
Abstract
Silica dust mainly attacks alveolar macrophages (AMs) and increases the apoptosis of AMs in silicosis patients. However, it is still unclear whether autophagy is affected. Autophagy mainly has defensive functions in response to stress, contributing to cell survival in adverse conditions, and conversely it has also been implicated in cell death. Lipopolysaccharide (LPS) induces autophagy and apoptosis in macrophages. The role of LPS in autophagy and apoptosis in AMs of silicosis patients is unknown. In this study, we collected AMs from 53 male workers exposed to silica and divided them into an observer (control) group, and stage I, II and III patient groups. We found increased levels of LC3B, SQSTM1/p62 and BECN1,whereas the phosphorylation of MTOR,and levels of LAMP2, TLR4, MYD88, TICAM1, as well as the number of lysosomes decreased with the development of silicosis. LPS stimulation triggered autophagy and increased levels of SQSTM1 in AMs. The autophagy inhibitor, 3-methyladenine (3MA), inhibited LPS-induced apoptosis in the AMs of silicosis patients. Moreover, 3MA reversed the LPS-induced decrease in BCL2 and the increase in BAX and CASP3 levels in AMs. These results suggest that autophagosomes accumulate in AMs during silicosis progression. LPS can induce the formation of autophagosomes through a TLR4-dependent pathway, and LPS may exacerbate the apoptosis in AMs. Blockade of the formation of autophagosomes may inhibit LPS-induced apoptosis via the intrinsic apoptotic pathway in AMs. These findings describe novel mechanisms that may lead to new preventive and therapeutic strategies for pulmonary fibrosis.
Abbreviations
| 3MA | = | 3-methyladenine |
| ACTB | = | actin beta |
| AMs | = | alveolar macrophages |
| BAX | = | BCL2-associated X protein |
| BECN1 | = | Beclin 1 autophagy-related |
| CASP3 | = | caspase 3 apoptosis-related cysteine peptidase |
| HTA125 | = | purified anti-human TLR4 (CD284) antibody |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| LPS | = | lipopolysaccharide |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MYD88 | = | myeloid differentiation primary response 88 |
| PI | = | propidium iodide |
| SQSTM1 | = | sequestosome 1 |
| TICAM1 | = | toll-like receptor adaptor molecule 1l |
| TLR | = | toll-like receptor |
Introduction
Silicosis is characterized by extensive nodular fibrosis within the lungs, and is caused by prolonged exposure to silica dust found in many industrial environments. Silicosis is one of the most common and serious types of pneumoconiosis; its progression is very complex and includes inflammation, immune response, structural damage and repair of cells and tissues, collagen hyperplasia, and fibrosis formation. Silicosis is the result of the interaction of many factors and constraints, and involves a variety of cells and biologically active substances.Citation1
Alveolar macrophages (AMs) are the main target cells of silica dust. Apoptosis of AMs and the release of fibrotic factors play an important role in the initiation and progression of pulmonary fibrosis.Citation2 There are 2 fundamental apoptosis signaling pathways, the extrinsic and intrinsic pathway.Citation3 In a previous study, we have found that slica-induced apoptosis of AMs is regulated by extrinsic pathway in silicosis.Citation2 However, mechanisms of silica-induced fibrosis remain unclear. Autophagy as an alternative cell death pathway is closely linked with apoptosis. We have also noted changes in autophagic activity during the progression of silicosis fibrosis in rats, suggesting autophagy may play a role in the pathogenesis of silicosis.Citation4
The main function of autophagy is to maintain an accurate balance of intracellular components during their synthesis, degradation, and recycling.Citation5,6 The possession of a double membrane is an integral part of the characteristic morphology of an autophagosome. Autophagosomes fuse with lysosomes to degrade substances contained within autophagosomes. The characteristic degradation of autophagy relies on lysosomal enzymes, so lysosomes are indispensable in the progression of autophagic degradation. Basic autophagy is necessary to maintain the dynamic balance of the intracellular environment and is strictly regulated. Changes in autophagy occur in response to the cell experiencing some sort of interference, otherwise it remains at a basal level.Citation7 Silica can be classed as a disruption to AMs, but the changes that occur in autophagy in AMs during silicosis remain unclear. We therefore investigated the role of autophagy in silicosis.
Lipopolysaccharide (LPS) is a characteristic component of the cell wall of Gram-negative bacteria of which many types have been detected in the air of coal mines in China.Citation8 LPS also exists in Asian sand dust (ASD).Citation9 We found LPS in silicosis patients' bronchoalveolar lavage fluid in this study (Fig. S1). LPS activates signal transduction pathways though toll-like receptor 4 (TLR4) in AMs,Citation10-12 and stimulates TLR4 in primary human macrophagesCitation13 and RAW 264.7 macrophages to activate autophagy.Citation13,14 TLR4-mediated autophagy reduces mycobacterial viability in mouse macrophages,Citation15 and LPS exacerbates silica-induced pulmonary fibrosis via inflammation and oxidative stress in mice.Citation16 However, while silica may disrupt the normal function of AMs, it remains unknown if LPS can induce autophagy in AMs during human silicosis, and whether this is mediated through TLR4. Meanwhile, it is unclear how LPS-induced autophagy regulates the apoptotic pathway in AMs of silicosis.
Our study shows that the formation of autophagosomes was increased, whereas autophagic degradation was suppressed, in the AMs of human silicosis patients. LPS may induce the formation of autophagosomes through a TLR4-dependent pathway, and exacerbate the apoptosis via the intrinsic pathway in the AMs of silicosis. Blockade of the formation of autophagosomes may inhibit apoptosis induced by LPS through intrinsic apoptotic pathway in AMs. These findings may help us develop new strategies to treat or prevent silicosis.
Results
Autophagosomes increase in alveolar macrophages of silicosis patients
Our earlier study had shown that the purification rate for AMs was 95% to 99%, and more than 95% of isolated cells expressed green fluorescence after the cells were stained with FITC anti-human CD68, a macrophage marker (Fig. S2A). After Wright-Giemsa staining, we found that silicosis patients' (stages I, II, III) AMs contained many multiple, dense cytoplasmic vesicles, with stage III group's AMs demonstrating a large number of silica-like foreign bodies (Fig. S2B).
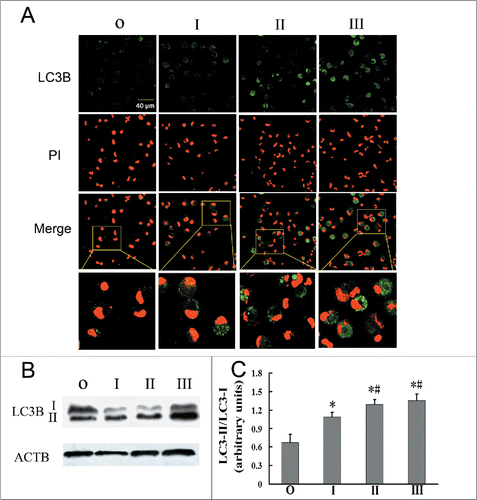
Autophagy by AMs examined by laser scanning confocal microscopy (LSCM) showed that AMs in the silicosis patient groups (stages I, II, III) demonstrated greater numbers of green fluorescent vesicles in the cytoplasm than the observer group when stained with a fluorescent-tagged LC3B antibody, an autophagy marker, and this showed a gradually increasing trend with silicosis stage (). During autophagy, LC3B-I is converted to the lower migrating form LC3B-II. The ratio of LC3B-II/LC3B-I in silicosis patient groups (stages I, II, III) was significantly higher than that of the observer group, indicating increased autophagy activity in silicosis patient groups (; P < 0.05 for all). Additionally, LC3B-II/LC3B-I ratios for the stage II and III groups were significantly higher than for the stage I group (; P < 0.05 for both), indicating increased autophagy activity in the former.
Figure 1. Autophagosomes increase in AMs with the development of silicosis. (A) A representative image of fixed alveolar macrophages from different groups stained for LC3B (original magnification ×600). Green, LC3B protein; Red, PI-labeled nuclei; PI was used for nuclear staining. Scale bar: 40 μm. (B) AMs from observers and different silicosis stages were analyzed for LC3B by western blot. ACTB/beta-actin protein was used as a loading control. (C) LC3-II/LC3-I ratios for each patient group. Significance was determined using one-way ANOVA (n = 11 for the observer group; n = 14 for stages I, II and III. *, P < 0.05 compared to the observer group,#, P < 0.05 vs. stage I patient group. O, observer group; I, stage I patient group; II, stage II patient group; III, stage III patient group).
To further examine autophagosomes, silica-exposed workers' AMs from each group were visualized by transmission electron microscopy (TEM; ). The AMs of the observer group displayed an intact structure with normal organelles within the cytoplasm. Autophagosomes were observed and lots of primary lysosomes were visualized in the observer group (, i and v). In AMs from the stage I group, the number of autophagosomes and autolysosomes was increased compared to those of the observer group, and postlysosomes were found in the cytoplasm (, ii and vi). The number of autophagosomes continued to increase, but lysosome numbers were reduced in AMs of the stage II group (, iii and vii); these results became more obvious in the stage III group, in which AM structures were incomplete and displayed some vacuolization (, iv and viii).
Figure 2. Autophagosomes increase in AMs of silicosis patients. (A) Representative transmission electron microscopic images. Upward arrows indicate autophagic vacuoles and rightward arrows indicate lysosomes in the AMs of different groups. Images i to iv show a macrophage of each patient group, and images v to viii show the same macrophage at a higher magnification. The observer (O) group displayed numerous lysosomes in the cytoplasm of AMs, and also fewer autophagosomes, than those of the silicosis patient groups (I, II, III). Groups I, II, III exhibited increased autophagic vacuoles and reduced lysosomes relative to the observer group. Top images (i to iv): ×7000; Bottom images (v to viii): ×15000. Scale bar: 1 μm (B) A graph of the number of autophagosomes per 25 AMs for each group. (C) A graph of the mean number of lysosomes per 15 AMs for each group. n = 6 for observer and stages I, II and III groups. *, P < 0.05 vs. observer group,#, P < 0.05 vs. stage I patient group. ▴, P < 0.05 vs. stage II patient group. O, observer group; I, stage I patient group; II, stage II patient group; III, stage III patient group.
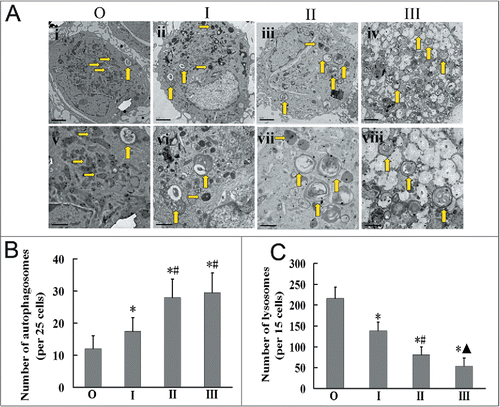
AMs were chosen randomly from each sample, and total numbers of autophagosomes and lysosomes were counted. The number of autophagosomes in the AMs of silicosis patient groups (stages I, II, III) were significantly greater than those of the observer group (, P < 0.05 for all). In turn, autophagosomes in AMs of the stage II and stage III groups were significantly greater in number than in those of the stage I group (; P < 0.05). In contrast, the number of lysosomes in AMs gradually decreased with the development of silicosis, with those of all silicosis groups significantly fewer than those of the observer group (; P < 0.05). These results suggest that silicosis fibrosis may lead to increased autophagosomes and decreased lysosomes in AMs of human silicosis.
Autophagic degradation is blocked in alveolar macrophages of silicosis patients
LAMP2 (lysosomal-associated membrane protein 2) is the main membrane protein of lysosomes; a defect in LAMP2 can lead to the blockade of autophagic degradation.Citation17 We found that LAMP2 expression in the observer group was significantly higher than that in the silicosis patient groups (stages I, II, III; ; P < 0.05 for all). Furthermore, LAMP2 expression gradually decreased with a progression in silicosis (). This further suggests that lysosome numbers decrease with the severity of silicosis, and that increased autophagosomes may be associated with lysosomal defects in the AMs of human silicosis patients.
Figure 3. Delivery of autophagosomes to lysosomes was blocked in AMs of silicosis patients. (A) The expression of LAMP2 and SQSTM1 proteins was analyzed by western blot. ACTB was used as a loading control. (B and C) LAMP2/ACTB and SQSTM1/ACTB ratios of each patient group. n = 11 for the observer group, n = 14 for stage I, II and III groups. *, P < 0.05 vs. observer group,#, P < 0.05 vs. stage I patient group, ▴, P < 0.05 vs. stage II patient group. O, observer group; I, stage I patient group; II, stage II patients group, III; stage III patient group.
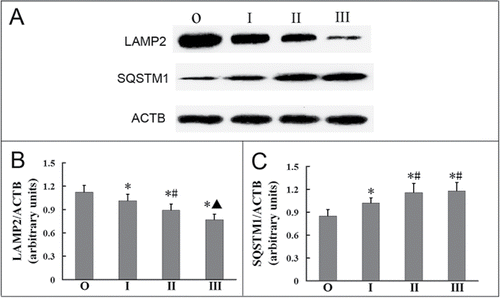
Serum concentration of SQSTM1 in silicosis patient groups were higher than that in the observer group (Fig. S3). SQSTM1 is a receptor protein that is involved in the delivery of cargo to the autophagosome.Citation18,19 We found that the expression of SQSTM1 in the observer group was significantly much lower than that in the silicosis patient groups (; P < 0.05 for all). Furthermore, the expression of SQSTM1 in the stage I group was lower than that in the stage II and III groups (; P < 0.05 for both). These results indicate that autophagic degradation is suppressed in alveolar macrophages of silicosis patients.
Decreased TLR4 in alveolar macrophages of human silicosis patients
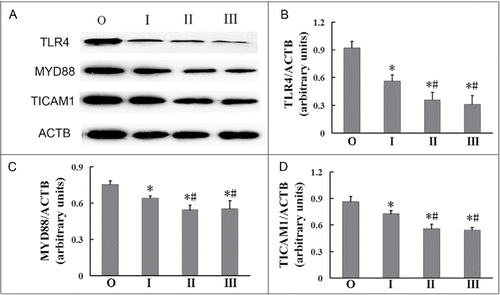
TLR4 is associated with autophagy and plays an important role in inflammation.Citation12 We investigated whether TLR4, MYD88 (myeloid differentiation primary response 88), an adaptor protein through which TLR4 signals, or TICAM1 (toll-like receptor adaptor molecule 1), the functional adaptor for TLR4, protein levels became modulated in AMs during the development of silicosis. We found that the expression of TLR4 was reduced in AMs with the development of silicosis. The expression of TLR4 in AMs of the observer group was significantly higher than those of all the silicosis patient groups (; P < 0.05 for all), with the expression of TLR4 in AMs of the stage I group higher than those of the stage II and III groups (; P < 0.05 for both). Similarly, the expressions of MYD88 and TICAM1 in AMs of the observer group were both significantly higher than those in AMs of the silicosis patient groups (; P < 0.05 for all), with the expression of these in AMs of the stage I group both higher than in AMs of the stage II and III groups (; P < 0.05 for both). These results suggest that silicosis fibrosis may lead to decreased TLR4 expression in AMs of human silicosis patients.
Figure 4. Decreased TLR4 in AMs of silicosis patients. (A) TLR4, MYD88 and TICAM1 proteins were analyzed by western blot. ACTB was used as a loading control. (B to D) Ratios of the indicated proteins to ACTB (n = 8 for observer, and stage I, II and III groups. *, P < 0.05 vs. observer group, #, P < 0.05 vs. stage I patient group. O, observer group; I, stage I patient group; II, stage II patient group; III, stage III patient group).
LPS can induce autophagy through a TLR4 signaling pathway in alveolar macrophages of silicosis patients
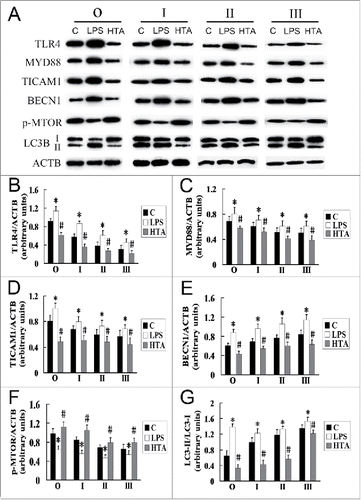
LPS, which can specifically activate the TLR4 signaling pathway, had previously been detected in the bronchoalveolar lavage fluid of the subjects of our study (Fig. S1). To investigate the role of LPS in TLR4-associated autophagy in AMs of silicosis subjects, we used LPS to activate TLR4, and the neutralizing antibody HTA125 to neutralize TLR4 (), and then measured the expression of LC3B and TLR4-related signaling proteins. For the observer and silicosis patient groups, the expression of MYD88, TICAM1 and BECN1 was significantly increased, and the phosphorylation of MTOR was significantly decreased after LPS stimulation of TLR4 in AMs (; P < 0.05 for all); in contrast, the expression of MYD88, TICAM1 and BECN1 was significantly decreased, and the phosphorylation of MTOR was significantly increased after blocking TLR4 in AMs (; P < 0.05 for all). Activation of TLR4 significantly increased the LC3-II/LC3-I ratio, while neutralization of TLR4 significantly reduced the ratio, in both the observer and silicosis patient groups (; P < 0.05 for all). These results suggest that LPS may induce the formation of autophagosomes in AMs of human silicosis subjects, via a TLR4-associated, autophagy signaling pathway.
Figure 5. LPS can increase the formation of autophagosomes in AMs at different stages of silicosis. (A) Lysates of LPS- and anti-HTA-treated AMs were separated by SDS-PAGE and western blot. C indicates AMs from lavage fluids of silicosis patients without intervention. LPS and HTA indicate AMs were untreated or treated with anti-human TLR4 antibody HTA125 prior to 10 μg/ml LPS exposure for 24 h, respectively. ACTB protein was used as a loading control. (B to G) Ratios of the indicated proteins to ACTB, or LC3-II/LC3-I, at different stages of human silicosis. n = 8 for observer, stage I, II and III groups. *, P < 0.05 vs. C; #, P < 0.05 vs. LPS.
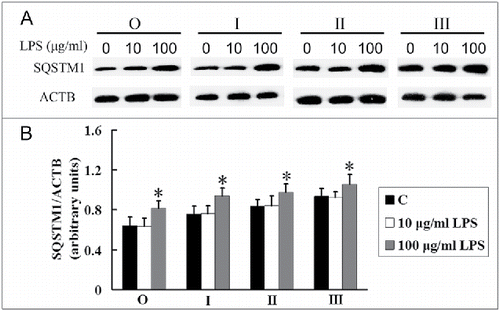
LPS increases the expression of SQSTM1 in alveolar macrophages of silicosis patients
Next we investigated the role of LPS in autophagic degradation in the AMs of silicosis patients. Although no significant difference between treated 10 μg/ml LPS and untreated AMs (C) was found in the expression of SQSTM1, the expression of SQSTM1 was significantly increased after 100 μg/ml LPS stimulation in AMs in both the observer and all silicosis patient groups (; P < 0.05). These results suggest that a high concentration of LPS may exacerbate the disorder of autophagic degradation in AMs of silicosis.
Figure 6. LPS increases the SQSTM1 levels in AMs at different stages of silicosis. (A) AMs were treated without (C) or with 10 or 100 μg/ml LPS exposure for 24 h, respectively. SQSTM1 protein was separated by SDS-PAGE and analyzed by western blot. ACTB protein was used as a loading control. (B) Ratios of SQSTM1 to ACTB at different stages of human silicosis (n = 8 for observer, stage I, II and III groups. *, P < 0.05 vs. C).
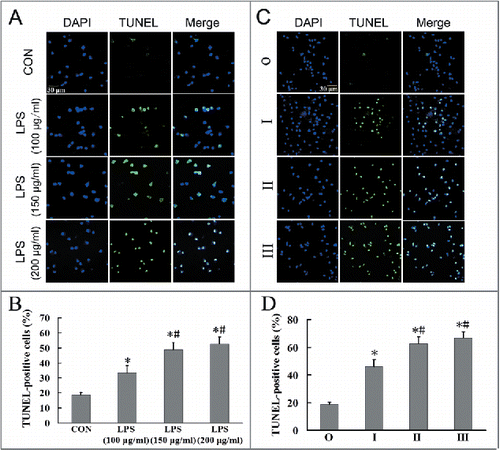
LPS-induced apoptosis may be dependent on the accumulation of autophagosomes in alveolar macrophages of silicosis patients
LPS-induced apoptosis was evaluated after treating observer group cells with LPS (100, 150, 200 μg/ml), and determining cell apoptosis after 24 h using TUNEL. LPS at 150 and 200 μg/ml significantly induced more TUNEL-positive cells than 100 μg/ml LPS (; P < 0.05 for both). We then used 150 μg/ml LPS in the following experiments. The numbers of LPS-treated TUNEL-positive cells observed in the stage II and III groups were significantly higher than those observed for the stage I and observer groups (; P < 0.05 for both). Additionally, the number of LPS-treated TUNEL-positive cells for the stage I group was higher than that noted for the observer group (; P < 0.05). These results suggest that apoptosis in AMs correlated with silicosis severity.
Figure 7. LPS can induce apoptosis in AMs of silicosis subjects. (A) AMs in the observer group were treated without or with LPS (100, 150, 200 μg/ml), and cell apoptosis determined after 24 h using TUNEL. Scale bar: 30 μm. (B) Quantification of TUNEL-positive apoptotic cells in AMs treated with different LPS concentrations. Six fields per sample were analyzed for each cell population. The bar graph shows the percentage of TUNEL-positive nuclei relative to DAPI-positive total nuclei (n = 5 each group. *, P < 0.05 vs. CON; #, P < 0.05 vs. 100 μg/ml LPS group). (C) Representative photographs of TUNEL staining in AMs at different stages of silicosis. Scale bar: 30 μm. (D) Quantification of TUNEL-positive apoptotic cells in AMs at different stages of silicosis. n = 5 for observer, stage I, II and III groups. *, P < 0.05 vs. observer group; #, P < 0.05 vs. stage I patient group).
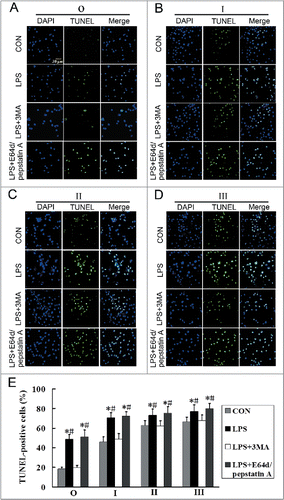
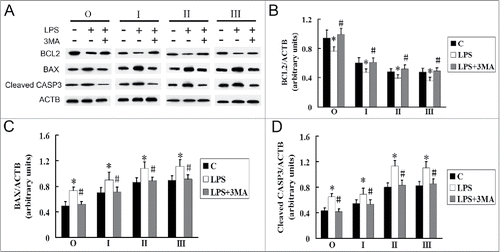
Our results show that LPS induced both autophagy and apoptosis. To detect the relationship between autophagy and LPS-induced apoptosis in AMs of silicosis subjects, we used the autophagy inhibitor, 3-methyladenine (3MA), and lysosomal protease inhibitors, E64d and pepstatin A, to inhibit autophagy activity (). 3MA significantly inhibited LPS-induced apoptosis, while E64d and pepstatin A did not significantly inhibit LPS-induced apoptosis, in the observer and stage I, II and III groups (; P < 0.05 for all). These results suggest that LPS-induced apoptosis may be associated with the accumulation of autophagosomes.
Figure 8. Inhibition of the formation of autophagosomes may reduce LPS-induced apoptosis in AMs at different stages of silicosis. (A–D) Representative photographs of TUNEL staining in AMs treated without or with 3MA (7.5 mM), or cotreated with or without E64d (10 μg/ml) and pepstatin A (10 μg/ml), and stimulated with LPS (150 μg/ml) for 24 h. Scale bar: 30 μm. (E) Ratios of TUNEL-positive apoptotic cells. 6 fields were analyzed for each sample from each cell population. The bar graph shows the percentage of TUNEL-positive nuclei relative to DAPI-positive total nuclei. n = 5 for observer, stage I, II and III groups. *, P < 0.05 vs. CON; #, P < 0.05 vs. LPS+3MA.
Inhibition of autophagy affects LPS-induced apoptosis through the intrinsic apoptotic pathway in alveolar macrophages of silicosis patients
To investigate the mechanism by which inhibition of autophagy protects AMs against LPS-induced apoptosis, we performed western blotting to measure the levels of BCL2, BAX, and cleaved CASP3. After LPS stimulation, the expression of BCL2 decreased, while the levels of BAX and cleaved CASP3 were elevated, in the observer and stage I, II and III groups. However, statistically significant increases in BCL2 and the decrease in both BAX and cleaved CASP3 were observed after 3MA intervention in the presence of LPS (; P < 0.05 for all). These results suggest that inhibition of autophagy may reverse the LPS-induced apoptosis through intrinsic apoptotic pathway in AMs.
Figure 9. Effects of LPS and 3MA on the expression of BCL2, BAX and cleaved CASP3 in AMs at different stages of silicosis. (A) C indicates AMs from lavage fluids of silicosis patients without intervention. AMs treated without or with 3MA (7.5 mM), and stimulated with LPS (150 μg/ml) for 24 h. BCL2, BAX and cleaved CASP3 proteins were analyzed by western blot. (B–D) Ratios of the indicated proteins to ACTB. n = 8 for observer, and stage I, II and III groups. *, P < 0.05 vs. C; #, P < 0.05 vs. LPS.
Discussion
Silicosis is a leading cause of, and one of the most serious types of, pneumoconiosis. Among the workers of industries that involve heavy exposure to SiO2, silicosis is a major health hazard and poses a serious burden to society. Some of the basic pathological changes seen in silicosis include the progressive development of silicon nodule formation and diffuse interstitial fibrosis. Previous research regarding the molecular mechanisms responsible for the development of silicosis has focused on cytokines and apoptosis, but these do not fully explain the pathogenic processes involved.
Autophagy as an alternative cell death pathway is a core pathway driving the scarring response in fibrogenic cells from lung, kidney and liver.Citation20 Autophagy has also been identified in myocardial, skin, liver, and renal fibrosis.Citation20-23 In contrast, a study of human fibrosis found that autophagy is not activated in a setting of idiopathic pulmonary fibrosis.Citation24 However, the potential role of autophagy in the AMs of silicosis is still unclear.
The basic autophagic activity of degrading proteins and damaged organelles plays an important role in maintaining a dynamic balance in the intracellular environment. Autophagy is induced rapidly in certain “crisis” situations, such as growth factor deficiency, starvation, cell remodeling, excessive cell damage, or the accumulation of intracellular metabolic wastesCitation25 and only very recently were silica nanoparticles found to induce autophagy.Citation26 In our previous study, we found that autophagy in lung tissue was modulated during the progression of silicosis fibrosis in a rat model.Citation4 Silica may thus be considered a “crisis” that can alter and modulate autophagy.
In this present study, the increased autophagic activity noted in the AM of silicosis patients may be associated with exposure to silica. The activation of autophagy may selectively degrade aggregation-prone proteins, invading pathogens, and damaged organelles in order to maintain cellular homeostasis.Citation27 However, silica dust may have its own peculiar characteristics that make it impossible to be degraded intracellularly, so that when AMs actively ingest silica dust to eliminate it, they might accelerate their own death. In support of this hypothesis, a study has shown that silica uptake leads to lysosomal rupture and the cell death of macrophages.Citation28 In this study, we noted reduced lysosome numbers and decreased LAMP2 levels in AMs of silicosis patients. These results suggest that silica may not only activate autophagy, but also attack lysosomes. Lysosomal rupture leads to decreased lysosomes, which may subsequently lead to the dysfunction of lysosomes and to impaired autophagic degradation.
Normally, higher autophagy results in the decreased expression of SQSTM1. An increase in SQSTM1 expression would suggest the delivery of autophagosomes to lysosomes is blocked.Citation29,30 In this study, the expression of SQSTM1 increased with the development of silicosis, indicating autophagic degradation may be inhibited, preventing the successful degradation of autophagosome contents. We also found that the concentration of SQSTM1 in the serum of silicosis patients was higher than that in the observers, which suggested that autophagy is involved in the progress of human silicosis. Silica-induced autophagy activity, lysosomal rupture and a defect in autophagic degradation may lead to the accumulation of autophagosomes in the AMs of human silicosis subjects. Furthermore, we found numerous autophagic vacuoles within the cytoplasm of silicosis patients' AMs, but found no typical characteristics of apoptosis, such as karyorrhexis or cell shrinkage; we therefore suspect autophagic cell death exists in silicosis patients' AMs, which may play a role in silicosis fibrosis.
Chronic inflammation is a major mechanism of pulmonary fibrosis. The lung exhibits systemic immune function abnormalities during the process of silicosis. TLRs play important roles in liver, lung, kidney, skin and myocardial fibrosis.Citation31 With regards to TLR4, it is not only the first TLR to be discovered, but also one of the most important. Many immune effector cells express TLRs, including monocyte-derived macrophages, as well as neutrophils, and also dendritic, epithelial and endothelial cells.Citation32 Studies have demonstrated that TLRs have at least 2 signaling pathways: a MYD88-dependent regulating pathway and a MYD88-independent regulating pathway. These 2 regulating pathways need 2 critical signaling proteins, MYD88 and TICAM1,Citation33,34 which can active downstream signaling molecules such as protein kinases, ubiquitin ligase, and transcription factors, among other molecules. In the liver, kidney, cysts, and intestine, fibrosis increases the expression of TLR4.Citation35-39 The expression of TLR4 mRNA is not significantly changed in bronchoalveolar lavage fluid cells of idiopathic pulmonary fibrosis patients.Citation40 However, in our study, TLR4 protein levels decreased with the development of silicosis. The mechanisms underlying TLR4 in silicosis may be more complex than fibrosis within other organs.
LPS (also known as endotoxin) is among the most potent of the immunostimulatory bacterial lipidsCitation41 and studies have demonstrated its presence in bronchoalveolar lavage fluid from lung infection patients.Citation42,43 We also found LPS within the bronchoalveolar lavage fluid of silicosis patients in our study. Autophagy is a self-protective mechanism with immune implications that responds to adverse environments, especially infection and inflammation. Autophagy is normally responsible for the elimination of LPS from cells, but its role in silicosis may differ. Silica decreases bactericidal activity in mouse macrophages,Citation44 and so we postulated silica-exposed workers would be more vulnerable to bacterial invasion. Macrophages are always sensitive to LPS stimulation in studies of their exposure to silica.Citation45-48 Without LPS stimulation, high doses of silica fail to activate the NLRP3 inflammasome or caspase-1 (CASP1), or to cause IL1B release.Citation45,46,49 Macrophages not prestimulated with LPS fail to activate CASP1 in response to inflammasome activators.Citation50,51 Exposure to innate immune stimuli, such as LPS in the environment, may exacerbate stable pulmonary fibrosis.Citation16 People affected by silicosis may be exposed to LPS in the coal mine work environment or even to silica dust, but the role of LPS in AMs of silicosis subjects is still unclear.
The identification of LPS by cells is mainly regulated by TLR4. The regulative relationship between LPS and autophagy has been demonstrated in many studies; for example, LPS activates autophagy though TLR4 in RAW264.7 cells and macrophagesCitation13 and induces autophagy in enterocytes.Citation52 However, when inhibiting the expression of TLR4 by TLR4 siRNA in human bronchial epithelial cells, the autophagy-related protein, LC3B, increased, suggesting that a defect in TLR4 may promote autophagy.Citation53 We wondered if LPS could affect autophagy in AMs of silicosis subjects and showed that LPS induced autophagy through a TLR4-mediated mechanism. Others have shown inhibition of TLR4 attenuates the autophagy-associated degradation of collagen in mice fibrotic lung tissues.Citation54 However TLR4 is not targeted to the lysosome in cystic fibrosis airway epithelial cells.Citation55 In our study, the level of SQSTM1 increased after LPS stimulation. It seems that exposure to LPS may exacerbate the disorder in autophagic degradation in AMs. This process may lead to cell death and be associated with lysosomal rupture in the AMs of silicosis.
The apoptosis of macrophages plays a significant role in the etiology of silicosis.Citation56,57 In vitro studies have shown that silica induced apoptosis in alveolar macrophages,Citation58,59 and this was associated with the FAS-FASLG signaling pathway and lysosomes in AMs.Citation2,60 However, LPS can also induce apoptosis in macrophages.Citation61,62 Recently, a study showed that intracellular LPS induced pyroptosis by stimulating CASP4 activation.Citation63 The mechanism underlying LPS-induced apoptosis in AMs of silicosis subjects is unknown. The intrinsic apoptotic pathway engages caspases via subclasses of the BCL2 protein family.Citation3 The intrinsic apoptotic pathway is mediated by hierarchical interactions between proapoptotic and antiapoptotic BCL2 proteins. Two main members of BCL2-related proteins exist, including antiapoptotic BCL2 and proapoptotic BAX.Citation64 The cytoprotective function of BCL2 comes from its ability to antagonize BAX and BAK1, and thus prevent apoptosis.Citation65 The higher BCL2/BAX ratio results in the decreased CASP3 activation.Citation66 Moreover, BCL2 has also been shown to inhibit autophagy.Citation67 In certain settings where there is uncontrolled upregulation of autophagy, autophagy can lead to cell death, possibly through increasing apoptosis.Citation68 In our study, the apoptosis of AMs increased with silicosis disease severity. LPS may induce apoptosis through accumulated autophagosomes in AMs of silicosis subjects. Inhibition of autophagy in AMs likely exerts its antiapoptotic effect by mitigating LPS-induced apoptosis through intrinsic apoptotic pathway. Our results suggest that foreign bodies, which could activate autophagy, might aggravate apoptosis through the intrinsic apoptotic pathway in AMs of silicosis subjects as well as accelerate silicosis progression. We postulate that LPS and other substances that induce autophagy may be risk factors for silicosis.
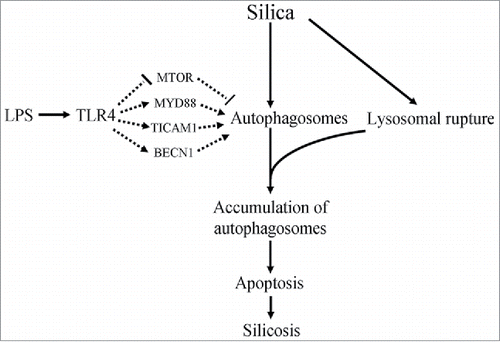
We also speculate that the accumulation of autophagosomes may be a potential mechanism of silicosis. Silica, as a “crisis” factor, might increase the number of autophagosomes in AMs, and LPS could also increase autophagosomes through a TLR4-mediated pathway. Silica dust can lead to lysosomal rupture to inhibit autophagic degradation, leading to the accumulation of autophagosomes. This would result in autophagy not playing a role in the clearance of silica and injured cells, with the accumulation of autophagosomes leading to apoptosis in AMs, resulting in aggravation of fibrosis (). Excessive accumulation of autophagosomes might inhibit TLR4 signaling in AMs. However, whether the reduction in TLR4 seen in AMs aggravated or attenuated inflammation and apoptosis in AMs is unknown, and remains the subject of further study.
Figure 10. A hypothetical diagram illustrates potential mechanisms in silicosis. Silica may lead to apoptosis via increasing the formation of autophagosomes in AMs. LPS regulates the formation of autophagosomes through a blockade of the MTOR signaling pathway and activation of MYD88, TICAM1 and BECN1 signaling pathways. However, silica engulfment results in lysosomal rupture, which may lead to the accumulation of autophagosomes. The excessive accumulation of autophagosomes may lead to apoptosis in AMs. Thus LPS or other substances that can activate autophagy may be risk factors for silicosis.
In conclusion, we have shown that decreased lysosome numbers lead to an autophagy disorder in alveolar macrophages of human silicosis subjects. LPS can increase the formation of autophagosomes through a TLR4-mediated pathway, and exacerbate the apoptosis in AMs of silicosis. Inhibition of autophagosomes can decrease LPS-induced apoptosis via intrinsic apoptotic pathway in AMs. This study highlights new pathogenetic mechanisms in AMs in silicosis, and suggests that the protection of lysosomes, to reduce the accumulation of autophagosomes and the consequent development of apoptosis in AMs, may be potential targets for silicosis prevention and treatment.
Patients and Methods
Subjects
Fifty-three male silica-exposed workers were selected for this study, including 11 observers, whose X-ray photographs showed uncertain silicosis-like changes, the nature and severity of which did not dynamically change within 5 y; 42 silicosis patients (stages I, II, III) were identified by X-rays. Occupational categories were determined by reviewing the occupational history of each subject.Citation69,70 All the selected workers' occupational category was tunneling work, during which they were exposed to silica only. Silicosis was diagnosed by a local pneumoconiosis diagnostic group, according to the GBZ70-2009 standard issued in China, and also according to ILO-2000 guidelines. The backgrounds of the subjects, as well as the occupational history of subjects, including the duration of silica exposure and the duration of cessation to silica exposure, were collected by questionnaire. All those subjects did not exhibit clinical signs or symptoms of autoimmune diseases such as sclerotic skin, Raynaud phenomenon, facial erythema, arthralgia, or malignancies. All subjects underwent massive whole-lung lavage at the Beidaihe Sanatorium for Chinese Coal Miners between May to September 2013. All subjects signed informed consent forms before the lavage. This study was approved by the Medical Ethics Committee of China Medical University (permit number CMU2009-TA-04).
All selected workers were divided into 4 groups: an observer (control) group (O), a stage I patient group (I), a stage II patient group (II) and a stage III patient group (III), based on their radiological changes. The mean age of selected subjects was 45.02 ± 7.34 (range 30 to 57) y, and the mean duration of silica exposure was 16.66 ± 7.01 (3 to 32) y. The mean duration, from the cessation of silica exposure to lung lavage, was 2.85 ± 3.55 (0 to 19) y, and the smoking rate was 83.0%. Information for all subjects is shown in . No statistically significant difference in mean age, duration of silica exposure, duration of cessation of silica exposure, and smoking rate was found among all groups.
Table 1. Subjects' basic information.
Reagents
3MA (M9281), E64d (E8640), pepstatin A (P4265), and lipopolysaccharide (LPS) (0111:B4, L3024) were purchased from Sigma-Aldrich Co. Purified anti-human TLR4 (CD284) antibody (HTA125; 312802) was purchased from BioLegend Inc. TLR4 (ab13867) and LAMP2 (ab13524) antibodies were purchased from Abcam. Anti-LC3B (3868), BECN1 (3738), phospho-MTOR (Ser2448) (5536), SQSTM1 (8025), MYD88 (4283), and TICAM1 (4596) antibodies were purchased from Cell Signaling Technology, Inc. Anti-BCL2 (AB112), BAX (AB026), and cleaved CASP3 (AC033) were purchased from Beyotime Biotechnology Co. The ACTB antibody (sc-130301) was purchased from Santa Cruz Biotechnology, Inc.
AMs isolation, purification and culture
All subjects underwent a large capacity lung lavage under general anesthesia. Lavage fluids were collected and filtered through a double-layered gauze to remove mucus, centrifuged at 1200 g, and washed 3 times with phosphate-buffered saline (PBS; Solarbio, P1010) buffer. After cells were counted using a hemocytometer, 5×106 cells were purified in Dulbecco's modified Eagle's medium (Gibco/Life Technologies/ThermoFisher Scientific; 11965-092) containing 10% fetal bovine serum (Invitrogen/Life Technologies/ThermoFisher Scientific, 26400-044) under 5% CO2 at 37ºC for 2 h based on their adherence. The medium was then changed to fresh medium, and nonadherent (non-AM) cells were washed.Citation2 Adherent, purified AMs left untreated were incubated at 37ºC for another 24 h as a control group. LPS group cells were incubated in medium with 10 μg/ml LPS for 24 h, while HTA-125 group cells were treated for 2 h with fresh media containing 10 μg/ml TLR4 blocking antibody (HTA-125) prior to 10 μg/ml LPS exposure for 24 h. Harvested AMs and supernatant liquids were stored at -80ºC until use.
Transmission electron microscopy
Purified AMs from 6 observers, and 18 silicosis patients (n = 6; stages I, II, III, respectively) were fixed with 2.5% glutaraldehyde, postfixed for 1 h in 1% osmium tetroxide, dehydrated in acetone, and embedded in Epon 812 (SPI Supplies, 02635-AB). Thin sections were stained with uranyl acetate/lead citrate and observed by a Hitachi H-7650 transmission electron microscope (Tokyo, Japan) operated at 80 KV.
Immunofluorescence microscopy
AMs from 6 observers, and 18 silicosis patients (n = 6; stages I, II, III, respectively) were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 (Solarbio, T8200) and blocked with 1% BSA (Solarbio, SW3015) for 15 min at room temperature. The cells were then incubated with a primary anti-LC3B antibody (1:200 in 5% BSA/PBS) overnight at 4ºC. Cells were washed with PBS and incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (1:500; Beyotime, China, A0423) for 1 h, then propidium iodide (PI; Sigma, P4170) was added for 10 min, followed by 3 washes of PBS. AM nuclei were stained red and target proteins were stained green. Images were observed with an Olympus FluoView FV1000 laser scanning confocal microscope (LSCM) (Tokyo, Japan) using FV10-ASW software.
TUNEL
TdT-UTP nick end labeling (TUNEL) assays were performed with a one step TUNEL kit (Beyotime, C1088) according to the manufacturer's instructions. Briefly, cells were permeabilized with 0.1% Triton X-100 for 2 min on ice, followed by TUNEL for 1 h at 37ºC in a dark, humidified chamber. The samples were examined and photographed under an Olympus FV1000 LSCM. Cells in the viewing field were identified by staining with 4′, 6-diamidino-2-phenylindole (DAPI; Beyotime, C1006), and the percentage of apoptotic cells was determined by counting TUNEL-positive cells and dividing the number by the total number of cells. Values obtained by counting 6 fields were averaged.
ELISA assay
Levels of LPS in lung lavage fluids and SQSTM1 in serum from subjects were measured by ELISA (Shanghai Tongwei Co.; ml022601; ml033879; respectively) according to the manufacturer's instructions. Briefly, blanks, standards, and samples were added separately to 96-well plates using 2 replicates per sample. After mixing by gentle shaking, plates were incubated for 30 min at 37ºC, washed 5 times, and 50 μl of HRP-conjugate reagent was added to each well. After incubation for 30 min at 37ºC and washing, chromogen solutions A (50 μl) and B (50 μl) were added and incubated for 10 min. Stop solution (50 μl) was then added to each well to stop the reaction. Blank wells were set to zero, and the optical density (OD) of each well at 450 nm was measured within 15 min.
Western blot
Proteins were extracted from the AMs of all subjects, lysed with cell lysis buffer (Cell Signaling Technology, 9803) containing 1 mM phenylmethylsulfonyl fluoride (PMSF; Solarbio, P8340), and quantified with a BCA Protein Assay kit (Beyotime, P0010). Total proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, IPVH00010) via a semi-dry electrophoretic transfer method. Membranes were blocked for 1 h with 7% powdered skimmed milk and exposed to primary antibodies against LC3B (1:1000), BECN1 (1:1000), phosphorylated MTOR (Ser2448; 1:1000), MYD88 (1:1000), TICAM1 (1:1000), SQSTM1 (1:1000), ACTB (1:1000),BCL2 (1:1000), BAX (1:1000), cleaved CASP3 (1:1000), TLR4 (1:500), or LAMP2 (1:500) at 4°C overnight. After washing, membranes were incubated with HRP-conjugated secondary antibody. Protein expression levels were visualized on X-ray film the following day using an ECL kit (Beyotime, P0018) and analyzed by Quantity One 7.0 imaging analysis software.
Statistical analysis
All values represent mean ± SD. SPSS v.18.0 software was used for all statistical analyses. The differences between values were evaluated using a Kruskal-Wallis test and one-way analysis of variance (ANOVA) followed by pairwise comparisons using a Student-Newman-Keuls post hoc test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1109765_Supplemental_Material.zip
Download Zip (721.9 KB)Acknowledgments
We would like to acknowledge staff from the Department of Pneumoconiosis at the Beidaihe Sanatorium for Chinese Coal Miners for their contribution to the collection of whole-lung lavage.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by the China Coal Miner Pneumoconiosis Prevention and Treatment Foundation (2011–05), Key Project of Science and Technology Support Program of Hebei Province (13277709D).
Related Research Data
References
- Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 2004; 55:395-417; PMID:14746528; http://dx.doi.org/10.1146/annurev.med.55.091902.103810
- Yao SQ, Rojanasakul LW, Chen ZY, Xu YJ, Bai YP, Chen G, Zhang XY, Zhang CM, Yu YQ, Shen FH, et al. Fas/FasL pathway-mediated alveolar macrophage apoptosis involved in human silicosis. Apoptosis 2011; 16:1195-204; PMID:21910009; http://dx.doi.org/10.1007/s10495-011-0647-4
- Ashkenazi A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J Clin Invest 2015; 125:487-9; PMID:25642709; http://dx.doi.org/10.1172/JCI80420
- Yulan Jin, Shi Chen, Saoqiao Yao, Xueyun Fan, Juxiang Yuan. Autophagy and silica-induced pulmonary fibrosis. FASEB J 2011; 25:999.1
- Klionsky DJ, Emr SD. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/10.1126/science.290.5497.1717
- Levine B, Klionsky DJ. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 2006; 27:411-25; PMID:16973212; http://dx.doi.org/10.1016/j.mam.2006.08.002
- Wang Zhen, Wang Naiqi, Li Zengzhi, Shen Wangtang, Qiao Jinshuang, Zhuang Lisheng, Ma Lanhui. The investigation of common pathogenic bacteria in environment of Tanshan Kailuan coal mine. Clinical Medicine of China 1980; Z1:14-8
- Ren Y, Ichinose T, He M, Song Y, Yoshida Y, Yoshida S, Nishikawa M, Takano H, Sun G, Shibamoto T. Enhancement of OVA-induced murine lung eosinophilia by co-exposure to contamination levels of LPS in Asian sand dust and heated dust. Allergy Asthma Clin Immunol 2014; 10:30; PMID:24982682; http://dx.doi.org/10.1186/1710-1492-10-30
- Hamann L, Stamme C, Ulmer AJ, Schumann RR. Inhibition of LPS-induced activation of alveolar macrophages by high concentrations of LPS-binding protein. Biochem Biophys Res Commun 2002; 295:553-60; PMID:12150986; http://dx.doi.org/10.1016/S0006-291X(02)00710-6
- Porter DW, Wolfarth M, Young SH, Rao MK, Meighan T, Barger M, Andrew ME, Huffman LJ. PGJ2 inhibition of LPS-induced inflammatory mediator expression from rat alveolar macrophages. J Toxicol Environ Health A 2007; 70:1967-76; PMID:17966068; http://dx.doi.org/10.1080/15287390701549260
- Lee JH, Del Sorbo L, Uhlig S, Porro GA, Whitehead T, Voglis S, Liu M, Slutsky AS, Zhang H. Intercellular adhesion molecule-1 mediates cellular cross-talk between parenchymal and immune cells after lipopolysaccharide neutralization. J Immunol 2004; 172:608-16; PMID:14688373; http://dx.doi.org/10.4049/jimmunol.172.1.608
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27:135-44; PMID:17658277; http://dx.doi.org/10.1016/j.immuni.2007.05.022
- He J, Wang Y, Xu LH, Qiao J, Ouyang DY, He XH. Cucurbitacin IIa induces caspase-3-dependent apoptosis and enhances autophagy in lipopolysaccharide-stimulated RAW 264.7 macrophages. Int Immunopharmacol 2013; 16:27-34; PMID:23541744; http://dx.doi.org/10.1016/j.intimp.2013.03.013
- Xu Y, Fattah EA, Liu XD, Jagannath C, Eissa NT. Harnessing of TLR-mediated autophagy to combat mycobacteria in macrophages. Tuberculosis (Edinb) 2013; 93 Suppl:S33-7; PMID:24388647; http://dx.doi.org/10.1016/S1472-9792(13)70008-8
- Brass DM, Spencer JC, Li Z, Potts-Kant E, Reilly SM, Dunkel MK, Latoche JD, Auten RL, Hollingsworth JW, Fattman CL. Innate immune activation by inhaled lipopolysaccharide, independent of oxidative stress, exacerbates silica-induced pulmonary fibrosis in mice. PLoS One 2012; 7:e40789; PMID:22815821; http://dx.doi.org/10.1371/journal.pone.0040789
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 2006; 27:495-502; PMID:16973206; http://dx.doi.org/10.1016/j.mam.2006.08.005
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001-4; PMID:19524504; http://dx.doi.org/10.1016/j.cell.2009.05.023
- Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HW 4th, Kyei GB, Johansen T, Vergne I, et al.Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 2010; 32:329-41; PMID:20206555; http://dx.doi.org/10.1016/j.immuni.2010.02.009
- Hernández-Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy 2012; 8:849-50; http://dx.doi.org/10.4161/auto.19947
- Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol 2011; 50:1035-43; PMID:21385586; http://dx.doi.org/10.1016/j.yjmcc.2011.03.002
- Aránguiz-Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C, Lara H, Lavandero S, Díaz-Araya G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta 2011; 1812:23-31; http://dx.doi.org/10.1016/j.bbadis.2010.07.003
- Oikarinen A. Hydroxychloroquine induces autophagic cell death of human dermal fibroblasts: implications for treating fibrotic skin diseases. J Invest Dermat 2009; 129:2333-5; http://dx.doi.org/10.1038/jid.2009.164
- Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One 2012; 7:e41394; PMID:22815997; http://dx.doi.org/10.1371/journal.pone.0041394
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88; PMID:16200202; http://dx.doi.org/10.1172/JCI26390
- Duan J, Yu Y, Yu Y, Li Y, Wang J, Geng W, Jiang L, Li Q, Zhou X, Sun Z. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int J Nanomedicine 2014; 9:5131-41; PMID:25395850; http://dx.doi.org/10.2147/IJN.S71074
- Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells 2010; 15:923-33; PMID:20670274; http://dx.doi.org/10.1111/j.1365-2443.2010.01433.x
- Kane AB, Stanton RP, Raymond EG, Dobson ME, Knafelc ME, Farber JL. Dissociation of intracellular lysosomal rupture from the cell death caused by silica. J Cell Biol 1980; 87:643-51; PMID:6161936; http://dx.doi.org/10.1083/jcb.87.3.643
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742
- Moscat J, Diaz-Meco MT. To aggregate or not to aggregate? A new role for p62. EMBO Rep 2009; 10:804; PMID:19648954; http://dx.doi.org/10.1038/embor.2009.172
- Huebener P, Schwabe RF. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim Biophys Acta 2013; 1832:1005-17; PMID:23220258; http://dx.doi.org/10.1016/j.bbadis.2012.11.017
- Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197-216; PMID:11861602; http://dx.doi.org/10.1146/annurev.immunol.20.083001.084359
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11:373-84; PMID:20404851; http://dx.doi.org/10.1038/ni.1863
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7:353-64; PMID:17457343; http://dx.doi.org/10.1038/nri2079
- Guo J, Loke J, Zheng F, Hong F, Yea S, Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ, et al. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology 2009; 49:960-8; PMID:19085953; http://dx.doi.org/10.1002/hep.22697
- Kazimierczak K, Kopec W, Klinger M. Toll-like receptors (TLR) in the pathogenesis of kidney diseases. Pol Merkur Lekarski 2007; 23:382-5; PMID:18361325
- Iredale JP. Regulating hepatic inflammation: pathogen-associated molecular patterns take their toll. Hepatology 2003; 37:979-82; PMID:12717378; http://dx.doi.org/10.1053/jhep.2003.50224
- Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL, Chizzolini C. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis Rheum 2008; 58:3913-23; PMID:19035500; http://dx.doi.org/10.1002/art.24049
- Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 2003; 124:1866-78; PMID:12806620; http://dx.doi.org/10.1016/S0016-5085(03)00403-7
- Samara KD, Antoniou KM, Karagiannis K, Margaritopoulos G, Lasithiotaki I, Koutala E, Siafakas NM. Expression profiles of Toll-like receptors in non-small cell lung cancer and idiopathic pulmonary fibrosis. Int J Oncol 2012; 40:1397-404; PMID:22344343
- Thompson PA, Gauthier KC, Varley AW, Kitchens RL. ABCA1 promotes the efflux of bacterial LPS from macrophages and accelerates recovery from LPS-induced tolerance. J Lipid Res 2010; 51:2672-85; PMID:20472936; http://dx.doi.org/10.1194/jlr.M007435
- Kollef MH, Eisenberg PR, Ohlendorf MF, Wick MR. The accuracy of elevated concentrations of endotoxin in bronchoalveolar lavage fluid for the rapid diagnosis of gram-negative pneumonia. Am J Respir Crit Care Med 1996; 154:1020-8; PMID:8887601; http://dx.doi.org/10.1164/ajrccm.154.4.8887601
- Flanagan P, Jackson S, Findlay G. Diagnosis of Gram negative, ventilator associated pneumonia by assaying endotoxin in bronchial lavage fluid. J Clin Pathol 2001; 54:107-10; PMID:11215277; http://dx.doi.org/10.1136/jcp.54.2.107
- Zimmerman BT, Canono BP, Campbell PA. Silica decreases phagocytosis and bactericidal activity of both macrophages and neutrophils in vitro. Immunology 1986; 59:521-5; PMID:3026960
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320:674-7; PMID:18403674; http://dx.doi.org/10.1126/science.1156995
- Kuroda E, Ishii KJ, Uematsu S, Ohata K, Coban C, Akira S, Aritake K, Urade Y, Morimoto Y. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity 2011; 34:514-26; PMID:21497116; http://dx.doi.org/10.1016/j.immuni.2011.03.019
- Davis MJ, Swanson JA. Technical advance: Caspase-1 activation and IL-1β release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J Leukoc Biol 2010; 88:813-22; PMID:20587739; http://dx.doi.org/10.1189/jlb.0310159
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847-56; PMID:18604214; http://dx.doi.org/10.1038/ni.1631
- Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol 2010; 40:620-3; PMID:20201015; http://dx.doi.org/10.1002/eji.200940185
- Schumann RR, Belka C, Reuter D, Lamping N, Kirschning CJ, Weber JR, Pfeil D. Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood 1998; 91:577-84; PMID:9427712
- Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM, Stacey KJ, Sweet MJ. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012; 217:1325-9; PMID:22898390; http://dx.doi.org/10.1016/j.imbio.2012.07.020
- Neal MD, Sodhi CP, Dyer M, Craig BT, Good M, Jia H, Yazji I, Afrazi A, Richardson WM, Beer-Stolz D, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol 2013; 190:3541-51; PMID:23455503; http://dx.doi.org/10.4049/jimmunol.1202264
- An CH, Wang XM, Lam HC, Ifedigbo E, Washko GR, Ryter SW, Choi AM. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol 2012; 303:L748-57; PMID:22983353; http://dx.doi.org/10.1152/ajplung.00102.2012
- Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, Yan J Li Z, Liu H Hua F, et al. TLR4 Activity Is Required in the Resolution of Pulmonary Inflammation and Fibrosis after Acute and Chronic Lung Injury. Am J Pathol 2012; 180:275-92; PMID:22062220; http://dx.doi.org/10.1016/j.ajpath.2011.09.019
- Kelly C, Canning P, Buchanan PJ, Williams MT, Brown V, Gruenert DC, Elborn JS, Ennis M, Schock BC. Toll-like receptor 4 is not targeted to the lysosome in cystic fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2013; 304:L371-82; PMID:23316065; http://dx.doi.org/10.1152/ajplung.00372.2011
- Borges VM, Lopes MF, Falcao H, Leite-Junior JH, Rocco PR, Davidson WF, Linden R, Zin WA, DosReis GA. Apoptosis underlies immunopathogenic mechanisms in acute silicosis. Am J Respir Cell Mol Biol 2002; 27:78-84; PMID:12091249; http://dx.doi.org/10.1165/ajrcmb.27.1.4717
- Wang L, Antonini JM, Rojanasakul Y, Castranova V, Scabilloni JF, Mercer RR. Potential role of apoptotic macrophages in pulmonary inflammation and fibrosis. J Cell Physiol 2003; 194:215-24; PMID:12494460; http://dx.doi.org/10.1002/jcp.10220
- Thibodeau M, Giardina C, Hubbard AK. Silica-induced caspase activation in mouse alveolar macrophages is dependent upon mitochondrial integrity and aspartic proteolysis. Toxicol Sci 2003; 76:91-101; PMID:12857937; http://dx.doi.org/10.1093/toxsci/kfg178
- Hu S, Zhao H, Al-Humadi NH, Yin XJ, Ma JK. Silica-induced apoptosis in alveolar macrophages: evidence of in vivo thiol depletion and the activation of mitochondrial pathway. J Toxicol Environ Health A 2006; 69:1261-84; PMID:16754540; http://dx.doi.org/10.1080/15287390500361875
- Thibodeau MS, Giardina C, Knecht DA, Helble J, Hubbard AK. Silica-induced apoptosis in mouse alveolar macrophages is initiated by lysosomal enzyme activity. Toxicol Sci 2004; 80:34-48; PMID:15056807; http://dx.doi.org/10.1093/toxsci/kfh121
- Albina JE, Cui S, Mateo RB, Reichner JS. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol 1993; 150:5080-5; PMID:7684418
- Sarih M, Souvannavong V, Adam A. Nitric oxide synthase induces macrophage death by apoptosis. Biochem Biophys Res Commun 1993; 191:503-8; PMID:7681667; http://dx.doi.org/10.1006/bbrc.1993.1246
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187-92; PMID:25119034; http://dx.doi.org/10.1038/nature13820
- Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015; 6:3519-30; PMID:25784482; http://dx.doi.org/10.18632/oncotarget.2792
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009; 16:966-75; PMID:19325568; http://dx.doi.org/10.1038/cdd.2009.33
- Li B, Zeng M, He W, Huang X, Luo L, Zhang H, Deng DY. Ghrelin protects alveolar macrophages against lipopolysaccharide-induced apoptosis through growth hormone secretagogue receptor 1a-dependent c-Jun N-terminal kinase and Wnt/β-catenin signaling and suppresses lung inflammation. Endocrinology 2015; 156:203-17; PMID:25337654; http://dx.doi.org/10.1210/en.2014-1539
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Liu H, Tang Z, Yang Y, Weng D, Sun G, Duan Z, Chen J. Identification and classification of high risk groups for Coal Workers' Pneumoconiosis using an artificial neural network based on occupational histories: a retrospective cohort study. BMC Public Health 2009; 29:366; http://dx.doi.org/10.1186/1471-2458-9-366
- Mukheriee AK, Bhattacharya SK, Saiyed HN. Assessment of respirable dust and its free silica contents in different Indian coalmines. Ind Health 2005; 43:277-84; PMID:15895842; http://dx.doi.org/10.2486/indhealth.43.277