
Abstract
Macroautophagy is an evolutionarily conserved cellular process involved in the clearance of proteins and organelles. Although the autophagy regulation machinery has been widely studied, the key epigenetic control of autophagy process still remains unknown. Here we report that the methyltransferase EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) epigenetically represses several negative regulators of the MTOR (mechanistic target of rapamycin [serine/threonine kinase]) pathway, such as TSC2, RHOA, DEPTOR, FKBP11, RGS16 and GPI. EZH2 was recruited to these genes promoters via MTA2 (metastasis associated 1 family, member 2), a component of the nucleosome remodeling and histone deacetylase (NuRD) complex. MTA2 was identified as a new chromatin binding protein whose association with chromatin facilitated the subsequent recruitment of EZH2 to silenced targeted genes, especially TSC2. Downregulation of TSC2 (tuberous sclerosis 2) by EZH2 elicited MTOR activation, which in turn modulated subsequent MTOR pathway-related events, including inhibition of autophagy. In human colorectal carcinoma (CRC) tissues, the expression of MTA2 and EZH2 correlated negatively with expression of TSC2, which reveals a novel link among epigenetic regulation, the MTOR pathway, autophagy induction, and tumorigenesis.
Abbreviations
| ChIP | = | chromatin immunoprecipitation |
| CRC | = | colorectal carcinoma |
| EZH2 | = | enhancer of zeste 2 polycomb repressive complex 2 subunit |
| IHC | = | immunohistochemistry |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β, MNase, micrococcal nuclease |
| MTA2 | = | metastasis associated 1 family, member 2 |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NuRD | = | nucleosome remodeling and histone deacetylase |
| PRC2 | = | Polycomb-Repressive Complex 2 |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70kDa, polypeptide 1 |
| SQSTM1/p62 | = | sequestosome 1 |
| TMA | = | tissue microarray |
| TSC2 | = | tuberous sclerosis 2 |
Introduction
Autophagy is a unique protein degradation process by which cytoplasmic constituents are delivered to the lysosome for digestion.Citation1-3 Therefore it is critical to provide energy by recycling macromolecules in response to nutrient and environmental stress. The kinase MTOR (mechanistic target of rapamycin [serine/threonine kinase]) is an important regulator of autophagy induction; active MTOR suppresses autophagy, and the inhibition of MTOR activity promotes it.Citation4,5 Autophagy involves a series of dynamic membrane rearrangements that are mediated by a core set of autophagy-related (ATG) proteins. Steps in this process include the sequestration of cytoplasm by the phagophore, autophagosome maturation and, ultimately, cargo degradation.Citation6,7 Although the cytoplasmic network leading to autophagy has been widely studied, the nuclear regulation that initiates and maintains the process remains poorly understood. In fact, while recent publications have just begun to suggest the role of transcription factors such as NFKB1/NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells),Citation8,9 E2F1 (E2F transcription factor 1),Citation10 and FOXO (forkhead box O) family membersCitation11-13 in autophagy induction, the epigenetic mechanisms that control chromatin reorganization for transcriptional initiation during autophagy regulation are still largely unknown.
Recently, several reports have shown that histone modifications play important roles in the regulation of autophagy process.Citation14 For example, serum starvation, a prominent inducer of autophagy, increases the number of cells that exhibit high levels of trimethylated H4 at Lys20/K20 (H4[K20me3]) in their constitutive heterochromatin.Citation15 Dimethylation of histone H3 at lysine 9 (H3[K9me2]) mediated by EHMT2/G9a (euchromatic histone-lysine N-methyltransferase 2) inhibits the expression of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3), WIPI1 (WD repeat domain, phosphoinositide interacting 1), and TP53INP2/DOR (tumor protein p53 inducible nuclear protein 2), which are essential for autophagosome formation.Citation16 Trimethylated H3 at Lys4/K4 (H3[K4me3]), in parallel with the deacetylation of H4[K16], was coupled to the induction of autophagy in both mammalian and yeast cells.Citation17
Chromatin modulation through covalent histone modifications is one of the main mechanisms of epigenetics regulation.Citation18-21 The functions of histone modifications rely largely on their effector proteins, and the activity of histone-modifying enzymes is modulated by several complexes.Citation22,23 In mammals, polycomb-repressive complex 2 (PRC2) is an important complex that is involved in various biological processes, including cell proliferation, stem cell pluripotency and cancer development.Citation24,25 PRC2 is composed of EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit), EED (embryonic ectoderm development) and SUZ12 (SUZ12 polycomb repressive complex 2 subunit).Citation26 EZH2 mainly catalyzes trimethylation of lysine 27 on histone H3 (H3[K27me3]), which spreads among the target genes to mediate gene repression.Citation27-29 Another important complex in gene repression is the nucleosome remodeling and histone deacetylase (NuRD) complex. The mammalian NuRD complex is composed of HDAC1/2 (histone deacetylase 1/2), CHD4 (chromodomain helicase DNA bingding protein 4), RBBP4/RbAp48 (retinoblastoma binding protein 4), RBBP7/RbAp46 (retinoblastoma binding protein 7), MBD3 (methyl-CpG binding domain protein 3) and metastasis-associated proteins (MTAs).Citation30-32 Recent studies have demonstrated that the NuRD complex is associated with the pluripotency of embryonic stem cells via MBD3,Citation33 and is also associated with DNA damage-response pathways via CHD4.Citation34-36 The NuRD complex plays a dual role in chromatin regulation and is widely recognized as an essential repressive regulator.Citation37 Functional correlations between the NuRD complex and other histone regulators are highly expected.
In the present work, we provide experimental evidence supporting the role of the methyltransferase EZH2 in the autophagy induction via transcriptional regulation of MTOR pathway-related genes, especially TSC2 (tuberous sclerosis 2). RNA interference (RNAi) of EZH2 resulted in increased TSC2 expression and consequently downregulation of MTOR activity. In addition, we demonstrate that EZH2 associates with the TSC2 gene promoters and represses gene expression dependent on MTA2, a component of the NuRD complex. Significantly, we further confirm a positive correlation between MTA2-EZH2 and SQSTM1/p62 (sequestosome 1), a hallmark of autophagy, in human colorectal carcinoma samples. Taken together, our findings have identified EZH2 as an epigenetic regulator of autophagy by regulating the MTOR signaling pathway.
Results
EZH2 knockdown induces autophagy
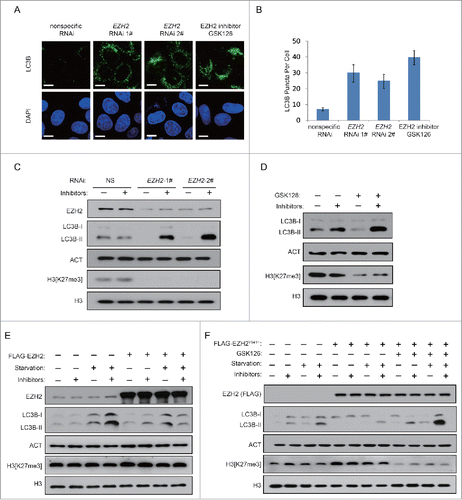
To test whether autophagy is induced by a deficiency of histone methyltransferase EZH2, we first examined the effect of EZH2 knockdown in HeLa cells. As shown in , the knockdown of EZH2 caused a substantial increase in the number of LC3B puncta. In contrast, few LC3B puncta were observed in nonspecific-siRNA-treated cells. In addition, we also compared LC3B-II accumulation as the biological marker of autophagy. As shown in and Figure S1A, inhibition of lysosomal proteases by E64 and pepstatin A led to a higher boost in the amount of LC3B-II, the lipid-conjugated form of LC3B, indicating that the knockdown of EZH2 causes greater turnover of LC3B. Similarly, inhibition of EZH2 by GSK126, a specific inhibitor of EZH2, resulted in autophagy induction ( and Fig. S1B). Next, we examined the role of EZH2 in serum starvation-induced autophagy. By testing LC3B-II accumulation, we found the autophagy induction upon serum withdrawal was obviously delayed in EZH2 overexpression cells ( and Fig. S1C). In addition, we also generate gain-of-function EZH2 mutant EZH2Y641H.Citation38 shows that overexpression of EZH2Y641H blocked the serum starvation-induced accumulation of LC3B-II. Moreover, the autophagy inhibition by EZH2Y641H could be reverted by EZH2 inhibitor GSK126 (), indicating that EZH2 is a negative regulator of autophagy.
Figure 1. Inhibition of EZH2 induces autophagy. (A) HeLa cells were transfected with NS (nonspecific) or EZH2-specific siRNA for 48 h with protease inhibitors (10 μM E64 and 10 μM pepstatin-A). Cells were stained with LC3B antibody (green) and DAPI, and observed under confocal microscopy for LC3B puncta. Scale bars: 10 μm. (B) Quantification of the number of LC3B puncta per cell in A. Data in B are means ± s.d. (n=50 , 3 experimental repeats). (C and D) HeLa cells were transfected with NS (nonspecific) or EZH2-specific siRNA (C) or treated with GSK126 (2 μM) (D) for 48 h in the presence or absence of protease inhibitors (E64 and pepstatin-A). Cell lysates were extracted and analyzed with immunoblotting as indicated. (E) A FLAG-tagged EZH2 or an empty plasmid was individually transfected into MCF-7 cells. 24 h after transfection, cells were then incubated in medium with or without serum for 24 h. LC3B-II accumulation was detected in the presence or absence of lysosomal protease inhibitors (E64 and pepstatin-A). (F) A FLAG-tagged EZH2Y641H mutant or an empty plasmid was individually transfected into MCF-7 cells with or without GSK126. 24 h after transfection, cells were incubated in medium with or without serum for 24 h. LC3B-II accumulation was detected in the presence or absence of lysosomal protease inhibitors (E64 and pepstatin-A).
EZH2 regulates autophagy via the MTOR signaling pathway
To identify the pathway responsible for the induction of autophagy by EZH2 deficiency, we next examined the involvement of the MTOR signaling pathway, which is well known for its critical role in autophagy induction. As shown in , phosphorylation of RPS6KB1 (Thr389) and phosphorylation of MTOR (Ser2448), the 2 markers of MTOR activity, were induced in the EZH2 (WT) and EZH2Y641H-overexpressing cells ( and Fig. S2A and B). However, similar results were not observed when the catalytically dead mutation EZH2H689A was overexpressed, which implies that the histone methyltransferase activity of EZH2 is indispensable in MTOR regulation ( and Fig. S2A). To further clarify the role of EZH2 in the MTOR signaling pathway, the EZH2Y641H plasmid was transfected into MCF-7 cells. As shown in , MTOR inhibition by serum starvation is also impaired with EZH2 gain-of-function mutation. In addition, the phosphorylation of RPS6KB1 and phosphorylation of MTOR were reduced in the EZH2 RNAi and EZH2-inhibitor treated cells ( and Fig. S2C and D), suggesting that EZH2 deficiency inhibits the MTOR pathway.
Figure 2 (see previous page). EZH2 regulates autophagy through the MTOR pathway. (A) A FLAG-tagged EZH2 (WT) or EZH2H689A mutant was individually transfected into MCF-7 cells for up to 48 h. Cell extracts were extracted and then analyzed with immunoblotting as indicated. (B) A FLAG-tagged EZH2Y641H mutant or an empty plasmid was individually transfected into MCF-7 cells. At 24 h post-transfection, cells were then treated with or without GSK126 for 48 h. Cell extracts were extracted and then analyzed with immunoblotting as indicated. (C) A FLAG-tagged EZH2Y641H mutant or an empty plasmid was individually transfected into MCF-7 cells with or without GSK126. 24 h after transfection, cells were incubated in medium with or without serum for 24 h. Cell extracts were extracted and then analyzed with immunoblotting as indicated. (D) HeLa cells were transfected with control (NS) or 2 independent EZH2-specific siRNAs. After 48 h, cells were further transfected with RNAi-resistant rescue form of wt-EZH2 to rescue the expression of EZH2. The cell lysate was extracted and then analyzed with immunoblotting as indicated. (E) HeLa cells were treated with or without 2 μM GSK126 for 48 h. Cell lysates were extracted and analyzed with immunoblotting as indicated. (F and G) HeLa cells were transfected with an empty plasmid or plasmids expressing active (RHEBQ64L-FLAG) or inactive (RHEBD60K-MYC) RHEB mutants. 24 h after transfection, cells were then transfected with control (NS) or EZH2-specific siRNA for 48 h with or without protease inhibitors (E64 and pepstatin-A). Cell extracts were analyzed by immunoblotting as indicated (F). Endogenous LC3B punctate signals were observed under a confocal microscope. Scale bars: 10 μm (G). (H) Quantification of the number of LC3B puncta per cell in (G). Data in (H) are means ± s.d. (n=50 , 3 experimental repeats).
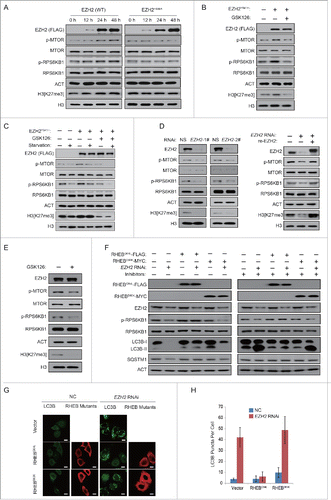
We then investigated whether the inhibition of MTOR is a necessary step for the induction of autophagy under such conditions. For this purpose, we overexpressed a constitutively active form of RHEB (RHEBQ64L), which positively regulates MTOR activity. As shown in , the expression of RHEBQ64L restored the activity of MTOR in EZH2 knockdown cells. Concurrently, it also suppressed the degradation of SQSTM1 and the increase in LC3B-II, indicating that the induction of autophagy is abolished. By using microscopy, we also found that RHEBQ64L eliminated the increase in the number of LC3B puncta (). In contrast, expression of an inactive form of RHEB (RHEBD60K) affected neither the activity of MTOR nor the level of autophagy (). Taken together, these results indicate that the inhibition of MTOR constitutes the definitive step in the induction of autophagy by EZH2 deficiency.
EZH2 regulates the expression of MTOR pathway-related genes
Since the above data pointed a role for EZH2 in the epigenetic regulation of MTOR activity, we decided to determine whether EZH2 might be involved in the regulation of genes related to the MTOR signaling pathway. Interestingly, we found that both EZH2 knockdown and EZH2 inhibitor increased the mRNA expression level of TSC2 ( and Fig. S3A). In addition, several other MTOR pathway related genes including RHOA, DEPTOR, FKBP11, RGS16 and GPI were also upregulated ( and Fig. S3A). Consistently, we noticed that the mRNA expression level of TSC2, but not TSC1, was reduced in MCF-7 and 293T cells overexpressing EZH2 ( and Fig. S3B). The protein expression level of TSC2 was also tested. As shown in and Figure S3C, we found that TSC2 was decreased following the overexpression of EZH2 (WT) and EZH2Y641H, but not EZH2H689A. In addition, we also found the negative correlation between the protein levels of EZH2 and TSC2 in several human cancer cell lines (Fig. S4).
Figure 3. EZH2 regulates the expression of TSC2 and other MTOR pathway-related genes. (A) HeLa cells were treated with 2 μM GSK126 for 48 h or transfected with 2 independent EZH2-specific siRNAs for 48 h. RNA was then extracted and RT-PCR was performed as indicated. (B) A FLAG-EZH2 (WT) or an empty plasmid was transfected into MCF-7 cells for 48 h. RNA was then extracted and analyzed with RT-PCR. mRNA expression level of the indicated genes is presented. (C) A FLAG-EZH2 (WT) or an empty plasmid was transfected into MCF-7 cells for 48 h. Western blots were performed using the indicated antibodies. (D) An empty plasmid, a FLAG-EZH2 (WT), a FLAG-tagged EZH2H689A mutant, or a FLAG-tagged EZH2Y641H mutant was transfected into 293T cells. 24 h later, cells transfected with EZH2Y641H were treated with GSK126 for 48 h. Western blotting was then performed using the indicated antibodies. (E and F) ChIP experiments were performed using anti-EZH2 and anti-H3[K27me3] antibodies, binding of EZH2 (E) or H3[K27me3] (F) to the indicated promoters was measured in HCT116 cells. CCND2 was used as a positive control. All error bars denote the s.d. (n=3 ).
![Figure 3. EZH2 regulates the expression of TSC2 and other MTOR pathway-related genes. (A) HeLa cells were treated with 2 μM GSK126 for 48 h or transfected with 2 independent EZH2-specific siRNAs for 48 h. RNA was then extracted and RT-PCR was performed as indicated. (B) A FLAG-EZH2 (WT) or an empty plasmid was transfected into MCF-7 cells for 48 h. RNA was then extracted and analyzed with RT-PCR. mRNA expression level of the indicated genes is presented. (C) A FLAG-EZH2 (WT) or an empty plasmid was transfected into MCF-7 cells for 48 h. Western blots were performed using the indicated antibodies. (D) An empty plasmid, a FLAG-EZH2 (WT), a FLAG-tagged EZH2H689A mutant, or a FLAG-tagged EZH2Y641H mutant was transfected into 293T cells. 24 h later, cells transfected with EZH2Y641H were treated with GSK126 for 48 h. Western blotting was then performed using the indicated antibodies. (E and F) ChIP experiments were performed using anti-EZH2 and anti-H3[K27me3] antibodies, binding of EZH2 (E) or H3[K27me3] (F) to the indicated promoters was measured in HCT116 cells. CCND2 was used as a positive control. All error bars denote the s.d. (n=3 ).](/cms/asset/8cd179e6-0db8-49f3-a6de-02021c5f35c7/kaup_a_1117734_f0003_c.gif)
To examine EZH2-dependent epigenetic regulation of these genes, we mapped the promoters of TSC2, RHOA, DEPTOR, FKBP11, RGS16 and GPI for EZH2 association in chromatin immunoprecipitation (ChIP) experiments. As shown in and F, EZH2 was highly enriched in the targeted gene promoters, and the enrichment of EZH2 was also associated with H3[K27me3]. Taken together, these results indicate that EZH2-dependent chromatin modifications occur at genes involved in the MTOR signaling pathway.
Gene-specific recruitment of EZH2 is dependent on MTA2 binding
Next, we explored how EZH2 is recruited to the MTOR pathway-related gene promoters. It has been reported that recruitment of EZH2 at its targeted loci is dependent on either the presence or activity of the NuRD complex.Citation39 Therefore we sought to investigate whether NuRD complex is involved in EZH2 regulated TSC2 expression. Firstly, by using coIP assays, we confirmed that EZH2 interacts with the main components of the NuRD complex including MTA2, MBD3 and HDAC1 (). We next identified the subunit that directly mediates the association. As indicated, His-EZH2 interacted with GST-MTA2, but not with the other GST-fusion NuRD subunits, suggesting that MTA2 and EZH2 bind directly in vitro (Fig. S5A). The MTA2 protein harbors several distinct structural domains: a BAH domain, an ELM2 domain and a SANT domain (Fig. S5B). As shown in Figure S5C, the N-terminal BAH domain of MTA2 was able to bind to EZH2 directly. EZH2 is composed of several domains including EID, D1, D2, CXC, and SET domain (Fig. S5B).Citation40 To identify the fragment of EZH2 responsible for the interaction with MTA2, His-MTA2 was incubated with the GST-EZH2 deletion mutant (A[a.a 69–746], B[a.a 160–746], C[a.a 330–746] or D[a.a 614–746]), and analyzed for GST affinity isolation and immunoblotting. As shown in Figure S5D, binding-domain mapping analysis showed that EZH2 bound to MTA2 via its D1 domain. Collectively, these results suggest that MTA2 is fundamental of the association between the NuRD complex and EZH2.
Figure 4 (see previous page). Gene-specific recruitment of EZH2 is dependent on MTA2 binding. (A and B) Nuclear proteins from HCT116 cells (A) or LOVO cells (B) were extracted and immunoprecipitated using anti-EZH2. Immunoprecipitation with rabbit IgG was used as a negative control. Western blotting was performed with the antibodies indicated. (C) HCT116 Cells were transfected with a nonspecific siRNA, or a siRNA targeting MTA2. ChIP assays were performed using the indicated antibodies. The enrichment of MTA2, EZH2, H3[K27me3] and H3[K27Ac] on the indicated promoter was measured. CCND2 was used as a positive control. (D) A FLAG-tagged MTA2 or an empty plasmid was individually transfected into HCT116 cells. ChIP assays were performed using the indicated antibodies. CCND2 was used as a positive control. (E) HCT116 cells transfected with MTA2 siRNA was followed by transfection of a plasmid encoding siRNA-resistant MTA2. Western blotting was performed as indicated. (F) A FLAG-tagged EZH2 or an empty plasmid was individually transfected into HCT116 cells, with or without the siRNA against MTA2. Western blotting was performed with the indicated antibodies.
![Figure 4 (see previous page). Gene-specific recruitment of EZH2 is dependent on MTA2 binding. (A and B) Nuclear proteins from HCT116 cells (A) or LOVO cells (B) were extracted and immunoprecipitated using anti-EZH2. Immunoprecipitation with rabbit IgG was used as a negative control. Western blotting was performed with the antibodies indicated. (C) HCT116 Cells were transfected with a nonspecific siRNA, or a siRNA targeting MTA2. ChIP assays were performed using the indicated antibodies. The enrichment of MTA2, EZH2, H3[K27me3] and H3[K27Ac] on the indicated promoter was measured. CCND2 was used as a positive control. (D) A FLAG-tagged MTA2 or an empty plasmid was individually transfected into HCT116 cells. ChIP assays were performed using the indicated antibodies. CCND2 was used as a positive control. (E) HCT116 cells transfected with MTA2 siRNA was followed by transfection of a plasmid encoding siRNA-resistant MTA2. Western blotting was performed as indicated. (F) A FLAG-tagged EZH2 or an empty plasmid was individually transfected into HCT116 cells, with or without the siRNA against MTA2. Western blotting was performed with the indicated antibodies.](/cms/asset/86bbf7c1-978f-4116-aa28-044b73c75313/kaup_a_1117734_f0004_b.gif)
To understand the elaborate coordination between the NuRD complex and EZH2, we further investigated the roles of MTA2 in recruiting EZH2 to the targeted genes. By using ChIP assays, we found that the enrichment of EZH2, as well as H3[K27me3] on the TSC2 promoter was significantly reduced after MTA2 depletion, while the enrichment of H3 acetylated at K27 (H3[K27Ac]) increased ( and Fig. S6). In addition, a FLAG-MTA2 plasmid was transfected into HCT116 cells (), and ChIP assays were performed to detect the enrichment of MTA2, EZH2, H3[K27me3] and H3[K27Ac] on the TSC2 promoter. As shown in , EZH2 and H3[K27me3] enrichment was increased in response to the increased MTA2 expression, while the enrichment of H3[K27Ac] reduced (). We also performed ChIP assays by using primers specific on the promoters of RHOA and DEPTOR, and got similar results (Fig. S7A to D), suggesting that MTA2 enrichment is a prerequisite for EZH2 recruitment.
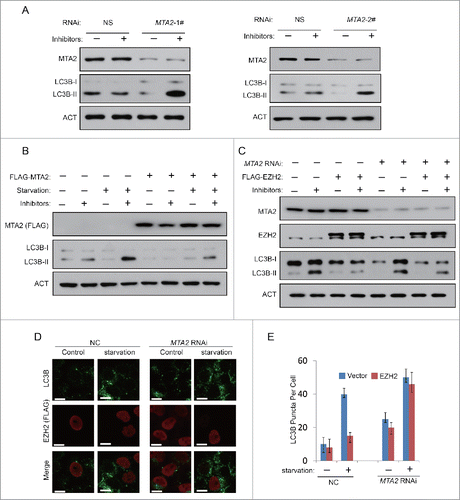
We next examined the role of MTA2 in TSC2 expression and MTOR pathway regulation. As expected, after MTA2 was knocked down, the expression level of TSC2 increased significantly, while the phosphorylation of RPS6KB1 and phosphorylation of MTOR were reduced (). Moreover, reduction of TSC2 expression and MTOR pathway upregulation induced by EZH2 were almost abolished in MTA2 RNAi cells (). Taken together, these data suggest that MTA2 is involved in coordinating EZH2 to regulate the MTOR pathway.
Since we have found MTA2 is required for EZH2-mediated gene repression of TSC2, we then tested whether MTA2 is also a regulator of autophagy. An MTA2 RNAi experiment was performed in HeLa cells to observe any changes of autophagic hallmarks. As shown in , the knockdown of MTA2 induced a significant increase in LC3B-II accumulation. In addition, the overexpression of MTA2 blocked serum starvation induced autophagy, indicating that MTA2 is also a negative regulator of autophagy (). Next, we examined the role of MTA2 in EZH2-repressed autophagy. As shown in , EZH2 cannot downregulate autophagy in MTA2-RNAi 293T and HeLa cells, implying that EZH2 reduced autophagy must occur via MTA2.
Figure 5. Inhibition of MTA2 induces autophagy. (A) HeLa cells were transfected with NS (nonspecific) or 2 independent MTA2-specific siRNAs for 48 h with or without protease inhibitors (E64 and pepstatin-A). Cell lysates were extracted and analyzed with immunoblotting as indicated. (B) A FLAG-tagged MTA2 or an empty plasmid was individually transfected into 293T cells. At 24 h after transfection, cells were then incubated in medium with or without serum for 24 h. LC3B-II accumulation was detected in the presence or absence of lysosomal protease inhibitors (E64 and pepstatin-A). (C) A FLAG-tagged EZH2 or an empty plasmid was individually transfected into 293T cells with or without the siRNA against MTA2. Cells were then incubated in serum-free medium for 24 h. LC3B-II accumulation was detected in the presence or absence of lysosomal protease inhibitors (E64 and pepstatin-A). (D) HeLa cells were transfected with A FLAG-tagged EZH2 or an empty plasmid, with or without the siRNA against MTA2, and then incubated in medium with or without serum for 24 h with protease inhibitors (E64 and pepstatin-A). Cells were then stained with LC3B antibody (green), FLAG antibody (red) and DAPI. LC3B puncta was observed under confocal microscopy. Scale bars: 10 μm. (E) Quantification of the number of LC3B puncta per cell in (D). Data in (E) are means ± s.d. (n=50, 3 experimental repeats).
MTA2 is a chromatin-binding protein
Because MTA2 is able to recruit EZH2 to the targeted genes, we further investigated the structural basis of the association between MTA2 and chromatin. Different cellular fractions were extracted (), as shown in , MTA2 was mainly located in the S3 and S4 fraction. HCT116 cells were transfected with a FLAG-MTA2 plasmid, and then the mononucleosomes were extracted with MNase treatment, immunoprecipitated with anti-FLAG and blotted with anti-H3[K4me3], anti-H3[K9me3], anti-H3[K27me3] or anti-H3. As shown in , MTA2 was able to associate with mononucleosomes, and MTA2-associated histones contained H3[K27me3] modifications, but not H3[K4me3] or H3[K9me3] modifications.
Figure 6. MTA2 is a chromatin-binding protein. (A) A schematic figure showing the procedures for isolating the different cellular fractions. Cell-equivalent amounts of fractions were probed by immunoblotting with anti-MTA2 to detect MTA2 enrichment in different fractions. (B) HCT116 cells were transfected with FLAG-MTA2 or an empty plasmid. The S4 fraction was immunoprecipitated with an anti-FLAG antibody. Western blotting was performed with the indicated antibodies. (C) GST, GST-MTA2 and GST-EZH2 were individually expressed, purified and incubated with reconstituted histone octamers in vitro. Histones precipitated were subjected to western blotting with anti-H3. (D) GST, GST-MTA2 and GST-EZH2 were individually expressed, purified and incubated with biotin-tagged histone peptides in vitro. (E) The GST-fusion mutant fragments of MTA2 were expressed, purified and incubated with biotin-tagged histone peptides.
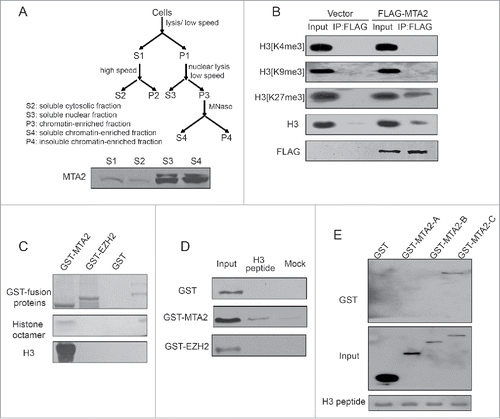
We next investigated whether MTA2 specifically recognized particular histone modification patterns. The direct binding of MTA2 to recombinant histones was examined in vitro with GST affinity isolation assays. As shown in , GST-MTA2 was able to directly bind to histone octamers, whereas GST-EZH2 and GST alone were not. In addition, GST-MTA2 was incubated with biotin-tagged H3 peptides, and GST-MTA2 was able to interact with H3 peptides directly, whereas GST-EZH2 and GST alone were not (). Moreover, as shown in , the binding of MTA2 to H3 relied on its SANT domain, whereas a similar binding capacity was not observed for the BAH or ELM2 domains. Together, these results suggest a possible structural basis by which MTA2 interacts with chromatin by recognizing unmodified H3.
EZH2 and MTA2 expression in colorectal carcinoma (CRC) tissues and its correlation with TSC2 and SQSTM1 expression
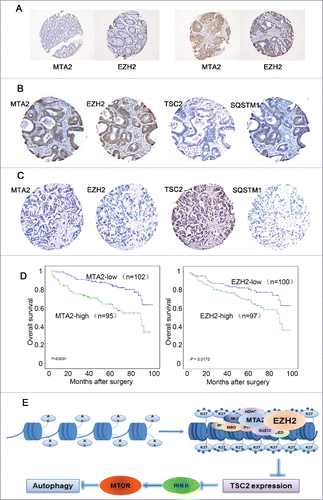
We further investigated the expression levels of MTA2, EZH2, TSC2 and SQSTM1 in CRC tissues. We firstly conducted an immunohistochemistry (IHC) staining for MTA2 and EZH2 in normal tissues and found that both proteins exhibited much lower levels of expression compared to cancer tissues (). Further IHC staining assays were conducted on a CRC-TMA containing 197 CRC specimens. The results of the IHC staining for MTA2, EZH2, TSC2 and SQSTM1 in representative samples of CRCs are shown in and C. Further correlation analysis demonstrated that the expression levels of MTA2 and EZH2 in CRCs were inversely correlated with the expression levels of TSC2 but were positively correlated with the expression levels of SQSTM1 (P < 0.05, Table S1). Correlation analysis also showed that high expression of MTA2 was positively correlated with CRC pT status, lymph node and distal metastasis and a more advanced clinical stage (P < 0.05, Table S2). In our previous study, we have shown that high SQSTM1 was significantly associated with the poor overall survival of CRC patients.Citation41 Consistently, Kaplan-Meier analysis showed that the mean survival time for patients with CRC who had a high level of MTA2 or EZH2 was significantly shorter than that of CRC patients with low levels of MTA2 or EZH2 (). Furthermore, a multivariate Cox regression analysis showed that MTA2 expression was an independent prognostic factor for the survival of CRC patients (Table S3). These data suggest that MTA2 may coordinate with EZH2 to regulate TSC2 expression in human cancer tissues, and thus are associated with activation of MTOR and inhibition of autophagy.
Figure 7 (see previous page). The expression levels of MTA2, EZH2, TSC2 and SQSTM1 in human CRC samples. (A) IHC analysis showing negative expression of MTA2 and EZH2 in normal tissues (left) and high expression of MTA2 and EZH2 in cancer tissues (right). (B) IHC analysis showing high expression levels of MTA2, EZH2 and SQSTM1, and low expression of TSC2 in tumor cells (CRC, case 33). (C) Another CRC (case 165) with high expression of TSC2 and low expression levels of MTA2, EZH2 and SQSTM1 in tumor cells. (D) Survival analysis of MTA2 and EZH2 expressions in a total of 197 patients with CRC. (E) A hypothetical model describing the regulatory mechanism by which EZH2 regulates MTOR and autophagy.
Discussion
In this study, we demonstrate that EZH2, a subunit of PRC2, represses the expression of MTOR pathway-related genes. Interestingly, knocking down EZH2 leads to the formation of LC3B-II and the formation of autophagosome (). Moreover, our data suggest that EZH2 represses the expression of genes involved in MTOR pathway inhibition under normal growth conditions. In addition, EZH2 associates with specific target gene promoters and catalyzes H3[K27] trimethylation via docking histones by MTA2 recruitment, revealing a transcriptional repressive model mediated by multiple types of histone modifications. These results may help us better decipher repressive epigenetic mechanisms on autophagy ().
Autophagy is the major cellular digestion process that removes damaged macromolecules and organelles. The MTOR signaling pathway plays a crucial role in regulating autophagy. Recent studies suggest that the MTOR pathway may be associated with epigenetic regulatory mechanisms.Citation42 For example, ADAMTS9 is a tumor suppressor; epigenetic inactivation of ADAMTS9 blocks its suppression of oncogenic MTOR signaling.Citation43 However, the mechanism for the epigenetic regulation of MTOR is far from being uncovered. In this study, we demonstrate that EZH2 regulates a common set of target genes including TSC2, RHOA, DEPTOR, FKBP11, RGS16, and GPI, the essential suppressors of the MTOR signaling pathway ( and Fig. S3), suggesting a possible link between the MTOR pathway and epigenetic regulation. EZH2 represses gene expression by creating a hypo-histone acetylated and hyper-H3[K27]-trimethylated repressive environment. Overexpression of EZH2 significantly suppresses the expression of TSC2, which activates the MTOR signaling pathway ().
Autophagy is one of the major biological processes regulated by the MTOR signaling pathway. It occurs at low basal levels in all living cells and is required for homeostatic functions.Citation14 In fact, a role for epigenetic regulation of autophagy has been strengthened in recent years. For example, we have previously demonstrated that in response to stress, FOXO1 is acetylated by dissociation from SIRT2, a histone deacetylase, to influence the autophagic process.Citation41,44 More recently it has been reported that histone post-translational modification during autophagy affects the transcriptional regulation of autophagy-related genes.Citation17 In this study, overexpression of EZH2 significantly delayed the induction of autophagy, and knockdown of EZH2 induces the autophagic process (), both of which are dependent on expression of the downregulators of MTOR. In addition, given that the NuRD complex and PRC2 are both involved in stem cell pluripotency, our research may elucidate new mechanisms governing autophagy in other biological processes.
The NuRD complex may be the key player in connecting PRC2 and in exerting suppression function on gene expression.Citation39 For example, recruitment of EZH2 at its targeted loci is dependent on either the presence or activity of NuRD (e.g. H3[K27] deacetylation), but loss of PRC2 has no effect on NuRD recruitment.Citation39 Our study added further information of the correlation that MTA2 directly interacts with EZH2, and the interaction is critical for EZH2 recruitment. It should be noted that we observed these correlations in CRC samples () and demonstrated the interactions in colon cancer cell lines (HCT116 and LOVO) (), which are different from the ES cells they used for functional research, suggesting that the discrepancy may be caused by tissue specificity. However, both studies uncovered the functional correlation between the NuRD complex and PRC2, implying the interaction may be a common phenomenon in mammalian cells.
PRC2 is involved in various cellular processes,Citation25,26 but the exact mechanism by which mammalian PRC2 is recruited to chromatin is still not clear. It is likely that YY1 and RYBP may be required for PRC1 and PRC2 recruitment.Citation45,46 However, the correlation between YY1 and PcG-targeted genes is not observed in the genome-wide analysis.Citation47 Recent reports also reveal that JARID2 and EED are linked to PRC2 recruitment in different manners. JARID2 contributes to PRC2 recruitment by recognizing and directly binding to specific DNA sequences within its target genes.Citation48,49 The carboxyl-terminal domain of EED, however, only recognizes and specifically binds to histone tails with H3[K27me] modifications.Citation50 In this study, we explored a new mechanism to explain the PRC2 recruitment: As a component of PRC2, EZH2 is recruited to chromatin by MTA2. Because MTA2 is able to associate with unmodified H3 peptides, MTA2 may be an initiator to recruit other chromatin regulators to the same region. In this case, EZH2 and other components of PRC2 may thus be recruited to chromatin after MTA2 initiation.
It has been hypothesized that the MTA family proteins distinguish NuRD from other histone deacetylase complexes and mediate the involvement of the NuRD complex in cancer.Citation51 For example, MTA1 expression increases during breast cancer progression,Citation52 and the overexpression of MTA1 induces mammary epithelial proliferation and carcinogenesis in mouse epithelial cells.Citation53 In contrast, MTA3 decreases during tumorigenesis and becomes silenced in late-stage invasive carcinomas.Citation54 MTA3 also represses Wnt4 signaling in mouse mammary epithelial cells.Citation55 However, compared with MTA1 and MTA3, the function of MTA2 in cancer development has not been fully uncovered. In this study, MTA1 and MTA2 were highly expressed in the CRC samples compared to MTA3, which is consistent with previous report.Citation56 However, the interaction between MTA1 and EZH2 was not observed, which may be resulted from the sequence difference of several amino acids of the BAH domain between MTA1 and MTA2. Also, we noticed that the expression of MTA2 in CRCs was inversely correlated with TSC2, but positively correlated with SQSTM1 (). In addition, high expression of MTA2 was positively correlated with CRC pT status, lymph node and distal metastasis and a more advanced clinical stage (Table S2). More significantly, the mean survival time for patients with high expression of MTA2 was markedly shorter than that for patients with low expression of MTA2 (). Our results may imply that MTA2 is a specific player in colon cancer cells, which differ from MTA1 and MTA3. Taken together, these results may indicate a possible role for autophagy in tumor development and open up the possibility that MTA2 may be a potential target for cancer treatment.
In conclusion, the physical association and functional integration between the NuRD complex and PRC2 illustrate new histone repressive machinery. This study also provides a possible mechanism to explain how these complexes dock to histones and establish a repressive status. More importantly, our work may establish a novel link between epigenetics and autophagy, which provides new insights for future cancer treatment.
Materials and Methods
Cells and reagents
Human cervical cancer cell line HeLa, human embryonic kidney cell line HEK293T, human osteosarcoma cell line U2OS, human breast adenocarcinoma cell line MCF-7, human prostate cancer cell line PC3, human lung cancer cell line H719, human colon cancer cell lines HCT116, HT29, SW480, SW620 and LOVO were maintained in DMEM (GIBCO, 12800–017) medium supplemented with 10% FBS (Hyclone, SV30087.02) in a humidified incubator containing 5% CO2. Pepstatin A (Sigma, P5318) and GSK126 (Selleck Chemicals, S7061) were maintained in DMSO at 10 mM and 5 mM stock concentrations respectively. E64 (Sigma, E3132) was maintained at a 10 mM stock in double-distilled water. The antibodies against EZH2 (ab3748), H3[K27me3] (ab6002), H3[K27Ac] (ab4729), H3 (ab1791), H3[K9me3] (ab8898), H3[K4me3] (ab8580), MBD3 (ab157464) and MTA3 (ab176346) were purchased from Abcam. The antibodies against MTA2 (H-170), MTA1 (H-166), HDAC1 (H-11) and ACT (C-11) were purchased from Santa Cruz Biotechnology. The antibodies against LC3B (2775S), TSC2 (3612S), RPS6KB1 (2709S), p-RPS6KB1 (Thr389) (9205S), MTOR (2972) and TSC1 (4906S) were purchased from Cell Signaling Technology. The antibodies against His (PM032), MYC (M407–3) and SQSTM1/p62 (PM045) were purchased from MBL. The antibody against p-MTOR (ser2448) (YP0176) was purchased from Immunoway. The antibody against FLAG (F1804) was purchased from Sigma-Aldrich.
Plasmids
The full-length or a fragment of MTA2 and full-length EZH2 were amplified by PCR from a human cell line and cloned into 3XFLAG-CMV-10, pGEX-4T-3, or pET-28b vectors. The H689A and Y641H mutants of EZH2 were constructed by using a site-directed mutagenesis Kit (Stratagene, 200517). The fragment of FLAG-EZH2 (A[a.a 69–746], B[a.a 160–746], C[a.a 330–746] and D[a.a 614–746]) was provided by Dr. Jae-Il Park (University of Texas MD Anderson Cancer Center). GST-EZH2 constructs were generated based on the FLAG fragment. re–MTA2 sequence, CCGGTACATACAGCAGAAG. re–EZH2 sequence, CCATGTCTACAACTACCAG. The underlines indicate the mutation sites in the re-MTA2 and EZH2 plasmids. The RHEBD60K-MYC and RHEBQ64L-FLAG plasmids were gifts from Dr. Kun-Liang Guan (University of California, San Diego).
RNA interference
All RNAi oligonucleotides were purchased from Shanghai GenePharma Company. These RNAi oligonucleotides were transfected into cells by using the Lipofectamine 2000 transfection kit (Invitrogen, 11668–019) according to the manufacturer's instructions. The final concentration of the siRNA molecules is 10 nM and cells were harvested 48 h or 60 h later according to the purposes of the experiments. Two different siRNAs were used. Sequences are: MTA2: 5′-CCGGUAUAUUCAGCAGAAA-3′ and 5′-CACC AAUCAACAGAAACCAGC-3' EZH2: 5′-CCAUGUUUACAACUAUCAA-3′ and 5′-GCCAAAUAUUGAACCUCCUGAG-3′ nonspecific: 5′-UUCUCCGAACGUG UCACGU-3′.
Coimmunoprecipitation and western blotting
For immunoprecipitation, cell extracts were prepared in lysis buffer (20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40 [Amresco, E109], 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [Merck Millipore, 52322] and protease inhibitor cocktail [Roche, 11873580001]) and incubated with appropriate primary antibodies or normal mouse/rabbit IgG overnight at 4oC. Protein A/G Sepharose beads (GE Healthcare, 17–0780–01/17–0618–01) were added and incubated with samples for 2 h at 4oC. Beads were washed with lysis buffer. The pellets were dissolved into 2xSDS loading buffer after centrifugation and boiled at 100oC for 5 min. Proteins were subjected to western blotting with the indicated antibodies.
RT-qPCR
Total RNA was isolated using Trizol reagent (Invitrogen, 15596018). cDNA was synthesized from 2 μg of RNA with oligo (dT)18 primers using the SuperScript kit (Invitrogen, 18080051). Relative gene expression was determined by real-time PCR using an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, CA, USA) according to the manufacturer's recommended protocol. Each analysis was performed in triplicate.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as described previously.Citation57 Briefly, 2×107 cells were cross-linked with 1% formaldehyde, resuspended in lysis buffer on ice for 15 min and fragmented by sonication. Soluble chromatin was diluted and subjected to immunoprecipitation with the indicated antibodies. Immune complexes were precipitated with protein A/G Sepharose beads, washed sequentially with low-salt (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.1% SDS [Sigma, L5750], 1% Triton X-100 [Amresco, 0694]), high-salt (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 500 mM NaCl, 0.1% SDS [Sigma, L5750], 1% Triton X-100 [Amresco, 0694]), LiCl (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 250 mM LiCl, 1% Nonidet P-40 [Amresco, E109], 1% sodium deoxycholate [Sigma, D6750]) and TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA), and eluted with elution buffer (1% SDS [Sigma, L5750] and 0.1 M NaHCO3). Cross-link was reversed and DNA was purified with DNA extraction kit (Qiagen, 28106). DNA was subjected to real-time PCR.
Isolation of cellular fractions
Micrococcal nuclease (MNase) has the capacity to digest chromatin to mono-nucleosomes.Citation58 The fractionation procedure and MNase treatment were performed as described.Citation59 Briefly, cells were collected, lysed in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose [AMRESCO, M117], 10% glycerol, 1 mM dithiothreitol [Bio-Rad, 1610610], protease inhibitor cocktail), with 0.1% Triton X-100 (Amresco, 0694), and incubated on ice for 8 min. Nuclei were collected by centrifugation (5 min, 1300 g, 4oC), and then lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, protease inhibitor cocktail) for 30 min on ice. Pellets were precipitated by centrifugation (5 min, 1700 g, 4oC) and resuspended in MNase digestion buffer containing 1 U MNase (Takara, 2910A). Samples were incubated at 37oC for 10 min, and reactions were stopped by EGTA (1 mM final concentration). Soluble/insoluble components were separated by centrifugation (5 min, 1700 g, 4oC). Half of the supernatant fraction was used for further immunoprecipitation. DNA was extracted by phenol-chloroform from the remainder of the supernatant fraction, precipitated by ethanol and analyzed by agarose gel electrophoresis.
GST affinity isolation assay
GST or GST-fusion proteins were expressed in bacteria induced with isopropyl-D-thio-galactoside (Merck Millipore, 420322) and purified by glutathione-Sepharose 4B beads (GE Healthcare, 17–0756–01) and then washed with TEN buffer (20 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 100 mM NaCl). Recombinant His-tagged proteins were expressed in and purified from bacteria by Ni (ii)-Sepharose (GE Healthcare, 17–3712–02) affinity isolation. Proteins were incubated at 4oC overnight. Beads were washed 3 times with TEN buffer and boiled with 2x SDS loading buffer. Proteins were analyzed by western blotting with anti-His or anti-GST antibody.
Histone peptide binding assay
Biotinylated histone peptides (Millipore, 12–403) were prebound with streptavidin beads (Sigma, 11205D), and then incubated with GST-fusion protein in binding buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.1% NP-40 [Amresco, E109]) overnight at 4oC. Streptavidin beads were precipitated, washed with binding buffer and subjected to western blotting analysis.
Patients and tissue specimens
In the present study, the paraffin-embedded pathologic specimens from 197 patients with colorectal carcinoma (CRC) were obtained from the archives of Department of Pathology, Cancer Center, Sun Yat-Sen University and Guangdong Provincial People's Hospital, Guangzhou, China, between January 2000 and November 2006. The cases selected were based on distinctive pathologic diagnosis of CRC, undergoing primary and curative resection for CRC, availability of resection tissue, follow-up data, and had not received preoperative anticancer treatment. These CRC cases included 124 (54.1%) men and 73 (45.9%) women, with mean age of 57.2 y Average follow-up time was 54.68 months (median, 60.0 months; range, 0.5 to 98 months). Patients whose cause of death remained unknown were excluded from our study. Clinicopathological characteristics for these patients were summarized in Table S2. This study was approved by the medical ethics committee of our institutes.
Tissue microarray (TMA) and immunohistochemistry (IHC)
TMAs were constructed in accordance with a previously described method.Citation60 Triplicate 0.6-mm diameter cylinders were punched from representative areas of an individual donor tissue block, and re-embedded into a recipient paraffin block in a defined position, using a tissue arraying instrument (Beecher Instruments, Silver Spring, MD).
The TMA blocks were cut into 5-μm sections and processed for IHC. TMA slides were incubated with anti-MTA2 (1:50 dilution), anti-EZH2 (1:50 dilution), anti-TSC2 (1:30 dilution) and anti-SQSTM1/p62 (1:200 dilution), respectively, and stored overnight at 4°C. Immunostaining was performed using the Envision System with diaminobenzidine (Dako, Glostrup, Denmark). A negative control was obtained by replacing the primary antibody with a normal rabbit IgG.
Positive expressions of MTA2 and EZH2 in CRC tissues were primarily observed in nuclear patterns, while TSC2 and SQSTM1 were shown cytoplasmic staining. For evaluation of the IHC staining of MTA2 and EZH2, we scored nuclear expression of the 2 proteins by recording the percentage of nuclei staining positive for MTA2 and EZH2, irrespective of staining intensity. In our study, the mean percentage of positive cells for MTA2 and EZH2 for all informative CRC samples was 39.3% and 51.2%, respectively. Hence, MTA2 and EZH2 immunoreactivity were classified into 2 groups: i.e., low expression, when positive cells were less than 39.3% and 51.2% for MTA2 and EZH2, respectively; and high expression, when at least 39.3% and 51.2% of the cells showed positive immunoreactivity in the nuclei for MTA2 and EZH2, respectively. For the evaluation of IHC staining of TSC2 and SQSTM1, a semiquantitative scoring criterion was used, in which both staining intensity and positive areas were recorded. A staining index (values 0 to 12), obtained as the intensity of TSC2 and SQSTM1 positive staining (negative=0 , weak=1 , moderate=2 , or strong=3 scores) and the proportion of immunostaining positive cells of interest (<25 %=1, 25 to 50%=2, >50% to <75%=3, ≥75%=4 scores) were calculated. The mean staining index of TSC2 and SQSTM1 in this CRC cohort was 6.2 and 6.5. Thus, categories of high expressions of TSC2 and SQSTM1 were defined as CRC cases with staining index of more than 6.2 and 6.5, respectively. Two independent pathologists (M-Y.Cai and D Xie) blinded to the clinicopathological information performed the scorings. The inter-observer disagreements were reviewed a second time, followed by a conclusive judgment by both pathologists.
Statistical analysis
The statistical analysis was performed using the SPSS statistical software package (standard version 13.0), SPSS, Chicago, IL. The correlations between molecular features detected with each other and the associations between expressions of MTA2, EZH2, TSC2 and SQSTM1 and CRC patient's clinicopathological features were assessed by the Chi-square test. For univariate survival analysis, survival curves were obtained with the Kaplan-Meier method. The Cox proportional hazards regression model was used to identify the independent prognostic factors. P values of < 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1117734_Supplemental_Material.zip
Download Zip (14.1 MB)Acknowledgments
We appreciate Dr. Jae-Il Park, MD Anderson, for kindly providing us the fragment plasmids of EZH2, and Dr. Kun-Liang Guan, for the RHEBD60K-MYC and RHEBQ64L-FLAG plasmids.
Supplemental Material
Supplemental data for this article can be accessed on the publisher’s website.
Funding
This study was supported by the "973 Projects" (2011CB910100, 2011CB504200 and 2013CB911000); National Natural Science Foundation of China (81222028, 81321003, 81472581, 81530074, 31570812 and 91319302), and grants (B70001) from the Ministry of Science and Technology of China.
Related Research Data
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Mizushima N. Autophagy in Protein and Organelle Turnover. Cold Spring Harb Symp Quant Biol 2011; 76:397-402; PMID:21813637; http://dx.doi.org/10.1101/sqb.2011.76.011023
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 2015; 25:354-63; PMID:25759175; http://dx.doi.org/10.1016/j.tcb.2015.02.002
- Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015; 125:25-32; PMID:25654547; http://dx.doi.org/10.1172/JCI73939
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963-6; PMID:9461583; http://dx.doi.org/10.1074/jbc.273.7.3963
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6:505-10; PMID:15928714; http://dx.doi.org/10.1038/nrm1666
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009; 29:2594-608; PMID:19289499; http://dx.doi.org/10.1128/MCB.01396-08
- Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21:105-20; PMID:22264792; http://dx.doi.org/10.1016/j.ccr.2011.12.006
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860-4; PMID:18408756
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/10.1016/j.cmet.2007.11.001
- Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D, Wang L, Zhang Y, Yin Y, et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012; 8:1712-23; PMID:22931788; http://dx.doi.org/10.4161/auto.21830
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 2014; 15:65-74; PMID:24326622; http://dx.doi.org/10.1038/nrm3716
- Kourmouli N, Jeppesen P, Mahadevhaiah S, Burgoyne P, Wu R, Gilbert DM, Bongiorni S, Prantera G, Fanti L, Pimpinelli S, et al. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J Cell Sci 2004; 117:2491-501; PMID:15128874; http://dx.doi.org/10.1242/jcs.01238
- Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, Herreros-Villanueva M, Gress TM, Ellenrieder V, Bujanda L, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 2013; 33:3983-93; PMID:23918802; http://dx.doi.org/10.1128/MCB.00813-13
- Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013; 500:468-71; PMID:23863932; http://dx.doi.org/10.1038/nature12313
- Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol 2013; 14:211-24; PMID:AMBIGUOUS; http://dx.doi.org/10.1038/nrm3545
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381-95; PMID:21321607; http://dx.doi.org/10.1038/cr.2011.22
- Jenuwein T, Allis CD. Translating the Histone Code. Science 2001; 293:1074-80; PMID:11498575; http://dx.doi.org/10.1126/science.1063127
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007; 128:707-19; PMID:17320508; http://dx.doi.org/10.1016/j.cell.2007.01.015
- Berger SL. The complex language of chromatin regulation during transcription. Nature 2007; 447:407-12; PMID:17522673; http://dx.doi.org/10.1038/nature05915
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 2007; 25:15-30; PMID:17218268; http://dx.doi.org/10.1016/j.molcel.2006.12.014
- Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene 2007; 26:5433-8; PMID:17694084; http://dx.doi.org/10.1038/sj.onc.1210611
- Schuettengruber B, Cavalli G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development (Cambridge, England) 2009; 136:3531-42; PMID:19820181; http://dx.doi.org/10.1242/dev.033902
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011; 469:343-9; PMID:21248841; http://dx.doi.org/10.1038/nature09784
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315-26; PMID:16630819; http://dx.doi.org/10.1016/j.cell.2006.02.041
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349-53; PMID:16625203; http://dx.doi.org/10.1038/nature04733
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science (New York, NY) 2002; 298:1039-43; http://dx.doi.org/10.1126/science.1076997
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 1998; 395:917-21; PMID:9804427; http://dx.doi.org/10.1038/27699
- Xue Y, Wong J, Moreno GT, Young MK, Côté J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 1998; 2:851-61; PMID:9885572; http://dx.doi.org/10.1016/S1097-2765(00)80299-3
- Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 1998; 95:279-89; PMID:9790534; http://dx.doi.org/10.1016/S0092-8674(00)81758-4
- Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol 2006; 8:285-92; PMID:16462733; http://dx.doi.org/10.1038/ncb1372
- Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Rendtlew Danielsen JM, Menard P, Sand JC, Stucki M, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol 2010; 190:731-40; PMID:20805324; http://dx.doi.org/10.1083/jcb.200912135
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 2010; 29:3130-9; PMID:20693977; http://dx.doi.org/10.1038/emboj.2010.188
- Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol 2010; 190:741-9; PMID:20805320; http://dx.doi.org/10.1083/jcb.201001048
- Feng Q, Zhang Y. The NuRD complex: linking histone modification to nucleosome remodeling. Curr Top Microbiol Immunol 2003; 274:269-90; PMID:12596911
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011; 117:2451-9; PMID:21190999; http://dx.doi.org/10.1182/blood-2010-11-321208
- Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J 2012; 31:593-605; PMID:22139358; http://dx.doi.org/10.1038/emboj.2011.431
- Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD, Park JI. PAF and EZH2 induce Wnt/β-catenin signaling hyperactivation. Mol Cell 2013; 52:193-205; PMID:24055345; http://dx.doi.org/10.1016/j.molcel.2013.08.028
- Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J, Fu W, Wei FZ, Wang L, Xie D, et al. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res 2013; 23:491-507; PMID:23277279; http://dx.doi.org/10.1038/cr.2013.2
- Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD, Zardo G, Nigro J, James CD, Fridlyand J, et al. Reversing HOXA9 Oncogene Activation by PI3K Inhibition: Epigenetic Mechanism and Prognostic Significance in Human Glioblastoma. Cancer Res 2010; 70:453-62; PMID:20068170; http://dx.doi.org/10.1158/0008-5472.CAN-09-2189
- Du W, Wang S, Zhou Q, Li X, Chu J, Chang Z, Tao Q, Ng EK, Fang J, Sung JJ, et al. ADAMTS9 is a functional tumor suppressor through inhibiting AKT/mTOR pathway and associated with poor survival in gastric cancer. Oncogene 2013; 32:3319-28; PMID:22907434; http://dx.doi.org/10.1038/onc.2012.359
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 2010; 12:665-75; PMID:20543840; http://dx.doi.org/10.1038/ncb2069
- Wilkinson FH, Park K, Atchison ML. Polycomb recruitment to DNA in vivo by the YY1 REPO domain. Proc Natl Acad Sci U S A 2006; 103:19296-301; PMID:17158804; http://dx.doi.org/10.1073/pnas.0603564103
- Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell 2010; 140:99-110; PMID:20085705; http://dx.doi.org/10.1016/j.cell.2009.12.022
- Xi H, Yu Y, Fu Y, Foley J, Halees A, Weng Z. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res 2007; 17:798-806; PMID:17567998; http://dx.doi.org/10.1101/gr.5754707
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Gen Dev 2010; 24:368-80; PMID:20123894; http://dx.doi.org/10.1101/gad.1886410
- Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010; 464:306-10; PMID:20075857; http://dx.doi.org/10.1038/nature08788
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, Voigt P, Martin SR, Taylor WR, De Marco V, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009; 461:762-7; PMID:19767730; http://dx.doi.org/10.1038/nature08398
- Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 2011; 11:588-96; PMID:21734722; http://dx.doi.org/10.1038/nrc3091
- Nicolson G, Nawa A, Toh Y, Taniguchi S, Nishimori K, Moustafa A. Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: Role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin Exp Metastasis 2003; 20:19-24; PMID:12650603; http://dx.doi.org/10.1023/A:1022534217769
- Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development (Cambridge, England) 2004; 131:3469-79; PMID:15226262; http://dx.doi.org/10.1242/dev.01213
- Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003; 113:207-19; PMID:12705869; http://dx.doi.org/10.1016/S0092-8674(03)00234-4
- Zhang H, Singh RR, Talukder AH, Kumar R. Metastatic tumor antigen 3 is a direct corepressor of the Wnt4 pathway. Gen Dev 2006; 20:2943-8; PMID:17050676; http://dx.doi.org/10.1101/gad.1461706
- Manavathi B, Kumar R. Metastasis Tumor Antigens, an Emerging Family of Multifaceted Master Coregulators. J Biol Chem 2007; 282:1529-33; PMID:17142453; http://dx.doi.org/10.1074/jbc.R600029200
- Wu LP, Wang X, Li L, Zhao Y, Lu S, Yu Y, Zhou W, Liu X, Yang J, Zheng Z, et al. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol Cell Biol 2008; 28:3219-35; PMID:18332107; http://dx.doi.org/10.1128/MCB.01516-07
- Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, Wei F-Z, Song B, Lu X, Yu Y, et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc Natl Acad Sci 2013; 110:5516-21; http://dx.doi.org/10.1073/pnas.1216596110
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 2005; 121:859-72; PMID:15960974; http://dx.doi.org/10.1016/j.cell.2005.03.036
- Xie D, Ma N-F, Pan Z-Z, Wu H-X, Liu Y-D, Wu G-Q, Kung H-F, Guan X-Y. Overexpression of EIF-5A2 is associated with metastasis of human colorectal carcinoma. Hum Pathol 2008; 39:80-6; PMID:17949776; http://dx.doi.org/10.1016/j.humpath.2007.05.011