characteristics indicative of a stable protein-protein complex (Table S1). This mode of interaction offers a conceptual explanation both for the association of ATG13 with ATG101 and their reciprocal stabilizing effects observed in cells. While the model appears plausible and is in accordance with available evidence, it should be considered a working hypothesis requiring further experimental validation, e.g. by mutational analysis in cell culture systems. In this respect, it is interesting to note that a Val-to-Asp mutation at position 152 of Drosophila Atg101 results in reduced autophagic flux, based on accumulation of the selective cargo p62/Ref(2)P in well-fed mutant adult flies (Gábor Juhász, personal communication). The equivalent residue in human ATG101, Ile152, is located in the central portion of helix αC, and its side chain is partly buried, interacting with Leu148 and Phe62 of the hydrophobic core. Exchange for a charged residue is therefore expected to locally disturb the helical structure or even cause a limited rotation of helix αC. Intriguingly, its neighbor Ile153 (identical in Drosophila Atg101 and human ATG101) is solvent-exposed and part of the ATG13 interface predicted by our model. It is therefore tempting to speculate that the mutation at position 152 will affect autophagy by altering ATG101-ATG13 complex formation.
Discussion
While this work was under review, the crystal structure of the Atg101-Atg13H complex from Schizosaccharomyces pombe was published.Citation14 The overall fold of fission yeast Atg101 is similar to its human ortholog, with a root-mean-square distance of 1.80 Å for 145 aligned Cα positions. Regarding the insertions relative to MAD2, however, we note intriguing differences. Specifically, ext1 is completely missing in S. pombe Atg101, while ext3 is significantly shorter, lacking both the helical segment and the capping loop. These features largely account for the overall difference in length (184 vs. 218 residues) between fission yeast Atg101 and human ATG101. Also note that ext2, being of similar size, assumes very different conformations in the 2 proteins. As indicated by sequence alignments, the 3 insertions are present not only in mammalian ATG101, but also in D. melanogaster and C. elegans ortholgs, and are thus likely to have appeared early during metazoan evolution, after divergence from a primordial MAD2-like protein. Generally, the extensive modification of the HORMA fold in a confined region of the ATG101 molecule suggests the emergence of an additional binding partner. The latter might be either a component of the ULK complex, including a second copy of ATG101, or a novel interacting protein. In contrast, the complex with the N-terminal domain of ATG13 is likely to be established by the core HORMA domain of ATG101, involving structural elements shared with MAD2. Indeed, the X-ray structure of the fission yeast Atg101-Atg13H complexCitation14 features an O-MAD2-C-MAD2-type mode of interaction, which is in excellent agreement with the model we are proposing for its human counterpart (Fig. S4). Ultimately, cocrystallization of ATG101 with the other constituents of the initiator complex will be required to unravel the precise network of interactions governing the function of this multiprotein assembly.
To conclude, the 3-dimensional structure of ATG101 determined in this study provides new insight into an enigmatic component of the mammalian autophagic machinery. Given the uniqueness of ATG101 in the human proteome, the availability of its 3-dimensional structure may inspire the rational design of highly specific pharmacologic modulators of autophagy, with possible applications not only in basic research, but also in therapy of human diseases.
Materials and Methods
Expression of soluble ATG101
The cDNA coding for human ATG101 (UniProt Q9BSB4) was amplified by polymerase chain reaction and ligated into a pET11a vector (Novagen, 69436-3). Following transformation of E. coli BL21(DE3) cells with the resulting plasmid, a preculture was grown overnight at 37°C in LB medium supplemented with 100 µg/mL ampicillin. It was then used to inoculate a 6-L culture in a fermenter, which was stirred at 30°C until the OD600 reached 0.3. While cooling down to 15°C, expression was induced by addition of 50 µM isopropyl β-D-thiogalactopyranoside/IPTG at an OD600 of 0.6, followed by further incubation for 24 h. For expression of 15N-labeled ATG101, cells were grown in a modified M9 minimal medium (46.6 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 2 mM MgSO4, 100 µM CaCl2, 10 µM Fe(III) citrate, 1 g/L NH4Cl, 4 g/L D-glucose, 348 nM ZnSO4, 152 nM MnCl2, 4.8 µM H3BO3, 840 nM CoCl2, 84 nM NiCl2, 58.7 nM CuCl2, 3.7 µM Na2MoO4, 116 nM Na2SeO3, 5 mg/L thiamine hydrochloride [Sigma-Aldrich, T4625], 1 mg/L each of D-biotin [Sigma-Aldrich, B4639], choline chloride [Sigma-Aldrich, C7017], folic acid [Sigma-Aldrich, F8758], nicotinamide [Sigma-Aldrich, N0636], calcium D-pantothenate [Sigma-Aldrich, P5710], pyridoxal hydrochloride [Sigma-Aldrich, 271748], 0.1 mg/L riboflavin [Sigma-Aldrich, 95170]). At an OD600 of 0.3, bacteria were harvested by centrifugation (4,000 × g for 20 min) and resuspended in 500 mL portions of minimal medium containing 1 g/L 15NH4Cl (Cambridge Isotope Laboratories, NLM-467-10) in place of the unlabeled compound, and 100 µg/mL ampicillin. After induction, expression was allowed to proceed for 48 h.
Expression of ATG101 in inclusion bodies
Initial steps were carried out as outlined for expression of soluble protein. Using the preculture, 500 mL batches of LB medium supplemented with 100 µg/mL ampicillin were inoculated and incubated with stirring at 37°C. Expression of ATG101 was induced at an OD600 of 0.6 by addition of 1 mM IPTG and the culture was grown for an additional 4 h. Isotope-labeled ATG101 was expressed in cells grown in minimal medium, as described above. Following sedimentation, bacteria were resuspended in the same amount of medium containing 1 g/L 15NH4Cl, 2 g/L (13C)glucose (Cambridge Isotope Laboratories, CLM-1396-10) in place of the unlabeled compounds, and 100 µg/mL ampicillin, and they were further grown at 37°C until the OD600 reached 0.6. Protein expression was carried out overnight at 37°C. For production of selenium-labeled ATG101, methionine-auxotrophic E. coli B834 cells (EMD Millipore, 69041-3) were used. Prior to induction, the culture contained 50 mg/L of L-methionine, which was replaced by L-selenomethionine (Anatrace, S2000) in the expression medium.
Purification of soluble ATG101
Wet cell paste was resuspended in ice-cold lysis buffer (20 mM Tris-HCl, pH 8.8, 50 mM NaCl, 10 mM β-mercaptoethanol [βME] supplemented with a protease inhibitor cocktail [Roche, 05056489001]). Cells were disrupted using an M-110P microfluidizer (Microfluidics, Westwood, USA) at 25,000 psi, followed by centrifugation of the lysate at 50,000 × g for 1 h at 4°C. The supernatant fraction was split into 50-mL portions and subjected to ion exchange chromatography on a HiPrep Q HP 16/10 column (GE Healthcare, 29-0181-82) equilibrated with lysis buffer without protease inhibitors. Applying a linear NaCl gradient, ATG101 was found to elute at approximately 190 mM. ATG101-containing fractions were pooled, concentrated to a maximum of 5 mg/mL by ultrafiltration and applied to size exclusion chromatography on a HiLoad 16/60 Superdex 75 pg column (GE Healthcare, 17-1068-01) equilibrated with 20 mM Tris-HCl, pH 8.8, 50 mM NaCl, 10 mM βME for crystallization trials or 150 mM sodium phosphate, pH 7.0 for NMR experiments.
Purification of ATG101 from inclusion bodies
Wet cell paste was resuspended in phosphate-buffered saline (10 mM Na2HPO4, 1.8 mM NaH2PO4, 137 mM NaCl) containing 10 mM βME and lysed with a microfluidizer at 25,000 psi. Following 2 washing-sedimentation cycles, the pellet fraction containing inclusion bodies was resuspended in 5 mL water and sonicated. Solid guanidinium chloride (AppliChem, A1499) was dissolved into the sample to a final concentration of 6 M; after addition of 50 mM sodium phosphate, pH 7.8, 10 mM βME, the sample was stirred for 30 min at room temperature. Solubilized ATG101 was cleared by centrifugation and the supernatant fraction subjected to denaturing size exclusion chromatography on a HiLoad 16/600 Superdex 200 pg column (GE Healthcare, 28-9893-35) equilibrated with 8 M urea, 50 mM sodium phosphate, pH 7.8, 10 mM βME. ATG101-containing fractions were pooled and the concentration was adjusted to 1 mg/mL with running buffer. Refolding was carried out by rapid dilution using 10 volumes of 20 mM Tris-HCl, pH 8.8, 50 mM NaCl, 10 mM βME. Refolded ATG101 was further purified by ion exchange chromatography and size exclusion chromatography, as outlined above. For selenomethionyl ATG101, incorporation of selenium was verified by mass spectrometry.
NMR spectroscopy
NMR experiments were performed with 15N-labeled ATG101 purified with or without a refolding step. 2D BEST (band-selective excitation short-transient)-TROSY (transverse relaxation-optimized spectroscopy) dataCitation15 were recorded with cryogenically cooled Z-pulse-field-gradient (PFG) 1H {13C, 15N} probes at 30°C on VNMRS spectrometers (Varian, Palo Alto, USA). Spectra shown in were acquired at a proton frequency of 900 MHz, with 150 complex points in the 15N time domain, 8 scans per t1 increment, and a 0.2 s recycle delay; those in Figure S1 were collected at a proton frequency of 800 MHz, with 180 complex points in the 15N time domain, 32 scans per t1 increment, and a 0.25 s recycle delay. Data were processed with NMRPipeCitation16 and analyzed with CcpNmr.Citation17
Dynamic light scattering
DLS data were recorded at 20°C on a SpectroSize 300 instrument (Xtal Concepts, Hamburg, Germany), using a 56 µM solution of ATG101. Ten acquisitions of 10 s each were jointly evaluated, and the distribution of hydrodynamic radii was determined from the autocorrelation of scattered light intensity using the software provided by the manufacturer. Calculation of diffusion coefficients from atomic coordinates was performed with HYDROPRO,Citation18 using a buffer density (ρ = 1.018 g/cm3) and viscosity (η = 1.084 × 10−3 Pa s) calculated with SEDNTERP (http://bitcwiki.sr.unh.edu/index.php/Main_Page).Citation19 The partial specific volume (vbar = 0.7359 cm3/g) of ATG101 was determined according to the method of Cohn and Edsall,Citation20,21 as implemented in SEDNTERP.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were carried out in a Beckman Optima XL-A centrifuge (Beckman-Coulter, Indianapolis, USA) equipped with absorption optics and a 4-hole titanium rotor. Protein samples of 120-µl volume were filled in standard aluminum double sector cells with quartz glass windows (Beckman-Coulter, A37301). Human ATG101 was analyzed at concentrations of 10 and 20 µM in 150 mM sodium phosphate buffer, pH 7.0, 2 mM tris(2-carboxyethyl)phosphine (Sigma-Aldrich, C4706) at 10°C. Following equilibration, concentration profiles were recorded with 20-µm radial resolution and averaging of 25 single registrations per radial value. Equilibria have been established at 50,400 × g, 93,220 × g, and 142,250 × g, respectively. Data evaluation was performed using SEDPHAT (http://www.analyticalultracentrifugation.com/sedphat/default.htm), using a buffer density (ρ = 1.019 g/cm3) and partial specific volume (vbar = 0.7317 cm3/g) of ATG101 calculated with SEDNTERP. Final results were subjected to Monte Carlo statistics with 1000 iterations.
Protein crystallization and diffraction data collection
Screening for crystallization conditions was performed at 20°C by sitting-drop vapor diffusion experiments, using a Freedom EVO robotic system (Tecan, Männedorf, Switzerland). Initial crystals were observed after 24 h with a reservoir solution containing 12% (w/v) PEG3350 and 50 mM Bis-Tris-HCl, pH 6.5 (condition 26 of Crystallization Low Ionic Strength Kit for Proteins [Sigma-Aldrich, 86684]), and a protein concentration of 5 mg/mL. This condition was subjected to refinement in a hanging-drop setup, finally yielding well-diffracting samples with 8% (w/v) PEG3350 (Sigma-Aldrich, 88276), 50 mM MES pH 5.6, 50 mM NaCl, 10 mM βME in the reservoir and a 2.2 mg/mL protein solution. In the case of selenomethionyl ATG101, a lower PEG3350 concentration (5% w/v) was found to give better results. Crystals usually reached their final size after 5 or 6 d. Prior to flash cooling, crystals were soaked in reservoir buffer containing up to 25% (v/v) glycerol or PEG400 (Sigma-Aldrich, 81172). Diffraction datasets were recorded at 100 K, using beamlines ID23-1, ID23-2 and ID29 of the European Synchrotron Radiation Facility (ESRF; Grenoble, France) equipped with PILATUS 6M (ID23-1 and ID29) and PILATUS2 3M (ID23-2) detectors (Dectris, Baden, Switzerland), respectively. For SAD phasing with selenomethionine-containing crystals, the wavelength was tuned to the f” peak of the selenium K edge, as determined by an X-ray fluorescence scan. Data processing was performed with XDS and XSCALE.Citation22 The native data set employed in final refinement showed significant diffraction anisotropy, with useful data (defined by a mean F/σ(F) > 3) extending to a resolution of 2.6 Å in the a* direction and 1.9 Å along b* and c*. It was therefore subjected to anisotropic truncation and scalingCitation23 implemented in the UCLA MBI Diffraction Anisotropy Server (http://services.mbi.ucla.edu/anisoscale/anisoscale_xds).
Structure determination
Initial phases were determined via SAD, making use of the selenium anomalous signal. Based on phasing statistics provided by SHELX,Citation24 the best dataset was selected as input to phenix.autosol,Citation25 applying HySSCitation26 for location of anomalous scatterers, PhaserCitation27 for calculation of experimental phases and RESOLVECitation28 for statistical density modification and polypeptide tracing. Out of 6 expected selenium sites, 5 were readily identified, and refinement of substructure parameters yielded starting phases with a figure of merit (FOM) equaling 0.33. Density modification improved these phases to a FOM of 0.71, allowing for an initial polypeptide trace to be built. This trace was further expanded by RESOLVE implemented in phenix.autobuild,Citation29 resulting in a preliminary model containing 164 residues. Missing parts of the structure were built manually using Coot,Citation30 and the model was improved by alternating reciprocal space refinement in phenix.refineCitation31 with rebuilding in Coot. Later cycles of refinement were carried out using a native data set. For statistics of data collection and refinement, refer to . The final model contains residues 1 to 210 of human ATG101. Validation with MolProbityCitation32 and Coot revealed good geometry with all of the residues in the allowed regions of the Ramachandran plot and no rotamer outliers. A search for proteins with similar fold was carried out using the SSM algorithm,Citation33 as implemented in PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm).
Table 1. Data collection and refinement statistics
Molecular graphics
Ribbon representations were generated with MOLSCRIPTCitation34 and RASTER3D,Citation35 applying secondary structure assignments provided by DSSP.Citation36 Solvent-excluded molecular surfaces were calculated with MSMS.Citation37
Accession codes
Atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank (accession code 4WZG).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1076605_Supplemental_material.docx
Download MS Word (2 MB)Acknowledgments
We are grateful to Pitter Huesgen for mass spectrometry analysis of ATG101 and seleno-methionyl ATG101 and to Ralf Biehl for helpful discussion. The authors acknowledge access to the Jülich-Düsseldorf Biomolecular NMR Center. Moreover, we acknowledge the ESRF for provision of synchrotron radiation facilities and would like to thank the staff for assistance in using beamlines ID23-1, ID23-2 and ID29.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID:19491929; http://dx.doi.org/10.1038/nrm2708
- Alers S, Löffler AS, Wesselborg S, Stork B. The incredible ULKs. Cell Commun Signal 2012; 10:7; PMID:22413737; http://dx.doi.org/10.1186/1478-811X-10-7
- Li F, Chung T, Vierstra RD. AUTOPHAGY-RELATED11 plays a critical role in general autophagy- and senescence-induced mitophagy in Arabidopsis. Plant Cell 2014; 26:788-807; PMID:24563201; http://dx.doi.org/10.1105/tpc.113.120014
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649-62; PMID:19287211; http://dx.doi.org/10.4161/auto.5.5.8249
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973-9; PMID:19597335; http://dx.doi.org/10.4161/auto.5.7.9296
- Liang Q, Yang P, Tian E, Han J, Zhang H. The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy 2012; 8:1426-33; PMID:22885670; http://dx.doi.org/10.4161/auto.21163
- Hegedűs K, Nagy P, Gáspári Z, Juhász G. The putative HORMA domain protein Atg101 dimerizes and is required for starvation-induced and selective autophagy in Drosophila. Biomed Res Int 2014; 2014:470482; PMID:24895579; http://dx.doi.org/10.1155/2014/470482
- Luo X, Yu H. Protein metamorphosis: the two-state behavior of Mad2. Structure 2008; 16:1616-25; PMID:19000814; http://dx.doi.org/10.1016/j.str.2008.10.002
- Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 2007; 131:730-43; PMID:18022367; http://dx.doi.org/10.1016/j.cell.2007.08.049
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 2004; 11:338-45; PMID:15024386; http://dx.doi.org/10.1038/nsmb748
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 2007; 372:774-97; PMID:17681537; http://dx.doi.org/10.1016/j.jmb.2007.05.022
- Jao CC, Ragusa MJ, Stanley RE, Hurley JH. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A 2013; 110:5486-91; PMID:23509291; http://dx.doi.org/10.1073/pnas.1220306110
- Suzuki H, Kaizuka T, Mizushima N, Noda NN. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struct Mol Biol 2015; 22:572-580; PMID is 26030876; http://dx.doi.org/10.1038/nsmb.3036
- Solyom Z, Schwarten M, Geist L, Konrat R, Willbold D, Brutscher B. BEST-TROSY experiments for time-efficient sequential resonance assignment of large disordered proteins. J Biomol NMR 2013; 55:311-21; PMID:23435576; http://dx.doi.org/10.1007/s10858-013-9715-0
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995; 6:277-93; PMID:8520220; http://dx.doi.org/10.1007/BF00197809
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 2005; 59:687-96; PMID:15815974; http://dx.doi.org/10.1002/prot.20449
- Ortega A, Amorós D, García de la Torre J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys J 2011; 101:892-8; PMID:21843480; http://dx.doi.org/10.1016/j.bpj.2011.06.046
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding S, Rowe A, Horton JC, editors. Analytical ultracentrifugation in biochemistry and polymer science. Cambridge: Royal Society of Chemistry; 1992. p. 90-125.
- Cohn EJ, Edsall JT. Proteins, amino acids and peptides. New York: Reinhold; 1943.
- Durchschlag H. Thermodynamic data for biochemistry and biotechnology. New York: Springer-Verlag; 1986.
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 2010; 66:125-32; PMID:20124692; http://dx.doi.org/10.1107/S0907444909047337
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2006; 103:8060-5; PMID:16690741; http://dx.doi.org/10.1073/pnas.0602606103
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A 2008; 64:112-22; PMID:18156677; http://dx.doi.org/10.1107/S0108767307043930
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr 2009; 65:582-601; PMID:19465773; http://dx.doi.org/10.1107/S0907444909012098
- Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Crystallogr D Biol Crystallogr 2003; 59:1966-73; PMID:14573951; http://dx.doi.org/10.1107/S0907444903018043
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst 2007; 40:658-74; PMID: 19461840; http://dx.doi.org/10.1107/S0021889807021206
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 2000; 56:965-72; PMID:10944333; http://dx.doi.org/10.1107/S0907444900005072
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr 2008; 64:61-9; PMID:18094468; http://dx.doi.org/10.1107/S090744490705024X
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486-501; PMID:20383002; http://dx.doi.org/10.1107/S0907444910007493
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010; 66:213-21; PMID:20124702; http://dx.doi.org/10.1107/S0907444909052925
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010; 66:12-21; PMID:20057044; http://dx.doi.org/10.1107/S0907444909042073
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 2004; 60:2256-68; PMID:15572779; http://dx.doi.org/10.1107/S0907444904026460
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 1991; 24:946-50; http://dx.doi.org/10.1107/S0021889891004399
- Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol 1997; 277:505-24; PMID:18488322; http://dx.doi.org/10.1016/S0076-6879(97)77028-9
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22:2577-637; PMID:6667333; http://dx.doi.org/10.1002/bip.360221211
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996; 38:305-20; PMID:8906967; http://dx.doi.org/10.1002/(SICI)1097-0282(199603)38:3<305::AID-BIP4>3.0.CO;2-Y