
Abstract
The epithelial-to-mesenchymal (-like) transition (EMT), a crucial embryonic development program, has been linked to the regulation of glioblastoma (GBM) progression and invasion. Here, we investigated the role of MIR517C/miR-517c, which belongs to the C19MC microRNA cluster identified in our preliminary studies, in the pathogenesis of GBM. We found that MIR517C was associated with improved prognosis in patients with GBM. Furthermore, following treatment with the autophagy inducer temozolomide (TMZ) and low glucose (LG), MIR517C degraded KPNA2 (karyopherin alpha 2 [RAG cohort 1, importin alpha 1]) and subsequently disturbed the nuclear translocation of TP53 in the GBM cell line U87 in vitro. Interestingly, this microRNA could inhibit autophagy and reduce cell migration and infiltration in U87 cells harboring wild-type (WT) TP53, but not in U251 cells harboring mutant (MU) TP53. Moreover, the expression of epithelial markers (i.e., CDH13/T-cadherin and CLDN1 [claudin 1]) increased, while the expression of mesenchymal markers (i.e., CDH2/N-cadherin, SNAI1/Snail, and VIM [vimentin]) decreased, indicating that the EMT status was blocked by MIR517C in U87 cells. Compared with MIR517C overexpression, MIR517C knockdown promoted infiltration of U87 cells to the surrounding structures in nude mice in vivo. The above phenotypic changes were also observed in TP53+/+ and TP53-/- HCT116 colon cancer cells. In summary, our study provided support for a link between autophagy and EMT status in WT TP53 GBM cells and provided evidence for the signaling pathway (MIR517C-KPNA2-cytoplasmic TP53) involved in attenuating autophagy and eliminating the increased migration and invasion during the EMT.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ATG | = | autophagy related |
| AV | = | autophagic vacuole |
| CDH2 | = | N, cadherin (cadherin 2, type 1 [neuronal]) |
| CDH13 | = | T-cadherin (cadherin 13) |
| CLDN1 | = | claudin 1 (senescence-associated epithelial membrane protein 1) |
| CQ | = | chloroquine |
| CTNNB1 | = | β-catenin (catenin [cadherin-associated protein], beta 1) |
| DMSO | = | dimethylsulfoxide |
| EMT | = | epithelial-to-mesenchymal (-like) transition |
| GBM | = | glioblastoma multiforme |
| GFP | = | green fluorescent protein |
| IP | = | immunoprecipitation |
| kn | = | knockdown |
| KPNA2 | = | karyopherin alpha 2 (RAG cohort 1, importin alpha 1) |
| LG | = | low glucose |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3B |
| MU | = | mutation |
| NC | = | negative control |
| NSC | = | neural stem cell |
| oe | = | overexpressing |
| OS | = | overall survival |
| PFS | = | progression-free survival |
| qRT-PCR | = | quantitative real-time reverse transcription polymerase chain reaction |
| SNAI2 | = | Slug (snail family zinc finger 2) |
| SNAI1 | = | snail (snail family zinc finger 1) |
| TEM | = | transmission electron microscopy |
| TMZ | = | temozolomide |
| VIM | = | vimentin |
| WT | = | wild type |
| ZEB1 | = | zinc finger E-box binding homeobox 1. |
Introduction
Although multimodality treatment, including surgical resection and ionizing radiation (IR) plus concomitant and adjuvant temozolomide (TMZ), has been widely accepted as a standard therapy for the treatment of glioblastoma (GBM), the prognosis of patients with GBM is still poor, with a median survival of 14.6 mo.Citation1 This poor prognosis may be explained by the intrinsic capacity of isolated tumor cells to invade the surrounding parenchyma, preventing complete surgical resection, or the acquisition of resistance to chemotherapy and radiotherapy. Both of these pathogenic phenotypes have been linked to the epithelial-to-mesenchymal (-like) transition (EMT). The EMT is a critical biological behavior involved in correct embryonic development and has recently been shown to mediate tumor invasion and metastasis in several types of epithelial tumors, including colon cancer, pancreatic cancer, and breast cancer.Citation2,3 Through the EMT, epithelial cells become mesenchymal-like cells, indicating that the cells obtain some features of mesenchymal cells, such as higher invasive ability and tolerance to chemoradiotherapy.Citation4-6 In GBM, members of the SNAI-family, e.g., ZEB1 and ZEB2, act as EMT activators, enhancing the invasiveness of GBM cells in vitro and in vivo.Citation7,8 Moreover, EMT processes in GBM cells are also linked to stem-like status,Citation8 which may cause cellular dedifferentiation of noncancer stem cells, allowing the cells to acquire self-renewal and tumor-initiating ability.Citation9,10
Increasing evidence has shown that IR and TMZ inhibit tumor growth in GBM by inducing a type of programmed cell death that is dependent of autophagy.Citation11-14 Autophagy is a survival-promoting pathway that captures, degrades, and recycles intracellular proteins and organelles in lysosomes. However, excessive levels of autophagy may also lead to cell death. Autophagy is involved in cancer either by suppressing tumorigenesis through its quality control function or by upregulating autophagy to survive microenvironmental stress and increase growth and aggressiveness.Citation15 Moreover, autophagy has recently been shown to be closely associated with EMT status in several epithelial carcinomas. In hepatocellular carcinoma cells, starvation-induced autophagy is critical for cellular invasion through induction of the EMT.Citation16 Furthermore, in mouse models of pulmonary metastasis of hepatocellular carcinoma cells, inhibition of autophagy has been shown to significantly decrease the incidence of pulmonary metastases in vivo.Citation17 Inhibition of autophagy via knockdown of autophagy related 12 (ATG12) in glioma cells does not affect cell viability, proliferation, or migration, but significantly reduces cellular invasion in a 3-dimensional (3D) organotypic model. Thus, autophagy may play a critical role in the benign-to-malignant transition that is central to the initiation of metastasis.Citation18 However, the precise contribution of autophagy to GBM invasion and the EMT is still unclear.
Our previous work reports the discovery of, the chr19q13.41 microRNA cluster (C19MC), which encodes 54 miRNAs, which exhibits a high DNA copy number in 11 out of 45 cases of supratentorial primitive neuroectodermal tumors.Citation19 Expression profiles have demonstrated significant enrichment of self-renewal and survival genes in C19MC-amplified tumors, which are significantly correlated to the poorer prognosis of patients. Among the miRNAs within this cluster, MIR512-3p, MIR517A, MIR517C, MIR519A, and MIR520G exhibit significantly elevated expression in supratentorial primitive neuroectodermal tumor samples with C19MC amplification. In preliminary experiments, MIR517C and MIR519A have been shown to be upregulated in the adult brain as compared with that in neural stem cells (NSCs) and the fetal brain, suggesting that MIR517C and MIR519A may have important biological functions in the embryonic development of the brain (Fig. S1A).
A literature review shows that, C19MC amplification is found in several other human tumors as well.Citation20 Moreover, the C19MC cluster is a novel prognostic biomarker in hepatocellular carcinoma by miRNA profiling.Citation21 In another publication, downregulation of MIR517A and MIR517C promotes the proliferation of hepatocellular carcinoma cells.Citation22 In estrogen receptor (ER)-positive breast cancer, about 50 of the miRNAs within the C19MC cluster were found to be significantly upregulated in chemoresistant cells. Among these miRNAs, MIR519A regulates cell viability and cell cycle progression.Citation23 Additionally, the expression of C19MC miRNAs is higher in villous trophoblasts than in extravillous trophoblasts, and both MIR517C and MIR519D regulate the migration of human trophoblasts.Citation24 These reports support that MIR517C and MIR519A harbored in C19MC may play an important role in tumorigenesis in several types of human tumors.
Since these 2 miRNAs may also have functions in the embryonic development of the brain, we aimed to determine whether the expression levels of these miRNAs were altered in gliomas. Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) analysis of 6 malignant glioma cell lines revealed that MIR517C and MIR519A were upregulated significantly in 2 lines (Fig. S1B), suggesting that these 2 miRNAs may also have functions in GBM. Moreover, as mentioned above, autophagy and the EMT are thought to be critical in the progression of GBM. Therefore, in this study, we sought to comprehensively analyze the precise biological functions of these 2 miRNAs in the regulation of autophagy and the EMT phenotype in GBM cells in vitro and in vivo.
Results
Low MIR517C expression was closely associated with poor prognosis in patients with GBM
In order to validate the relevance of MIR517C in the prognosis of patients with glioma, we examined the expression of MIR517C and MIR519A in patients with newly diagnosed GBM who had undergone surgery plus concomitant and adjuvant TMZ chemoradiotherapy (Stupp regimen: radiotherapy plus concomitant TMZ 75 mg/m2; adjuvant TMZ 150 mg/m2 5/28 d for 6 cycles)Citation1 as recommended by the National Comprehensive Cancer Network (NCCN) Clinical Practice Guideline for Central Nervous System Cancer (Version 2, 2014). Forty-six clinical samples were used for analysis of MIR517C and MIR519A by qRT-PCR, as shown in . The expression level of the 2 miRNAs together with the other variables, such as the patient’s age, gender, tumor volume and location (i.e., functional area or not), and degree of tumor resection were recorded (Table S1). Notably, in 23 cases (50%), MIR517C was expressed at a level higher than the median; we defined this as the high-expression group or MIR517C (+). The other 23 cases were included in the low-expression group or MIR517C (-).
Figure 1. Low MIR517C expression was associated with poor prognosis in patients with GBM. (A) MIR517C and MIR519A expression levels in 46 GBM samples were detected by qRT-PCR (using the 2-ΔCT method); 23 cases (50%) exhibited high expression of these miRNAs, for which the expression was higher than the median (indicated by the red arrows; see also Tables S1 and S2). (B) Kaplan-Meier tumor-free survival analysis according to MIR517C levels. Patients with low expression of MIR517C, for which the expression was lower than the median (n = 23), had significantly poorer outcomes than those with high expression of MIR517C (P = 0.0014). (C) Kaplan-Meier overall survival analysis. The 23 patients with low MIR517C expression had significantly poorer outcomes than those with high MIR517C expression (P = 0.0083).
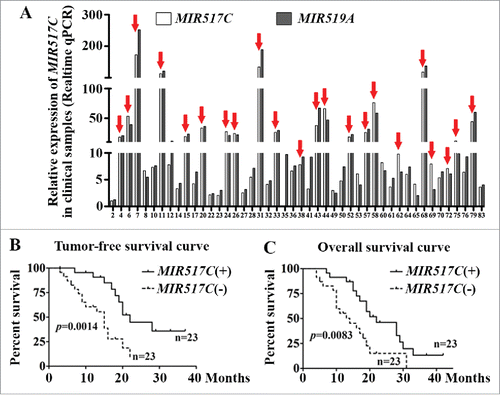
By Kaplan-Meier analysis of each variable, tumor volume, functional area, MIR517C expression level, and MIR519A expression level were selected for Cox multivariate regression analysis of overall survival (OS). The functional area, degree of tumor resection, and expression levels of MIR517C and MIR519A were selected for Cox regression analysis of progression-free survival (PFS). The final Cox multivariate regression analysis indicated that MIR517C expression level affected the prognosis of OS (P = 0.023) and PFS (P = 0.003; Table S2). Moreover, PFS and OS curves consistently showed that patients in the MIR517C (+) group exhibited better prognoses than patients in the MIR517C (-) group ().
Induction of autophagy by TMZ was dependent on the status of TP53
Before investigating the biological function of MIR517C in GBM, we first tested several metabolic and genetic stress inducers, such as low-glucose (LG) conditions and TMZ treatment in parental U87 and U251 cells, which harbored wild-type (WT) or mutant (MU) TP53, respectively. As the concentration of TMZ was increased, we observed accumulation of LC3B-II, a commonly used marker of autophagic activity, by western blot analysis in U87 cells (WT TP53), but not in U251 cells (MU TP53), even after treatment with 300 μM TMZ (). Next, the autophagy inhibitors 3-methyladenine (3-MA) and chloroquine (CQ) were used to interrupt autophagy in the early and late stages, respectively, to further confirm the induction of autophagy by TMZ.Citation25 Western blot analysis in U87 cells showed that the accumulation of LC3B-II induced by TMZ was decreased by 3-MA, but increased by CQ (). CQ inhibits autophagy by blocking lysosome acidification and degradation of autolysosomes, promoting the accumulation of the lipidated form of LC3B (LC3B-II). However, TMZ did not induce any significant changes in LC3B-II accumulation in U251 cells. Notably, for densitometric analysis of western blot results, LC3B-II levels were normalized to the level of ACTB/β-actin rather than the level of LC3B-I because LC3B-I is high relative to LC3-II in brain tissues, and the LC3-I signal can be overwhelming; this is consistent with recommendations presented in the recent guidelines for monitoring autophagy.Citation25 Immunofluorescence assays were further used to examine the autophagic induction of TMZ in U87 cells. The numbers of LC3B fluorescence particles were evaluated by ImageJ software. The result indicated that the punctate staining of LC3B significantly increased after 24 h of treatment with TMZ (), and this effect was suppressed by 3-MA. However, in U251 cells harboring MU TP53, TMZ had no significant autophagy-inducing effects within 48 h (Fig. S2E). Transmission electron microscopy (TEM) further supported the induction of autophagy by TMZ in U87 cells, but not in U251 cells ().
Figure 2. Induction of autophagy by TMZ depended on the phenotype of TP53. (A) Western blot analysis of LC3B-II in U87 and U251 cells after exposure to different concentrations of TMZ. The lower histogram shows densitometric analysis of western blot results (#, P > 0.05; *, P < 0.05; **, P < 0.001). (B) Effects of TMZ, 3-MA, and CQ on the conversion of LC3B-II in U87 and U251 cells. The lower histogram shows densitometric analysis of western blot results (#, P > 0.05; *, P < 0.05; **: P < 0.001). (C) Immunofluorescence analysis of LC3B in U87 cells after treatment with TMZ, 3-MA, and CQ. The LC3B particles were quantized by NIH ImageJ software (*, P < 0.05; **, P < 0.001). (D) Representative transmission electron microscope (TEM) images of U87 and U251 cells after exposure to 0 or 300 μM TMZ. Red arrows indicate formation of autophagic vacuoles. (E) Expression of autophagy-related proteins in U87 cells treated with TMZ and TP53 siRNA. The histogram shows densitometric analysis of western blot results (#, P > 0.05; *, P < 0.05; **, P < 0.001). (F) TEM images of U87 cells after exposure to TMZ and TP53 siRNA. Red arrows indicate formation of autophagic vacuoles.
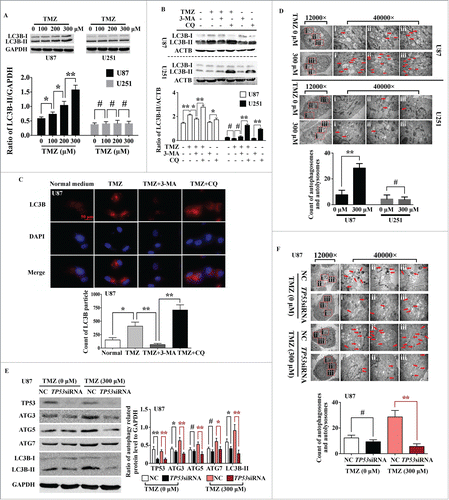
Next, we confirmed the presence of WT TP53 in U87 cells and MU TP53 (R273H mutant) in U251 cells by DNA sequencing (Fig. S3). Then, using TP53 siRNA to knockdown the expression of WT TP53 in U87 cells, we found that key autophagy-related proteins, such as ATG3, ATG5, and ATG7 were significantly downregulated, and the accumulation of LC3B-II was also decreased (). TEM further demonstrated that the induction of autophagy by TMZ was abolished by transfection with TP53 siRNA (). Taken together, these data demonstrated that the induction of autophagy by TMZ in GBM cells was dependent on the status of TP53.
Induction of autophagy increased cell migration and infiltration through the EMT in the context of WT TP53
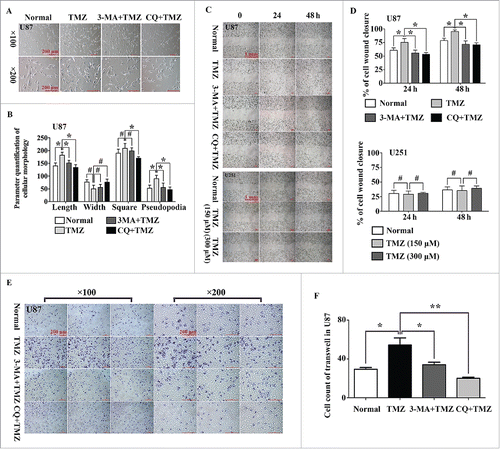
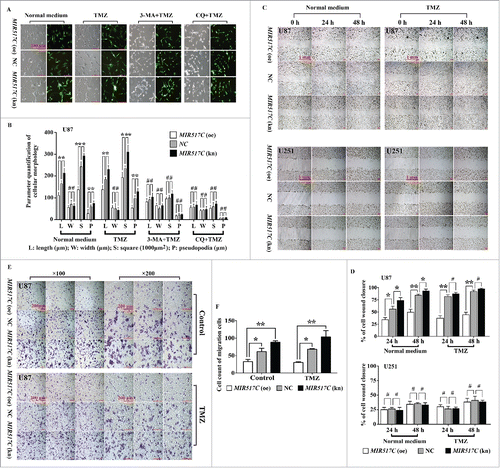
After treatment with LG or 150 μM TMZ for 24 h, morphological changes were observed in U87 cells (harboring WT TP53) compared with that in untreated cells. Cell parameters were quantified using ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov); this analysis showed that the lengths or cellular projections (pseudopodia) in the cell body cultures were longer after treatment with 150 μM TMZ compared with those after culture in normal medium (). When 3-MA (2.5 mM) and CQ (50 μM) were used in combination with TMZ, the morphological changes disappeared, suggesting that these morphological changes may be related to the onset of autophagy. We further evaluated whether induction of autophagy could alter the migration of glioma cells using wound healing assays; ImageJ software was used to calculate the area of cell wound closure, as recommend by Yue et al.Citation30 The results indicated that TMZ-induced U87 cell migration was blocked by treatment with 3-MA and CQ (). However, the autophagy-inducing effects of TMZ were not observed in U251 cells (harboring MU TP53), even when the concentration of TMZ was increased to 300 μM. In cell invasion assays using transwell chambers, U87 cell invasion was stimulated by 150 μM TMZ, and this effect was again blocked by treatment with 3-MA and CQ (). Cell counting of 6 high-resolution fields indicated that TMZ significantly increased the infiltrative capacity of U87 cells (P = 0.002). Similar results were observed when U87 cells were cultured in LG medium (Fig. S2A to D). Moreover, as shown in Fig. S4A and B, TMZ inhibited the proliferation of U87 cells, and additional treatment with 3MA and CQ did not alter these effects of TMZ.
Figure 3. Induction of autophagy increased cell migration and infiltration in U87 cells harboring WT TP53, but not in the U251 cells harboring MU TP53. (A) Representative phase-contrast microscope images of U87 cells treated with normal medium, TMZ, TMZ plus 3-MA, and TMZ plus CQ. (B) The related parameters (length, width, and area of cells and length of pseudopodia) were measured by NIH ImageJ software (#, P > 0.05; *: P < 0.05). (C) Wounding healing assay of U87 cells (top) and U251 cells (bottom) in the presence or absence of autophagy inducers or inhibitors. (D) Quantification of the percentage of cell wound closure in U87 and U251 cells (#, P > 0.05; *: P < 0.05). (E) The infiltrative ability of U87 cells was measured following treatment with TMZ, TMZ plus 3-MA, or TMZ plus CQ. (F) The histogram represents the cell count of infiltrative cells in 6 high-resolution fields for 3 independent experiments. Error bars represent SDs (*, P < 0.05; **, P < 0.001).
Next, siRNA targeting the autophagic key protein ATG7 was used to further investigate whether the effects on the above phenotypes were achieved through autophagy. The morphological changes induced by TMZ were interrupted by transfection with ATG7 siRNA (). Similar results were also obtained in wound healing and transwell assays ().
Figure 4. ATG7 siRNA inhibited cell migration, infiltration, and the expression of EMT-related proteins in U87 cells but not in U251 cells. (A) Representative phase-contrast microscope images of U87 cells with ATG7 siRNA and NC, which were cultured in TMZ (150 μM) medium. The related parameters (length, width, and area of cells and length of pseudopodia) were measured by NIH ImageJ software. (B) Wounding healing assay of U87 cells with ATG7 siRNA and NC in normal medium (top) and in TMZ (bottom). Quantification of the percentage of cell wound closure in U87 and U251 cells (*, P < 0.05;, **: P < 0.001). (C) The infiltrative ability of U87 cells with ATG7 siRNA and NC was measured. The right histogram represents the cell count of infiltrative cells in 6 high-resolution fields for 3 independent experiments. Error bars represent SDs. (D) Western blotting was used to detect the expression of autophagy- and EMT-related proteins in U87 cells transiently transfected with ATG7 siRNA in normal medium or in the presence of TMZ. Densitometric analysis of the western blots shown in the right histogram (#, P > 0.05; *, P < 0.05; **, P < 0.001). (E) Western blotting was used to detect the expression of autophagy- and EMT-related proteins in U251 cells transiently transfected with ATG7 siRNA in normal medium or in the presence of TMZ (150 or 300 μM). Densitometric analysis of the western blots shown in the right histogram (#, P > 0.05; **, P < 0.001). (F) siRNAs targeting ATG7 and TP53 were cotransfected into U87 cells, and LC3B conversion was detected by western blotting. Densitometric analysis is shown in the right histogram (#, P > 0.05; *, P < 0.05; **, P < 0.001).
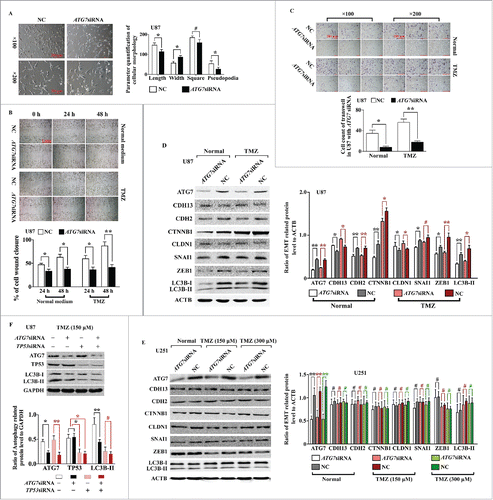
Western blot analysis was then used to measure the levels of LC3B and EMT-related proteins. In ATG7-knockdown cells with or without TMZ treatment, the ratio of LC3B-II/ACTB was substantially decreased, confirming the inhibition of autophagy (). Moreover, levels of epithelial markers, such as CDH13/T-cadherin (cadherin 13) and CLDN1 (claudin 1), were increased, while levels of mesenchymal markers, such as CDH2 (cadherin 2, type 1, N-cadherin [neuronal]), CTNNB1/β-catenin (catenin [cadherin-associated protein], beta 1, 88kDa), and SNAI1/Snail (snail family zinc finger 1), were decreased. The expression levels of other EMT-related regulators, such as ZEB1 (zinc finger E-box binding homeobox 1), were also decreased. However, in U251 cells, which express MU TP53, ATG7 siRNA did not alter the accumulation of LC3B-II, even in the presence of 150 or 300 μM TMZ. Correspondingly, the protein levels of EMT-related markers did not change significantly compared with those in NC cells (). Next, U87 cells were cotransfected with TP53 and ATG7 siRNA to examine whether knockdown of TP53 altered the function of siATG7. As shown in , siATG7 did not further reduce autophagy in TP53-knockdown cells. Similar morphological changes were also observed following transfection with siRNA targeting BECN1/Beclin-1 (Fig. S5).
Taken together, these data demonstrated that autophagy induced by TMZ and LG in U87 cells (harboring WT TP53) could promote U87 cell migration and infiltration; this effect could be blocked by the autophagy inhibitors 3-MA and CQ. ATG7 and BECN1 siRNA could inhibit autophagy and interrupt the above effects of TMZ. Furthermore, this alteration may change the EMT status of GBM cells. However, no such tendencies were observed in U251 cells (harboring MU TP53).
MIR517C prevented the activity of autophagy inducers in U87 cells through inhibition of autophagy and the EMT phenotype
Since low MIR517C expression was associated with poor prognoses in patients with GBM, and based on the results of our previous study,Citation13 we further investigated whether MIR517C played a functional role in U87 and U251 cells. Interestingly, lentiviral-mediated introduction of MIR517C, negative control (NC), or a MIR517C inhibitor caused morphological differences in U87 cells. Cells overexpressing MIR517C, U87 MIR517C (oe) cells, had shorter cell bodies and with fewer short cell projections, while cells harboring the NC construct or the MIR517C inhibitor, termed U87 MIR517C (kn) cells, had larger cell bodies and more frequent long cell projections. Moreover, the autophagy inhibitors 3-MA and CQ could alter the influence of MIR517C (), indicating that the morphological changes induced by MIR517C depended on these changes in autophagic status. However, no morphological changes were observed in U251 cells (Fig. S6).
Figure 5. MIR517C regulated cell morphology, migration, and infiltration. (A) Representative phase-contrast microscope images of U87 cells cultured in normal medium or in the presence of 150 μM TMZ, TMZ plus 3MA, or TMZ plus CQ, following lentiviral-mediated overexpression (oe) or knockdown (kn) of MIR517C. All cells expressed GFP for detection of fluorescence. (B) The related cellular parameters (length, width, and area of cells and length of pseudopodia) were measured by NIH ImageJ software. (C) Wounding healing assays in U87 (top) and U251 (bottom) cells to compare cell migration between MIR517C (oe), NC, and MIR517C (kn). (D) Quantification of the percentage of cell wound closure (#, P > 0.05; *, P < 0.05; **, P < 0.001). (E) Transwell assays were used to evaluate cell infiltration capacity following overexpression (oe) or knockdown (kn) of MIR517C in U87 cells cultured in normal medium or in the presence of 150 μM TMZ. NC cells were used as a control. (F) Histogram representing the number of infiltrative cells in 6 high-resolution fields for 3 independent experiments. Error bars represent SDs.
Based on these morphological changes, we next investigated the biological functions of MIR517C in U87 and U251 cells. Cell proliferation assays revealed that MIR517C had no role in regulating U87 cell proliferation when cells were treated with TMZ. Additionally, no significant differences in apoptosis were observed in U87 MIR517C (oe), NC, and MIR517C (kn) cells, as demonstrated by flow cytometry analysis. Western blotting for CASP3/caspase-3 and CASP9/caspase-9 confirmed these results. Further analysis by flow cytometry also showed that there were no changes in cell cycle distribution among U87 MIR517C (oe), NC, and MIR517C (kn) cells (Fig. S7).
Next, we used wound healing assays to examine whether MIR517C regulated cell migration in U87 cells. As shown in , cell migration was increased in NC and MIR517C (kn) cells compared with that in MIR517C (oe) cells, suggesting that MIR517C inhibited TMZ-induced migration in U87 cells. However, no differences were observed in U251 cells, suggesting that this phenotype may be dependent on TP53. We also assessed the infiltration capacity of cells stably expressing MIR517C, NC, or MIR517C shRNA using a transwell apparatus. After an 8-h incubation, the percentage of migrated cells was significantly less in cells overexpressing MIR517C (P < 0.001), regardless of the presence of TMZ (). Importantly, there were no significant differences in cell proliferation between groups in this experiment, as determined using Cell Counting Kit-8 (CCK-8) assays (Fig. S4C, D).
To further investigate the mechanism through which MIR517C inhibited cell migration in U87 cells, we used western blotting and immunofluorescence assays to examine the expression of key proteins in U87 and U251 cells. The results showed that overexpression of MIR517C caused downregulation of BECN1, ATG3, ATG7, ATG5, and ATG12 in U87 cells (harboring WT TP53), but not in U251 cells (harboring MU TP53). Decreased conversion of LC3B was also observed, indicating that MIR517C inhibited autophagy (). Immunofluorescence assays also confirmed the decreasing numbers of LC3B protein spots in U87 cells overexpressing MIR517C (). Moreover, overexpression of MIR517C in U87 cells also caused upregulation of epithelial marker proteins, such as CDH13 and CLDN1, as compared with NC and MIR517C (kn) cells. Conversely, mesenchymal marker proteins, including CDH2, CTNNB1, and VIM, were significantly downregulated. Similarly, the expression of EMT-related regulators, such as ZEB1, SNAI2/Slug (snail family zinc finger 2), and SNAI1 were downregulated in U87 cells overexpressing MIR517C. Interestingly, these changes were not observed in U251 cells (). These changes in EMT-related proteins indicated that MIR517C interfered with the acquisition of the EMT-like phenotype induced by TMZ and LG in U87 cells.
Figure 6. MIR517C inhibited TMZ-induced autophagy and the EMT. (A) Western blot analysis was used to detect the expression of key autophagy-related proteins in U87 and U251 cells following overexpression (oe) or knockdown (kn) of MIR517C. NC cells were used as a control. (B) Levels of EMT-related proteins were detected by western blotting in U87 and U251 cells following overexpression (oe) or knockdown (kn). NC cells were used as a control. (C) Densitometric analysis of the western blots shown in (A). #, P > 0.05; *, P < 0.05; **, P < 0.001. (D) Densitometric analysis of the western blots shown in (B). #, P > 0.05; *, P < 0.05; **, P < 0.001. (E) Immunofluorescence analysis of LC3B expression in U87 and U251 cells following overexpression (oe) or knockdown (kn) of MIR517C. LC3B particle data were subjected to statistical analysis (#, P > 0.05; *, P < 0.05; **, P < 0.001). (F) Representative transmission electron microscope images of U87 and U251 cells following overexpression (oe) or knockdown (kn) of MIR517C. NC cells were used as a control. Red arrows indicate formation of autophagosomes, and green arrows indicate autolysosomes.
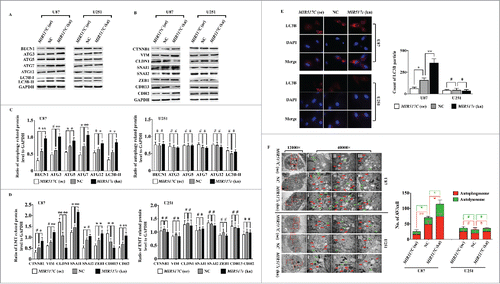
To confirm the inhibition of autophagy by MIR517C, we next performed TEM examination of GBM cells. From TEM images, we observed autophagosomes and autolysosomes. Among the 3 groups, cells overexpressing MIR517C exhibited reduced numbers of both autophagosomes and autolysosomes, providing direct evidence for the inhibition of autophagy. However, in U251 cells, no significant differences were observed in the numbers of autophagic vacuoles among groups, indicating that MIR517C did not influence autophagy in U251 cells ().
Collectively, these data showed that MIR517C inhibited autophagy and the EMT process induced by TMZ in U87 cells expressing WT TP53, but not in U251 cells expressing MU TP53. Therefore, we speculated that the target genes of MIR517C may play important roles in the regulation of autophagy and the EMT in GBM cells in a TP53-dependent manner.
Targeting of KPNA2 by MIR517C disrupted the nuclear translocation of TP53 protein, thereby inhibiting autophagy
How does MIR517C inhibit autophagy and the EMT in U87 cells, and why is this phenotype not observed in U251 cells? To answer these questions and to identify target genes of MIR517C, we used 2-dimensional (2D) electrophoresis of cell lysates from U87 cells transfected with MIR517C mimics, mimics NC, AntagomiR, or Nontarget AntagomiR. Several distinct spots were identified as altered (Table S3). As shown in , we identified one protein spot that was significantly altered in cells overexpressing MIR517C as compared to NC cells. Using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS), we identified this spot as KPNA2/importin subunit alpha-2 (karyopherin alpha 2 [RAG cohort 1, importin alpha 1]). Western blotting confirmed that KPNA2 protein was downregulated in U87 and U251 cells overexpressing MIR517C as compared with NC and MIR517C (kn) cells in both normal medium and TMZ culture ().
Figure 7. MIR517C inhibited autophagy through targeting of KPNA2. (A) Two-dimensional electrophoresis analysis of U87 cells transfected with MIR517C mimics or NC. Distinct protein spots could be observed (red dotted area, red arrow). The target spot was confirmed as KPNA2 by mass spectrometry analysis. (B) Western blotting confirmed the expression of KPNA2 protein in U87 and U251 cells following overexpression (oe) or knockdown (kn) of MIR517C. NC cells were used as a control. The right histogram shows densitometric analysis (#, P > 0.05; *, P < 0.05; **, P < 0.001). (C) U87 and U251 cells exhibiting KPNA2 overexpression or knockdown were established, and autophagy-related protein levels were measured by western blotting. (D) Densitometric analysis of the western blots shown in (C). #, P > 0.05; *, P < 0.05; **, P < 0.001. (E) Immunofluorescence analysis of LC3B expression in U87 cells following overexpression or knockdown of KPNA2. LC3B particle data were subjected to statistical analysis (*, P < 0.05; **, P < 0.001). (F) Sequence matching between MIR517C and the 3′-UTR of KPNA2 is shown. U87 cells were transfected with MIR517C mimic, a mimic control (NC), AntagomiR, or nontargeting AntagomiR (AntagomiR NC). The level of MIR517C was assayed by Taq Man qRT-PCR. (G) Luciferase assays in U87 cells cotransfected with pGL3-control reporter constructs containing the KPNA2 3′-UTR MIR517C-binding site (pGL3-KPNA2-UTR) or a mutated binding site (pGL3-KPNA2-UTRmt) and MIR517C mimic, mimic control, AntagomiR, or nontargeting AntagomiR for 72 h. Samples were run in triplicate, and 3 independent experiments were performed.
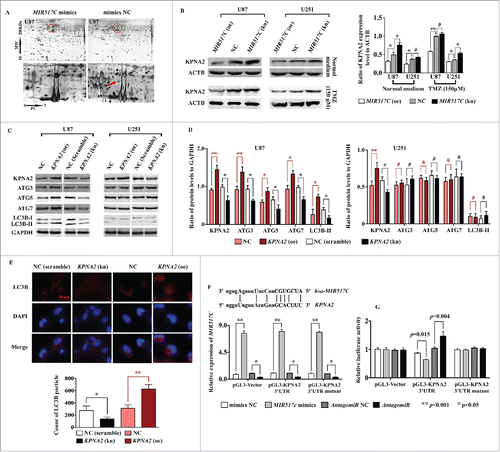
We then asked whether the autophagic and EMT-related functions of MIR517C were achieved through regulation of KPNA2 expression. Interestingly, compared with NC cells, the knockdown of KPNA2 in U87 cells led to downregulation of ATG3, ATG5, and ATG7, whereas overexpression of KPNA2 led to upregulation of these proteins ( ). Consistent with this, overexpression of KPNA2 increased the fluorescence intensity of LC3B punctate staining, whereas knockdown of KPNA2 weakened the intensity of LC3B staining, as compared with the NC group (). However, no changes were observed among the groups in U251 cells.
To determine whether MIR517C directly targeted the 3′-untranslated region (UTR) of KPNA2 mRNA, dual luciferase reporter plasmids were constructed by inserting WT and MU 3′-UTRs of KPNA2 mRNA bearing MIR517C putative binding sites into the pGL3-control vector immediately downstream of the luciferase coding sequence. The above constructed plasmids were transfected into U87 cells by electroporation. MIR517C mimic, mimic NC, AntagomiR, and Nontarget AntagomiR were also cotransfected into cells. The expression level of MIR517C was verified by qRT-PCR, and the results are shown in . After detection of luciferase reporter assays, the luciferase activity was significantly inhibited by MIR517C in the reporter bearing the WT KPNA2 3′-UTR, but not in the plasmid with a mutated KPNA2 3′-UTR (). In contrast, MIR517C (kn) significantly increased the luciferase activity of the reporter containing the WT KPNA2 3′-UTR.
KPNA2 binds to the nuclear location signal (NLS) of cargo protein and acts as an adaptor to KPNB. Importantly, KPNA2 is involved in the nuclear transport of several important transcriptional factors, including TP53. Because the expression status of TP53 is different between U87 and U251 cells, we speculated that MIR517C may function through KPNA2, disturbing the nucleocytoplasmic transport of WT TP53 protein and consequently inhibiting autophagy and the EMT. To test this hypothesis, we used immunofluorescence analysis, as shown in . While overexpression of MIR517C in U87 cells caused accumulation of TP53 protein in the cytoplasm compared to NC and MIR517C (kn) U87 cells, no significant differences were observed in U251 cells. The interaction between KPNA2 and TP53 was confirmed using immunofluorescence analysis and coimmunoprecipitation (IP) in U87 cells (). Western blot analysis of the nuclear and cytoplasmic fractions () further demonstrated that MIR517C functioned to interrupt the nuclear translocation of TP53, which occurred not only in U87 cells (harboring WT TP53), but also in U251 cells (harboring MU TP53).
Figure 8. MIR517C inhibited autophagy by interfering with the nucleocytoplasmic transport of TP53 through KPNA2 in U87 cells. (A) Representative confocal immunofluorescence images of TP53 in U87 and U251 cells following overexpression (oe) or knockdown (kn) of MIR517C. NC cells were used as a control. (B) Immunofluorescence analysis was used to determine the colocalization of KPNA2 and TP53. (C) CoIP assays were used to detect interactions between KPNA2 and TP53. (D) Cells exhibiting stable knockdown of TP53 were established, and TP53 protein levels were analyzed by western blotting. (E) Densitometric analysis of the western blots shown in (D) (#, P > 0.05; *, P < 0.05; **, P < 0.001). (F) Cells exhibiting stable knockdown of TP53 were established, and the expression level of TP53 was detected by western blot. (G) MIR517C mimics, NC, and AntagomiR were transfected into TP53(kn) cells, and the expression levels of autophagy-related proteins were detected by western blotting and densitometric analysis. (H) Densitometric analysis of the western blots shown in (G). #, P > 0.05.
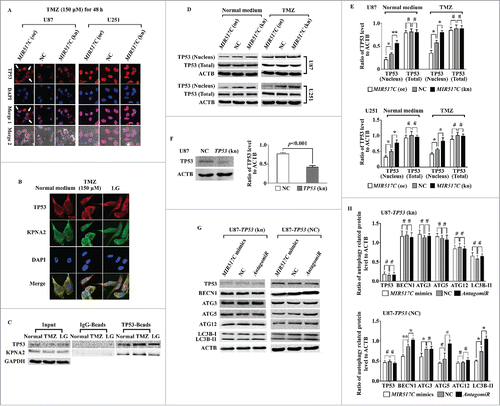
To further explore the requirement for TP53, we established a TP53 stable knockdown cell line in U87 cells (TP53 kn) and verified TP53 knockdown by western blotting (). Importantly, we found that knockdown of TP53 protein in U87 cells abolished the previously observed changes in ATG3, ATG5, and ATG12 levels as well as the conversion of LC3B protein, indicating that the autophagy inhibitory function of MIR517C depended on the function of WT TP53 ().
Validation of the MIR517C-KPNA2-TP53 pathway in TP53−/− and TP53+/+ colon carcinoma cells
To further validate the function of MIR517C through KPNA2 and WT TP53, human TP53+/+ and TP53-/- HCT116 cells were used for western blotting. As shown in , we first confirmed the levels of TP53 protein in the 2 cell types. Then, MIR517C mimic, NC, and AntagomiR were transfected into the cells, and levels of KPNA2 and autophagy-related proteins were determined. Interestingly, overexpression of MIR517C led to significant downregulation of KPNA2 and autophagy-related proteins in these cells, similar to the effects observed in U87 glioma cells. The conversion of LC3B-II also decreased. However, in TP53-/- cells, although KPNA2 protein levels were downregulated following overexpression of MIR517C, the levels of autophagy-related proteins did not differ among groups ().
Figure 9. Validation of the role of MIR517C and KPNA2 in inhibition of autophagy in TP53+/+ and TP53-/- HCT116 colon cancer cells. (A) Western blotting was used to measure TP53 protein levels in TP53+/+ and TP53-/- HCT116 cells. The histogram on the right shows the results of densitometric analysis. (B) MIR517C mimics, NC, or siRNA were transfected into colon cancer cells, and the expression levels of KPNA2 and autophagy-related proteins were detected by western blotting. (C) Densitometric analysis of the western blots shown in (B). #, P > 0.05; *, P < 0.05; **, P < 0.001.
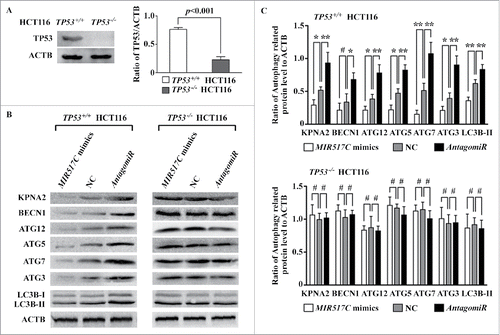
MIR517C reduced tumor invasion and inhibited autophagy and the EMT phenotype in a mouse xenograft model
Along with the in vitro cellular data, we also tested whether MIR517C could reduce tumor invasion and inhibit autophagy and the EMT phenotype in vivo. Therefore, we analyzed the role of MIR517C in vivo in a nude mouse model (see Materials and Methods). U87 MIR517C (kn) cells formed larger tumors than MIR517C (oe) and NC cells (). Moreover, as compared to the MIR517C (oe) tumors, NC and MIR517C (kn) tumors exhibited clear characteristics of invasion (). The surfaces of tumors derived from MIR517C (oe) cells were smooth, without significant adhesions to periphery tissue, except in one case in which the tumor was found to be firmly adhered to the connective tissue and periphery fasciae. Conversely, tumors derived from U87 NC and MIR517C (kn) cells exhibited frequent invasion into connective tissue, with severely obscured boundaries (). Some cells even began to infiltrate into the neighboring muscle tissue, without any evident interface. Observations by fluorescence microscopy and H&E staining indicated that these differences in invasive characteristics were quite obvious; the borders of MIR517C (oe) U87 cell-derived tumors were clear, and the edges between the tumor and fasciae were distinct. However, U87-NC tumor interfaces were irregular, and some tumor parenchyma had infiltrated into neighboring connective and muscle tissue (). In some MIR517C (kn) U87 cell-derived tumors, tumor cells accumulated in the surrounding muscle, exhibiting extensive infiltration and the formation of islets within the muscle.
Figure 10. Inhibition of autophagy and the EMT by MIR517C in a mouse model. (A) MIR517C (oe), NC, and MIR517C (kn) U87 cells were subcutaneously injected into the flanks of nude mice, with MIR517C (oe) and MIR517C (kn) U87 cells injected into the left side, and NC U87 cells injected into the right side. Tumor volumes were measured every 4 d for 24 d, and mice were then sacrificed. Fluorescence images of tumors are shown. (B) Tumors were dissected, and tumor invasion and infiltration was examined. The blue arrow indicates tumor invasion into the surrounding fascia and connective tissue, while the red arrow shows invasion into the surrounding muscle tissue. (C) Fluorescence microscope images and H&E staining of tumor samples. The red arrow shows invasion of the tumor cells with GFP expression into the muscle fibers. (D) Representative transmission electron microscope images of tumors derived from MIR517C (oe), NC, and MIR517C (kn) U87 cells. Red arrows indicate formation of autophagic vacuoles. (E) Immunohistochemical staining of EMT-related proteins in tumors derived from MIR517C (oe), NC, and MIR517C (kn) U87 cells. The higher-magnification images are shown at the upper-right corner of each image.
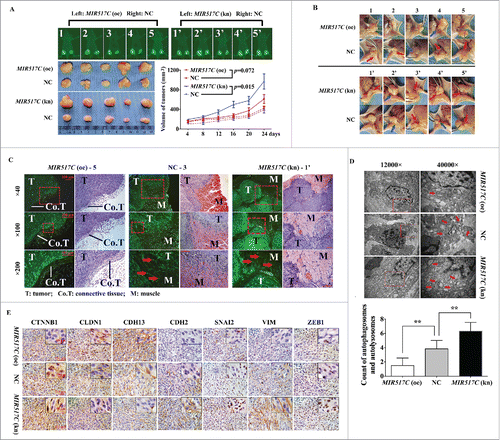
TEM observations demonstrated that the numbers of autophagic vacuoles differed among the 3 groups (), with MIR517C (oe) U87 cell-derived tumors exhibiting lower numbers of autophagic vacuoles than tumors derived from NC and MIR517C (kn) cells. This finding was consistent with our in vitro results described above. Additionally, immunohistochemical staining () revealed that nuclear expression of CTNNB1 was upregulated, supporting the induction of the EMT.Citation4 As the most important components of epithelial tight junctions, CLDN1 (a membrane protein) had lower expression in tumors derived from MIR517C (kn) cells. Moreover, MIR517C inhibited the conversion of CDH13 to CDH2, which is thought to provide a mechanism for transendothelial migration.Citation31 Other EMT markers, such as VIM (cytoplasmic) and the EMT regulators SNAI2 (nuclear) and ZEB1 (nuclear), were downregulated in tumors derived from MIR517C (oe) cells.
Taken together, these data indicated that MIR517C played an important role in determining the EMT phenotype through interfering with the autophagy status in GBM cells expressing WT TP53.
Discussion
In this study, we examined the role of MIR517C in autophagy and the EMT in GBM. We found that MIR517C expression was associated with patient prognosis in GBM and that MIR517C modulated autophagy and the EMT through a pathway involving TP53 and KPNA2. Therefore, MIR517C may represent a novel therapeutic target for the treatment of GBM.
The EMT represents the molecular reprogramming and phenotypic changes involved in the conversion of polarized immotile epithelial cells to motile mesenchymal cells. This process allows the remodeling of tissues during embryonic development and is also implicated in the promotion of tumor invasion and metastasis. Several recent studies have identified the MIR200 family and MIR205 as key regulators of the EMT and enforcers of the epithelial phenotype.Citation32,33 Interestingly, in this article, MIR517C overexpression in U87 GBM cells inhibited migration either in vitro or in vivo, concomitant with induction of epithelial features. We observed the upregulation of SNAI2, SNAI1, VIM, CTNNB1, and CDH2 in U87 cells, which was surprisingly associated with CLDN1 and CDH13 downregulation. This observation implied that there may be an alternative cadherin switch distinct from the classical CDH1/E-cadherin to CDH2 switch in epithelial cells.Citation34 Western blot analysis of 2 panels of human GBM cell lines confirmed that CDH1 expression was rare as it is often replaced by CDH13.Citation35 In GBM cells, overexpression of CDH13 suppresses proliferation and migration,Citation36 consistent with our observations in nude mice. In addition, we found that MIR517C overexpression suppressed CDH13, suggesting that MIR517C inhibited EMT signals to affect CDH13 transcription. Our finding further supported a role for CDH13 in the acquisition of the EMT-like phenotype in GBM, consistent with the results of a previous study.Citation31
Our study further indicated that inhibition of the EMT by MIR517C occurred via the inhibition of autophagy. Autophagy is a process of dynamic flux involving sequestration of phagophores, generation of autophagosomes or amphisomes, and fusing to lysosomes (autolysosomes). In this study, we monitored autophagy based on recently published guidelinesCitation19 using western blotting, immunofluorescence, and TEM. Moreover, the lipidated form of LC3B (LC3B-II) has been used as a reliable marker of completed autophagosomes. Using these tools, we found that TMZ upregulated the conversion of LC3B-II and increased LC3B expression, demonstrating that autophagy was stimulated. Moreover, 3-MA and CQ, common autophagic inhibitors, can alter autophagic flux in early and late stages, respectively.Citation25 3-MA treatment decreased LC3B-II conversion and the punctate expression of LC3B, which reflected the inhibition of autophagosome formation. However, CQ conversely increased LC3B-II expression by blocking the fusion of autophagosomes to lysosomes and reducing the degradation of autophagosomes. We also found that overexpression of MIR517C caused a significant reduction in the numbers of both autophagosomes (early autophagic vacuoles) and autolysosomes (late autophagic vacuoles), indicating that MIR517C blocked the progression of autophagy. Therefore, these data suggested that MIR517C may interfere with the formation of autophagosomes rather than blocking the fusion of the autophagosomes to lysosomes. We found that induction of autophagy increased cell migration and infiltration in GBM cells; these effects were subsequently blocked by inhibitors of autophagy (i.e., 3-MA and CQ). Importantly, knockdown of ATG7, an important autophagy-related protein, blocked the acquisition of the EMT phenotype. In order to confirm the role of ATG7 in TP53 function as reported by Lee et al.,Citation37 we used BECN1 siRNA; these results further revealed the link between autophagy and the EMT in GBM cells. Previous studies have established that autophagy is an evolutionarily conserved, multistep lysosomal degradation process in which a cell digests intracellular organelles when it needs to ‘self-cannibalize’.Citation38 This may serve to regulate normal turnover and maintain homeostasis by removing damaged organelles and broken DNA under metabolic stress. However, in cancer, the current consensus is that autophagy has a dual role. On the one hand, autophagy functions as a tumor-suppressor mechanism by preventing the accumulation of damaged organelles and aggregated proteins. On the other hand, autophagy is a key cell survival mechanism for established tumors.Citation15 Interestingly, when cells with defects in apoptosis cannot undergo autophagy, this creates the inability to tolerate metabolic stress, reduces cellular fitness, and activates a necrotic pathway to cell death. This necrosis in tumors is associated with inflammation and enhancement of tumor growth, due to the survival of a small population of surviving, but injured, cells in a microenvironment that favors oncogenesis.Citation39 Moreover, recent studies have suggested that autophagy is also an important mediator in the invasion of cancer cells. For example, autophagy accelerates the invasion of cancer cells during starvation or hypoxia.Citation18,40 Some new studies have revealed the close relationship between autophagy and the EMT. For example, Li et al.Citation16 report that starvation-induced autophagy is critical for cellular invasion through induction of the EMT in hepatocellular carcinoma cells. Furthermore, in breast carcinoma, Akalay et al.Citation41 show that EMT-induced tumor cell resistance to cytotoxic T-lymphocytes (CTLs) is correlated with the induction of autophagy. In another study,Citation42 inhibition of autophagy enhances transcriptional activation of the CD24 gene, potentiating the epithelial-like phenotype of CD44 (+) CD24 (+) cells versus the mesenchymal CD44 (+) CD24 (-/low) progeny, which is also evidence that induction of autophagy promotes the EMT. In glioma cells, inhibition of autophagy using ATG12 shRNA reduced cellular invasion in a 3-dimensional (3D) organotypic model, suggesting that there may be an interaction between the EMT and autophagy in glioma.Citation12 However, the specific contribution of autophagy to GBM invasion and the EMT is still unclear.
Our results indicated that induction of autophagy promoted the occurrence of the EMT phenotype only in GBM cells expressing WT TP53. Notably, we did not establish a dominant mutation of TP53 in U87 cells owing to the many complicated domains of functional TP53. Establishment of an effective dominant mutation may provide more direct evidence of the mechanism through which autophagy alters the EMT status in glioblastoma, and further studies are required to achieve this in the future. In this study, we established TP53 stable knockdown cells to prove that the relationship between autophagy and EMT was dependent on the TP53 phenotype, which was further confirmed in TP53+/+ and TP53-/- HCT116 colon cancer cells.
Moreover, we found that this mechanism proceeded through a pathway involving MIR517C, KPNA2 and TP53. As a member of the karyopherin family, KPNA2 has been investigated as a promising biomarker and prognostic factor in several types of solid tumors, including breast cancer, ovarian cancer, prostate cancer, and melanoma.Citation43-46 In our study, KPNA2 was found to be a direct target of MIR517C, and overexpression of KPNA2 blocked the function of MIR517C. Although we do not have direct data showing the precise role of KPNA2 in glioma, based on the improved outcomes in patients exhibiting elevated expression of MIR517C, we hypothesize that KPNA2 may act as an oncogene in GBM. These findings are supported by a study by Gousias et al.,Citation46 who show that patients with astrocytoma who exhibit higher expression of KPNA2 have poorer prognoses and increased resistance to chemotherapy, suggesting that KPNA2 may be a diagnostic and prognostic marker for astrocytoma.
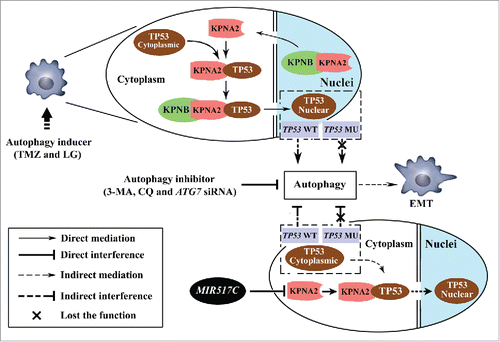
KPNA2 is a key adaptor protein involved in the nucleocytoplasmic transition of several important transcription factors, such as TP53, NFKB/NF-κB (nuclear factor kappa-B), and MAPK1. In this study, cytoplasmic accumulation of TP53 was observed in U87 cells overexpressing MIR517C, suggesting that MIR517C interrupted the nuclear translocation of TP53 by inhibiting the expression of KPNA2. Our results are consistent with studies by KimCitation47 and Moll,Citation48 in which the expression of a truncated form of karyopherin isolated from a breast cancer cell line impairs TP53 nuclear import and constitutive cytoplasmic localization of TP53. This, in turn, results in increased expression of genes involved in cell proliferation. Our study provided additional evidence for the interaction between KPNA2 and TP53 in U87 cells. In another recent study in non-small cell lung cancer, the association between KPNA2 and TP53 is also demonstrated,Citation49 and TP53 downstream targets are also affected by KPNA2 knockdown. TP53, known as the “guardian of the cellular genome” and the “cellular gatekeeper," plays a fundamental role in the regulation of autophagy according to its cellular location. In the nucleus, TP53 functions as a proautophagic factor in a transcription-dependent or -independent manner. However, in the cytoplasm, TP53 suppresses the induction of autophagy.Citation50 Thus, downregulation of KPNA2 can interfere with autophagy through disruption of the nuclear import of TP53, which was also shown in our current study. Therefore, we suggest that a pathway involving MIR517C, KPNA2 and TP53 is involved in the regulation of autophagy and the EMT (). Of note, cytoplasmic TP53 is sensitive to proteasomal degradation. In terms of the autophagy-lysosomal system and ubiquitin-proteasomal system, the 2 most important components of cellular mechanisms for protein degradation, growing evidence supports that there is crosstalk between the 2 systems.Citation51,52 Under conditions inducing autophagy, proteasomal degradation may be downregulated.Citation53 Results similar to the cytoplasmic TP53 accumulation observed in our study are also observed by Giorgi and coworkers, who find that TP53 accumulates in the cytoplasm of HCT116 cells treated with doxorubicin or hydrogen peroxide.Citation54 Although the details of these mechanisms are still unclear, cytoplasmic TP53 has been widely reported to inhibit autophagy through extranuclear, transcription-independent mechanisms.Citation50
Figure 11. Schematic drawing of the MIR517C-KPNA2-cytoplasmic TP53 pathway. In TP53 wild-type GBM cells, autophagy inducers (TMZ and LG) stimulated autophagy dependent on the cytoplasmic-nuclear translocation of TP53 mediated by KPNA2. The EMT status of glioblastoma cells was altered to increase cell migration and invasion; this could be blocked by treatment with autophagy inhibitors, i.e., 3-MA and CQ, and by ATG7 siRNA. In contrast, MIR517C was able to directly target and degrade the KPNA2 protein and could interrupt the nuclear translocation of functional TP53 protein. As result of accumulation of cytoplasmic TP53, the autophagy induced by TMZ and LG was attenuated, eliminating the increases in migration and invasion observed during the EMT.
Although in this study we only used U87 cells harboring WT TP53 and U251 harboring MU TP53 as GBM model cells, TP53+/+ and TP53-/- HCT116 cells were used to demonstrate the WT TP53-dependent function of MIR517C and KPNA2 in the single altered cell background. Furthermore, we are planning to design several mutants in the functional domains of TP53 in order to investigate the details of the MIR517C-KPNA2-TP53 pathway in the regulation of autophagy and the EMT in GBM. Finally, a recent study shows that downregulation of MIR517C facilitates hepatocellular carcinoma cell growth.Citation22 In our present study, we provided further evidence that MIR517C could act as an “autophagamiR” in GBM; perhaps its function will not only regulate autophagy but also the EMT. Further studies are required to examine these relationships and for the development of potential therapeutic strategies for the treatment of GBM. In particular, potent MIR517C mimetics may selectively inhibit glioma cell migration, which may therefore represent an attractive target for novel molecular therapies for malignant glioma.
Materials and Methods
Ethics statement
Written informed consent was provided from all patients and their families for the use of clinical information and samples for research purposes. The protocol was approved by the Ethics Committees of Nanfang Hospital.
Clinical tumor sample collection
Forty-six patients (26 men and 20 women; median age, 49.7 ± 9.7 y [range, 32 to 68 y]) with newly diagnosed GBM who had undergone surgery plus standard chemoradiotherapy were included. Gliosarcoma variant cases and cases in patients younger than 18 y of age were excluded from this cohort. All specimens had confirmed pathological diagnosis and were classified according to the World Health Organization (WHO) criteria. DNA and RNA were extracted by standard methods. All samples were collected from the Nanfang Hospital of Southern Medical University (Guangzhou, China) at the time of surgery before any other therapies. Fresh samples were immediately preserved in liquid nitrogen. Tumor samples and clinical information were collected with written informed consent for each patient. The protocols were approved by the ethics committee of Southern Medical University.
RNA isolation and real-time qPCR
To validate the miRNA expression data, qPCR was performed using an ABI StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Total RNA from clinical glioma samples and glioma cell lines was isolated using TRIzol reagent (Invitrogen, 15596-026) according to the manufacturer’s instructions. Single-stranded cDNA was synthesized from 10 ng of RNA using an miR-specific stem-loop reverse-transcription RT-primer and the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, 4366596). Noncoding controls for RNU6, RNU44, RNU48, and RNU6B were expressed at similar levels in all tumor samples and normal fetal brain samples (data not shown); therefore, all miRNA expression levels were determined relative to RNU6B using the formula 2-ΔCT. Independent experiments were performed in triplicate.
Reagents
TMZ (3-methyl-4-oxo-8-imidazolo [5,1-d] [1,2,3,5] tetrazinecarboxamide) was purchased from Sigma Chemical Co. (T2577) and was dissolved in DMSO (Sigma, D2650). The dissolved TMZ was then added to complete culture medium at final concentrations of 150 and 300 μM. 3-MA (Sigma, M9281) was dissolved in DMEM at 60°C and diluted to 2.5 mM before use. CQ (Sigma, C6628) was dissolved in sterile phosphate-buffered saline (Sigma, 08057) for a final concentration of 50 μM.
Cell lines and culture conditions
U87, U251, and HEK293T cells were purchased from the Chinese Academy of Sciences (Shanghai, China). TP53+/+ and TP53-/- HCT116 cells were a kind gift from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, containing 4.5 g/L glucose; Gibco, 11995-065) with 10% fetal bovine serum (Gibco, 16140-071), 100 U/mL penicillin, and 100 mg/mL streptomycin (Gibco, 15140-122). All cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2. For LG culture, the DMEM contained only 1.0 g/L glucose (Gibco, 11885-084).
Use of NIH ImageJ (Bethesda, MD, USA; http://rsb.info.nih.gov) to measure the length, width, and area of cells
For measurement of cell parameters, including length, width, and area, we used ImageJ. In the image to be analyzed, the measurement scale was set, and a line was drawn between 2 points of known distance, i.e., using a ruler on the photograph. This was used to set the scale under the Analyze tab. Additionally, in the Set Scale window the length of the line, in pixels, will be displayed. The known distance and units of measure should be typed into the appropriate boxes. A line should then be drawn between the 2 points to show the cell length or width. The status bar will show the angle (from horizontal) and the length. The Measure button under the Analyze tab could then be used to transfer the values to a data window. The area selection tool, wand, or Analyze Particles function was then used to surround the area of the whole cell. These data could be transferred to the data window as the measurement of area. The NeuronJ plug-in for NIH ImageJ software was further used to measure the length of pseudopodia according to a previous report. Citation55
TEM imaging
MIR517C (oe), MIR517C-NC, and MIR517C (kn) U87 and U251 cells after TMZ treatment and freshly harvested tumors from mice were fixed with 2.5% glutaraldehyde, washed, and postfixed in osmium. Following dehydration by serial dilutions of ethanol, cells were stained with uranyl acetate and embedded in culture dishes. Thin sections of the cured blocks were cut with a diamond knife, stained with lead citrate, and then observed and photographed by TEM (HITACHI H-7650, Tokyo, Japan).
Antibodies and western blotting
Cells were lysed in RIPA Buffer (Cell Signaling Technology, 9800), and protein concentrations were determined by using BCA assays (Cell Signaling Technology, 7780). Total protein (40 μg) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 10% gel and electrotransferred to polyvinylidene fluoride membranes. Membranes were then blocked with 5% nonfat dry milk in Tris-buffered saline (Sigma, T7777, pH 7.6) and were incubated overnight at 4°C with rabbit monoclonal anti-LC3B, anti-BECN1/Beclin, anti-ATG3, anti-ATG5, anti-ATG7, anti-ATG12, anti-CTNNB1/β-catenin (detecting total CTNNB1), anti-VIM/Vimentin, anti-CLDN1/Claudin, anti-SNAI1/Snail, anti-SNAI2/Slug, anti-ZEB1, and anti-CDH2/N-cadherin antibodies (Cell Signaling Technology, Autophagy Antibody Sampler Kit 4445 and EMT Antibody Sampler Kit 9782). Alternatively, blots were incubated with anti-CDH13/T-cadherin antibodies (Millipore, ABT121), mouse polyclonal anti-ACTB/β-actin antibodies (Cell Signaling Technology, 4967), rabbit monoclonal anti-CASP9/caspase-9 and anti-CASP3/caspase-3 antibodies (Cell Signaling Technology, Apoptosis Antibody Sampler Kit 9915), rabbit polyclonal anti-KPNA2 (Sangon Biotech, D154103-0200), or rabbit monoclonal anti-TP53 antibodies (Cell Signaling Technology, 9282). HRP-conjugated anti-rabbit and anti-mouse IgG antibodies were obtained from Cell Signaling Technology (7074 and 7076).
Analysis of LC3B particles using ImageJ software
For analysis of LC3B particles, the image was opened and converted to gray scale (8 or 16 bit). The threshold was adjusted to highlight the target structures. The background was subtracted using the rolling ball feature to delete “noisy” particles. “Apply” was then chosen to create a binary version of the image. After setting the proper parameters for the particles, “analyze particles” was used to obtain the particles of interest.
Cell wound healing and cell migration assays
Confluent cell monolayers were grown in 6-well plates and gently scratched with a plastic pipet tip to create a uniform, cell-free wound area. Fresh or conditioned medium was then applied with or without TMZ, CQ, or 3-MA, as indicated. Photomicrographs of the wound area were taken at 0, 12, 24, and 48 h.
For cell migration assays, 1 × 104 cells in 100 μL DMEM without fetal bovine serum were seeded on fibronectin-coated polycarbonate membrane inserts in a transwell apparatus (Corning, 3422). In the lower chamber, 500 μL of conditioned medium was added as a chemoattractant. After the cells were incubated for 8 h at 37°C in a 5% CO2 atmosphere, the inserts were washed with PBS, and cells on the top surface of the inserts were removed with cotton swabs. Cells adhering to the lower surface were fixed with methanol, stained with 0.1% crystal violet solution, and counted under a microscope in 6 high-resolution predetermined fields. All assays were independently repeated at least 3 times.
Wound healing image capture and data analysis Citation56
Images at time zero (t = 0 h) were captured to show the initial area of the wounds, and the recovery of the wounded monolayers by cell migration toward the wound was analyzed at 24 and 48 h (t = Δ h). The area of the wound was quantified using ImageJ software with the polygon selection mode. The cells’ migration area toward the wounds was expressed as percentage of wound closure: % of wound closure =[A(t = 0 h) – A(t = Δh)] / A(t = 0 h) × 100%, where A(t = 0 h) is the area of wound measured immediately after scratching, and A(t = Δh) is the area of the wound measured 24 or 48 h after scratching.
Transient knockdown of ATG7 in U87 and U251 cells
siRNA targeting ATG7 was synthesized (GenePharma). Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, 11668-019) 24 h after plating. Transfection complexes were prepared according to the manufacturer’s instructions and added directly to the cells to a final oligonucleotide concentration of 100 nM. Transfection medium was replaced 8 h post-transfection. The sequence of ATG7 siRNA was 5′-ACUAAAAGGGGCAAACUGCAG-3′.
Stable miRNA expression
U87 and U251 cells were infected with Lv3 pGLV-MIR517C mimetic, Lv3 pGLV-MIR517C inhibitor, or control Lv3 pGLV-NC (GenePharma, 130122AZ). The infection efficiency was evaluated by GFP expression, as observed under a fluorescence microscope (DP71, Olympus, Japan). The stable cell lines were selected in puromycin (1 mg/mL; Sigma, P8833) and tested for miRNA expression by qRT-PCR.
The sequences contained in the viruses were as follows: Lv-MIR517C mimetic: 5′-AUCGUGCAUCCUUUUAGAGUGU-3′; Lv-MIR517C inhibitor: 5′-ATCGTGCATCCTTTTAGAGTGT-3′; LV-GFP-NC: 5′-ACTACCGTTGTTATAGGTG-3′.
Oligonucleotide transfection (MIR517C mimics, mimics-NC, AntagomiR, and Nontarget AntagomiR)
Prepared cells were transfected with chemical synthetized RNA oligonuclotides: MIR517C mimic, negative control (mimic-NC), or MIR517C AntagomiR or Nontarget AntagomiR purchased from GenePharma (Shanghai, China). Sequences of the oligonuclotides were as follows: the MIR517C mimic was a synthetic RNA duplex with a sense strand sequence of 5′-AUCGUGCAUCCUUUUAGAGUGU-3′; mimic-NC was a synthetic RNA duplex with a sense strand sequence of 5′-UUCUCCGAACGUGUCACGUTT-3′; MIR517C AntagomiR (inhibitor) was a signal strand with strand sequence of 5′-CAGUACUUUUGGGUAUGACAA-3′; and the Nontarget AntagomiR (negative control) had the sequence 5'-CAGUACUUUUGUGUAGUACCA-3′. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, 11668-019) 24 h after plating. Transfection complexes were prepared according to the manufacturer’s instructions and added directly to the cells to a final oligonucleotide concentration of 100 nM. Transfection medium was replaced 8 h post-transfection.
Mutational analysis of the TP53 gene by PCR
See Supplemental Data.
Cell proliferation, apoptosis, and cell cycle analysis
See Supplemental Data.
Construction of the luciferase reporter of the KPNA2 3′-UTR
To generate luciferase reporter vectors containing a putative MIR517C binding site, sense and antisense strands of the oligonucleotides of the KPNA2 3′-UTR were synthesized, annealed, and subcloned into the pGL3-control vector (Promega, E1741) in KpnI and XhoI sites immediately downstream of the stop codon of the luciferase gene. A mutated form of the KPNA2 3′-UTR was used to replace the complementarity to nucleotides 4 to 7 of the MIR517C seed region. Cloning orientation was verified by sequencing.
Luciferase reporter assay
Luciferase activity assays were performed using the Dual-Luciferase Reporter Assay System (Promega, E1910) according to the manufacturer’s instructions. For the KPNA2 3′-UTR luciferase assay, U87 cells were cotransfected with wild-type or mutant pGL3-KPNA2-3′UTR constructs and miRNA oligonucleotide (MIR517C mimetic, mimetic-NC, AntagomiR, or Nontarget AntagomiR). Cells were lysed 48 h after transfection, and firefly luciferase and Renilla luciferase activities were determined with using the Dual-Luciferase Reporter System.
Two-dimensional electrophoresis
Proteins were extracted from cells and lysed in 1 mL of lysis buffer containing 7 M urea, 2 M thiourea, 4% CHAPS (Sigma, C3023-25G), 65 mM DTT, and 2% Pharmalyte (pH 4–7; GE Healthcare, 17-6000-86) followed by sonication on ice. The lysates were centrifuged at 12,000g for 1 h at 4°C. Subsequently, the protein concentrations of the supernatants were determined by the modified Bradford method, and aliquots of protein samples were stored at -80°C. Prior to 2-DE, the protein samples were purified using a 2D Clean-Up Kit (GE Healthcare, 80648451) according to the manufacturer’s instructions. Differentially expressed proteins were identified using 2D gel electrophoresis and mass spectrometry; 2D gel electrophoresis was performed using 4–7 pI Immoboline strips, and proteins were separated according to charge and then according to molecular weight. The gels then underwent silver staining in order to visualize proteins, and protein spots of interest were cut from the gels. Proteomics analysis was repeated 3 times.
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/TOF MS)
MALDI-TOF/TOF MS measurements were performed on a Bruker Ultraflex III MALDI-TOF/TOF MS (Bruker Daltonics, Germany) operating in reflectron mode with 20 kV accelerating voltage and 23 kV reflecting voltage. A saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 0.1% trifluoroacetic acid was used as the matrix. One microliter of the matrix solution and sample solution at a ratio of 1:1 was applied onto the Score 384 target well. A standard peptide calibration mix in the mass range of 800 to 3200 Da (Bruker Daltonics) was analyzed for external calibration of the mass spectrometer. The calibration mix contained Angiotensin II, Angiotensin I, Substance P, Bombesin, ACTH clip 1 to 17, ACTH clip 18 to 39, and Somatostatin 28. A series of 8 samples was spotted around one external calibration mixture. The SNAP algorithm (S/N threshold: 5; quality factor threshold: 30) in FlexAnalysis 3.0 was used to identify the 100 most prominent peaks in the mass range m/z 700 to 4000. The subsequent MS/MS analysis was performed in a data-dependent manner, and the 5 most abundant ions fulfilling certain preset criteria (S/N higher than 3 and quality factor higher than 30) were subjected to high-energy CID analysis. The collision energy was set to 1 keV, and nitrogen was used as the collision gas.
Data interpretation and database searching
Peptide mass fingerprints (PMFs) were analyzed using Mascot 2.1 (Matrix Science Ltd.) against the SwissProt database. The search parameters were as follows: trypsin digestion with one missed cleavage; carbamidomethyl modification of cysteine as a fixed modification and oxidation of methionine as a variable modification; peptide tolerance maximum, ± 0.3 Da; MS/MS tolerance maximum, ± 50 ppm; peptide charge, +1; monoisotopic mass. P < 0.05 was used for the local PMF search. For unambiguous identification of proteins, more than 5 peptides must be matched for a PMF search.
KPNA2 overexpression and knockdown
For the KPNA2 overexpression vector, the coding region of human KPNA2 (NM_002266.2) was inserted into a pcDNA3 vector (Invitrogen, V790-20) by electroporation. To construct shRNA expression vectors, oligonucleotides with a hairpin structure, overhanging sequences, and terminator were synthesized, annealed, and then inserted into the pSilencer vector.
MIR517c/KPNA2 luciferase reporter assays
The sense and antisense strands of the oligonucleotides of the KPNA2 3′-UTR were synthesized, annealed, and subcloned into the pGL3-basic vector (Promega, E1751). A mutated form of the KPNA2 3′-UTR (“ugguUuguuAcuGuaGCACUUU” was mutated to “ugguUuguuAcuGuaCAGCUUU”) was inserted into the pGL-3-basic vector, and the pGL-3 control vector was used as a control. Luciferase activity assays were performed using the Dual-Luciferase Reporter Assay System (Promega, E1910).
Colocalization (immunofluorescence) and coIP
The details of immunofluorescence analysis are described as previous. For colocalization, after incubating cells for 2 h at 37°C with primary mouse anti-TP53 antibodies (diluted 1:2000 in KB; Cell Signaling Technology, 2524) or rabbit anti-KPNA2 antibodies (diluted 1:100 in KB; Sangon Biotech, D154103-0200), cells were washed with KB twice for 2 min each wash. After incubating for 1.5 h at 37°C with FITC-conjugated goat anti-rabbit IgG antibodies (diluted 1:200 in KB; Keygen Biotech, KGAA26), and Cy3-conjugated goat anti-mouse IgG antibodies (diluted 1:500 in KB; Keygen Biotech, KGAB020), cells were washed with KB twice for 2 min each wash and stained with 0.2 mg/mL DAPI (Keygen Biotech, 17511). Dishes were stored at 4°C in the dark and were observed with a confocal laser-scanning microscope (Olympus, FLUOVIEW FV10i, Tokyo, Japan).
For coIP, 4 × 105 cells were cultured in 10-cm dishes for 2 d and then treated with normal medium, medium containing TMZ (150 μM), or LG medium. Twenty-four h later, the cells were lysed in 10 mL precooled RIPA buffer containing 100 μL PMSF (Cell Signaling Technology, 8553). Anti-TP53 antibodies and IgG antibodies were conjugated to Protein A+G beads (Beyotime, P2012) according to the manufacturer’s instructions. The conjugated beads were rinsed 3 times with RIPA buffer and then added into 3 tubes of each cell lysate. The immunoprecipitates were washed 3 times with lysis buffer and then used for detecting TP53 and KPNA2 by western blotting, as described above.
Construct of TP53-knockdown U87 cells
To construct a TP53-knockdown cell line in U87 cells (TP53 kn), pSilencer vector (Invitrogen, AM5764) was used to construct the TP53 short hairpin RNA (shRNA) expression vector. Gene-specific shRNAs targeting TP53 were ligated downstream of the CMV promoter. The pSilencer-TP53 plasmid was transfected into U87 cells by electroporation, and the U87 (TP53 kn) cell line was established by continuous selection with G418 (Invitrogen, 10131-027). The sequences of TP53 shRNA were as follows: forward, 5′-GACTCCAGTGGTAATCCTAC-3′; reverse: 5′-GTAGGATTACCACTGGAGTCTT-3′.
Tumor xenograft assays in nude mice
The protocol for the mouse xenograft model was approved by the Experimental Animal Center of Southern Medical University. In brief, GBM cells were subcutaneously injected in the flanks of nude mice (BALB/c-nu, 21–28 d old), with cells stably overexpressing MIR517c or expressing MIR517c shRNA injected into the left flank, and NC cells injected into the right flank. Tumor size was measured every 4 d for 24 d, and mice were then euthanized. Mice were subjected to whole-body imaging using an Image Station 2000MM (Kodak, Rochester, USA). Tumor and neighboring tissues were carefully dissected, and samples from core and peripheral regions were dissected for paraffin embedding according to standard procedures. Tissue blocks were used for histochemistry, immunohistochemistry, and transmission electron microscopy study.
Immunohistochemistry and immunofluorescence assay
Tumor specimens from nude mice were embedded by paraffin as routine methods. The tissue slices were obtained for immunohistochemistry according to manufacturer’s protocol. Anti-human CTNNB1/β-catenin (1:200, total CTNNB1), anti-VIM/vimentin (1:200), anti-CDH2/N-cadherin (1:200), anti-CLDN1/Claudin (1:125), anti-SNAI2/slug (1:200), anti-ZEB1 (1:200; Cell Signaling Technology, EMT Sampler Kit, 9782), and anti-CDH13/T-cadherin (1:500; Millipore, ABT121) were used. Stained tissue sections were reviewed and scored separately by 2 pathologists blinded to the clinical parameters.
For immunofluorescence assay, a total of 1 × 105 cells was seeded on glass-bottomed dishes (35-mm dishes with a 10-mm bottom well) and cultured overnight. Subsequently, cells were washed with KB (1.21g Tris [hydroxymethyl] methyl aminomethane, 8.8g NaCl and 3g BSA was dissolved completely in 100ml ddH2O as 10×KB-solution which was kept in 4°C and will be diluted to 1×KB-solution and adjusted its pH to 7.4 before usage) twice then fixed in 4% paraformaldehyde and permeabilized in KB solution and 0.2% Triton X-100 (Sigma, 9002-93-1) at room temperature. After the blocking solution was removed, cells were incubated with primary antibodies targeting LC3B (1:200; Cell Signaling Technology, 4445) or TP53 (1:1500; Cell Signaling Technology, 9282) diluted in KB for 60 min at 37°C. Cells were then washed with KB twice. After incubating for 30 to 45 min at 37°C with Alexa Fluor 594 Goat Anti-Rabbit IgG (H+L) Antibody (diluted 1:2000 in KB), cells were washed with KB again, then stained with 0.2 mg/mL DAPI. Dishes were stored at 4°C in the dark and observed under a fluorescence microscope (Leica, DMI3000, Wetzlar, German) or confocal laser-scanning microscope (Olympus, FLUOVIEW FV10i, Tokyo, Japan).
Statistical analyses
Results are shown as means; error bars represent the standard error of the mean (SEM) or the standard deviation (SD) as indicated. Correlations between MIR517c expression and survival were assessed using the Kaplan Meier method and log-rank (Mantel-Cox) test. Analysis of variance (ANOVA) was used to determine the significance of difference in CCK-8, wound healing, and migration assays. Luciferase reporter assays were statistically analyzed by unpaired, 2-tailed Student t tests. Paired, 2-tailed t tests were used to determine the significance of differences in tumor volumes in the xenograft study. Differences were considered significant when P values were less than 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher’s website.
1108507_Supplemental_Material.zip
Download Zip (8.6 MB)Acknowledgments
We thank Peter Dirk for the generous gift of neural stem cells and technical assistance for stem cell culture. We also thank Shengli An for helping the statistical analysis.
Funding
This project was supported by the National Natural Science Foundation of China (Grant Nos. 81101921), Natural science Foundation of Guangdong Province (Grant Nos. 2014A030313298) and the National Key Clinical Specialist Construction Program of China.
Related Research Data
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987-96; PMID:15758009; http://dx.doi.org/10.1056/NEJMoa043330
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98:10356-61; PMID:11526241; http://dx.doi.org/10.1073/pnas.171610498
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 2005; 5:744-9; PMID:16148886; http://dx.doi.org/10.1038/nrc1694
- Kahlert UD, Nikkhah G, Maciaczyk J. Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett 2013; 331:131-8; PMID:23268331; http://dx.doi.org/10.1016/j.canlet.2012.12.010
- Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012; 119:5782-94; PMID:22394600; http://dx.doi.org/10.1182/blood-2011-09-380410
- Martinez-Orozco R, Navarro-Tito N, Soto-Guzman A, Castro-Sanchez L, Perez Salazar E. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur J Cell Biol 2010; 89:476-88; PMID:20207443; http://dx.doi.org/10.1016/j.ejcb.2009.12.005
- Han SP, Kim JH, Han ME, Sim HE, Kim KS, Yoon S, Baek SY, Kim BS, Oh SO. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell Mol Neurobiol 2011; 31:489-96; PMID:21225336; http://dx.doi.org/10.1007/s10571-010-9643-4
- Kahlert UD, Maciaczyk D, Doostkam S, Orr BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J, Niedermann G, et al. Activation of canonical WNT/beta-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett 2012; 325:42-53; PMID:22652173; http://dx.doi.org/10.1016/j.canlet.2012.05.024
- Brabletz T. To differentiate or not–routes towards metastasis. Nat Rev Cancer 2012; 12:425-36; PMID:22576165; http://dx.doi.org/10.1038/nrc3265
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011; 108:7950-5; PMID:21498687; http://dx.doi.org/10.1073/pnas.1102454108
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448-57; PMID:14713959; http://dx.doi.org/10.1038/sj.cdd.4401359
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548-58; PMID:16946731; http://dx.doi.org/10.1038/sj.cdd.4402030
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359-68; PMID:24991840; http://dx.doi.org/10.4161/auto.28984
- Palumbo S, Pirtoli L, Tini P, Cevenini G, Calderaro F, Toscano M, Miracco C, Comincini S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J Cell Biochem 2012; 113:2308-18; PMID:22345070; http://dx.doi.org/10.1002/jcb.24102
- White E. The role for autophagy in cancer. J Clin Investig 2015; 125:42-6; PMID:25654549; http://dx.doi.org/10.1172/JCI73941
- Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013; 34:1343-51; PMID:23430956; http://dx.doi.org/10.1093/carcin/bgt063
- Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, Zhou J, Qiu SJ, Dai Z, Fan J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013; 9:2056-68; http://dx.doi.org/10.4161/auto.26398
- Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012; 11:2022-9; PMID:22580450; http://dx.doi.org/10.4161/cc.20424
- Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C, Collins VP, Van Meter T, Picard D, Zhou L, et al. Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 2009; 16:533-46; PMID:19962671; http://dx.doi.org/10.1016/j.ccr.2009.10.025
- Flor I, Bullerdiek J. The dark side of a success story: microRNAs of the C19MC cluster in human tumours. J Pathol 2012; 227:270-4; PMID:22374805; http://dx.doi.org/10.1002/path.4014
- Augello C, Vaira V, Caruso L, Destro A, Maggioni M, Park YN, Montorsi M, Santambrogio R, Roncalli M, Bosari S. MicroRNA profiling of hepatocarcinogenesis identifies C19MC cluster as a novel prognostic biomarker in hepatocellular carcinoma. Liver Int 2012; 32:772-82; PMID:22429613; http://dx.doi.org/10.1111/j.1478-3231.2012.02795.x
- Liu RF, Xu X, Huang J, Fei QL, Chen F, Li YD, Han ZG. Down-regulation of miR-517a and miR-517c promotes proliferation of hepatocellular carcinoma cells via targeting Pyk2. Cancer Lett 2013; 329:164-73; PMID:23142219; http://dx.doi.org/10.1016/j.canlet.2012.10.027
- Ward A, Shukla K, Balwierz A, Soons Z, Konig R, Sahin O, Wiemann S. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER plus breast cancer. Journal of Pathology 2014; 233:368-79; PMID:24752803; http://dx.doi.org/10.1002/path.4363
- Xie L, Mouillet JF, Chu T, Parks WT, Sadovsky E, Knofler M, Sadovsky Y. C19MC microRNAs Regulate the Migration of Human Trophoblasts. Endocrinology 2014; 155(12):4975-85: en20141501
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy 2009; 5:1155-65; PMID:19844161; http://dx.doi.org/10.4161/auto.5.8.10090
- Noack J, Choi J, Richter K, Kopp-Schneider A, Regnier-Vigouroux A. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis 2014; 5:e1425; PMID:25255218; http://dx.doi.org/10.1038/cddis.2014.384
- Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg 2003; 99:1047-52; PMID:14705733; http://dx.doi.org/10.3171/jns.2003.99.6.1047
- Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, Hemminki O, Diaconu I, Pesonen S, Koski A, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther 2013; 21:1212-23; PMID:23546299; http://dx.doi.org/10.1038/mt.2013.51
- Yue PY, Leung EP, Mak NK, Wong RN. A simplified method for quantifying cell migration/wound healing in 96-well plates. J Biomol Screen 2010; 15:427-33; PMID:20208035; http://dx.doi.org/10.1177/1087057110361772
- Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ, Cheresh DA, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012; 22:21-35; PMID:22789536; http://dx.doi.org/10.1016/j.ccr.2012.05.037
- Leng S, Bernauer AM, Zhai R, Tellez CS, Su L, Burki EA, Picchi MA, Stidley CA, Crowell RE, Christiani DC, et al. Discovery of common SNPs in the miR-205/200 family-regulated epithelial to mesenchymal transition pathway and their association with risk for non-small cell lung cancer. Int J Mol Epidemiol Genet 2011; 2:145-55; PMID:21686129
- Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther 2010; 10:219-22; PMID:20592490; http://dx.doi.org/10.4161/cbt.10.3.12548
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420-8; PMID:19487818; http://dx.doi.org/10.1172/JCI39104
- Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN, Anastasiadis PZ. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS One 2010; 5:e13665; PMID:21060868; http://dx.doi.org/10.1371/journal.pone.0013665
- Huang ZY, Wu Y, Hedrick N, Gutmann DH. T-cadherin-mediated cell growth regulation involves G2 phase arrest and requires p21(CIP1/WAF1) expression. Mol Cell Biol 2003; 23:566-78; PMID:12509455; http://dx.doi.org/10.1128/MCB.23.2.566-578.2003
- Lee IH, Kawai Y, Fergusson MM, Rovira, II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012; 336:225-8; PMID:22499945; http://dx.doi.org/10.1126/science.1218395
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/10.1038/nrm2529
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3:28-31; PMID:16969128; http://dx.doi.org/10.4161/auto.3269
- Indelicato M, Pucci B, Schito L, Reali V, Aventaggiato M, Mazzarino MC, Stivala F, Fini M, Russo MA, Tafani M. Role of hypoxia and autophagy in MDA-MB-231 invasiveness. J Cell Physiol 2010; 223:359-68; PMID:20112292
- Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, Chouaib S. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 2013; 9:1104-6; PMID:23635487; http://dx.doi.org/10.4161/auto.24728
- Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011; 10:3871-85; PMID:22127234; http://dx.doi.org/10.4161/cc.10.22.17976
- Zheng M, Tang L, Huang L, Ding H, Liao WT, Zeng MS, Wang HY. Overexpression of karyopherin-2 in epithelial ovarian cancer and correlation with poor prognosis. Obstet Gynecol 2010; 116:884-91; PMID:20859152; http://dx.doi.org/10.1097/AOG.0b013e3181f104ce
- Dahl E, Kristiansen G, Gottlob K, Klaman I, Ebner E, Hinzmann B, Hermann K, Pilarsky C, Dürst M, Klinkhammer-Schalke M, et al. Molecular profiling of laser-microdissected matched tumor and normal breast tissue identifies karyopherin alpha2 as a potential novel prognostic marker in breast cancer. Clin Cancer Res 2006; 12:3950-60; PMID:16818692; http://dx.doi.org/10.1158/1078-0432.CCR-05-2090
- van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer 2009; 124:1829-40; PMID:19117056; http://dx.doi.org/10.1002/ijc.24146
- Mortezavi A, Hermanns T, Seifert HH, Baumgartner MK, Provenzano M, Sulser T, Burger M, Montani M, Ikenberg K, Hofstädter F, et al. KPNA2 expression is an independent adverse predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res 2011; 17:1111-21; PMID:21220479; http://dx.doi.org/10.1158/1078-0432.CCR-10-0081
- Kim IS, Kim DH, Han SM, Chin MU, Nam HJ, Cho HP, Choi SY, Song BJ, Kim ER, Bae YS, et al. Truncated form of importin alpha identified in breast cancer cell inhibits nuclear import of p53. J Biol Chem 2000; 275:23139-45; PMID:10930427; http://dx.doi.org/10.1074/jbc.M909256199
- Moll UM, LaQuaglia M, Benard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A 1995; 92:4407-11; PMID:7753819; http://dx.doi.org/10.1073/pnas.92.10.4407
- Wang CI, Chien KY, Wang CL, Liu HP, Cheng CC, Chang YS, Yu JS, Yu CJ. Quantitative proteomics reveals regulation of karyopherin subunit alpha-2 (KPNA2) and its potential novel cargo proteins in nonsmall cell lung cancer. Mol Cell Proteomics 2012; 11:1105-22; PMID:22843992; http://dx.doi.org/10.1074/mcp.M111.016592
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181-5; PMID:20044243; http://dx.doi.org/10.1016/j.ceb.2009.12.001
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 2008; 131:1969-78; PMID:18187492; http://dx.doi.org/10.1093/brain/awm318
- Rubinsztein DC. Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron 2007; 54:854-6; PMID:17582326; http://dx.doi.org/10.1016/j.neuron.2007.06.005
- Du Y, Yang D, Li L, Luo G, Li T, Fan X, Wang Q, Zhang X, Wang Y, Le W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 2009; 5:663-75; PMID:19337030; http://dx.doi.org/10.4161/auto.5.5.8377
- Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di Virgilio F, Zito E, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A 2015; 112:1779-84; PMID:25624484; http://dx.doi.org/10.1073/pnas.1410723112
- Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ 3rd, Clark RS. Autophagy in Neurite Injury and Neurodegeneration: In Vitroand In Vivo Models. Methods Enzymol 2009; 453:217-49; PMID:19216909; http://dx.doi.org/10.1016/S0076-6879(08)04011-1
- Yue PY, Leung EP, Mak NK, Wong RN. A Simplified Method for Quantifying Cell Migration/Wound Healing in 96-Well Plates. J Biomol Screen. 2010 Apr; 15(4):427-33; http://dx.doi.org/10.1177/10870571103-61772