
ABSTRACT
Lysosomal impairment causes lysosomal storage disorders (LSD) and is involved in pathogenesis of neurodegenerative diseases, notably Parkinson disease (PD). Strategies enhancing or restoring lysosomal-mediated degradation thus appear as tantalizing disease-modifying therapeutics. Here we demonstrate that poly(DL-lactide-co-glycolide) (PLGA) acidic nanoparticles (aNP) restore impaired lysosomal function in a series of toxin and genetic cellular models of PD, i.e. ATP13A2-mutant or depleted cells or glucocerebrosidase (GBA)-mutant cells, as well as in a genetic model of lysosomal-related myopathy. We show that PLGA-aNP are transported to the lysosome within 24 h, lower lysosomal pH and rescue chloroquine (CQ)-induced toxicity. Re-acidification of defective lysosomes following PLGA-aNP treatment restores lysosomal function in different pathological contexts. Finally, our results show that PLGA-aNP may be detected after intracerebral injection in neurons and attenuate PD-related neurodegeneration in vivo by mechanisms involving a rescue of compromised lysosomes.
Introduction
Alterations of the autophagy-lysosomal pathways (ALPs), which are essential to maintain proper protein and organelle quantity and quality within cells, have been reported in Parkinson disease (PD)-derived samples, including fibroblasts,Citation1,2 induced pluripotent stem cell-derived dopaminergic neuronsCitation3 and post-mortem brain tissuesCitation4-7 as well as in several toxin and genetic models of PD.Citation4,5 To date, genetic research in PD has identified 18 loci (PARK1-18) of which mutation results in either dominant, recessive inheritance or increased risk for developing PD.Citation8 All genes that have been positively associated with PD have also been connected to ALPs. In particular, mutations in 2 genes that encode lysosomal proteins, including the enzyme GBA/glucocerebrosidase (glucosidase, β, acid) and the lysosomal type 5 P-type ATPase ATP13A2 (ATPase type 13A2), have been linked to PD—the former as an important risk factor for PD,Citation9 and the latter through linkage in Kufor-Rakeb syndrome and juvenile forms of PD.Citation10 Mechanistic studies have previously reported that loss of ATP13A2 function is associated with impaired lysosomal acidification, decreased proteolytic processing of lysosomal enzymes, reduced degradation of lysosomal substrates, and decreased lysosomal-mediated clearance of autophagosomes (AP) in ATP13A2 PD patient-derived fibroblasts,Citation1,2 indicating that lysosomal impairment may play a primary pathogenic role in this disease.
Overall, growing evidence now indicates that lysosomes, involved at the late steps of the ALPs, by fusing with AP and digesting their content, are impaired,Citation11 which stress the need to focus therapeutic development on this target. While pharmacological or genetic enhancement of autophagy might be beneficial in experimental models of PD,Citation12-14 the restoration/enhancement of lysosomal function itself represents a novel, precise, localized, and promising therapeutic strategy for PD.
Nanoparticles prepared from biodegradable polymers, are considered an attractive way of drug and gene delivery due to their nontoxic nature and their ability to internalize into mammalian cells,Citation15 in particular in neurons.Citation16 Interestingly, acidic nanoparticles (aNP) of poly(DL-lactide-co-glycolide) PLGA, which are approved by the US FDA,Citation17 have been reported to (i) traffic to lysosomes and (ii) act on the lysosomal pH.Citation18 However, whether PLGA-aNP can acidify sick lysosomes and restore lysosomal/autophagic function in pathological contexts remains to be determined. In addition, it is unknown whether such nanotechnology-based strategy can be translated to an in vivo situation.
Results
aNP are delivered to lysosomes
PLGA-aNP adopt a spherical shape and their diameter ranges from 50 to 100 nm (Fig. S1A), as previously reported in the literature.Citation19,20 We used several markers of the endocytic pathway fused to the green fluorescent protein (GFP) to determine the subcellular localization of PLGA-aNP: RAB5A (early endosome), RAB7A (late endosome) and LAMP1 (lysosomal-associated membrane protein 1) (lysosome). Human dopaminergic neuroblastoma BE-M17 cells were transfected prior to incubation with PLGA-aNP at 180 ng/µl containing a red fluorescent dye, Nile red. We then observed PLGA-aNP entrance at several time points (15, 30, 60 min and 24 h). Cellular uptake of PLGA-aNP was already observed 15 min after exposure (). No colocalization could be seen with early endosomes at 15 min () neither at later time points (). In contrast, we observed partial colocalization with RAB7A-positive compartments beginning at 15 min () and reaching a maximum at 60 min after exposure (). PLGA-aNP remained in the cells at least during 24 h and were ultimately localized within lysosomes, as shown by colocalization with LAMP1-GFP (). PLGA-aNP appear as discrete puncta surrounded by LAMP1-GFP signal as observed with a deconvolution of confocal microscopy image and 3-dimensional (3D) reconstruction (). Similar results were obtained in these cells by immunostaining for LAMP2 (Fig. S1B to E). Of note, as PLGA-aNP size (i.e., 50 to 100 nm) is below the resolution limit of the microscope, we cannot decipher between single PLGA-aNP and clusters of PLGA-aNP in cells. Electron microscopy examination showed the presence of PLGA-aNP into lysosomal-related vesicles as identified on the basis of their size and morphology, in BE-M17-treated cells, compared with untreated cells (). Of interest, PLGA-aNP were not localized to other organelles, as evidenced by electron microscopy (EM) () and a total absence of colocalization with mitochondrial, Golgi apparatus and endoplasmic reticulum markers, analyzed by confocal fluorescence microscopy (Fig. S2). Taken together, our results indicate that aNP are taken up into cells and appropriately targeted to lysosomes.
Figure 1. Acidic nanoparticles are delivered to lysosomes. Localization of PLGA-aNP containing a red fluorescent dye, Nile red (red, 180 ng/µl) in M17 cells in endosomes or lysosomes after 15 min (A,E,I), 30 min (B,F,J), 1 h (C,G,K) and 24 h (D,H,(L)to N) incubation. ((A) to D) Fluorescent signal of RAB5A-GFP (i.e., early endosomes, green) and PLGA-aNP at the selected time points. ((E) to H) Fluorescent signal of RAB7A-GFP (i.e., late endosomes, green) and PLGA-aNP at the selected time points. Colocalization was observed after 15 min (E) and reached a maximum after 1 h (G). ((I) to M) Fluorescent signal of LAMP1-GFP (i.e., lysosomes, green) and PLGA-aNP at the selected time points. Colocalization was observed after 24 h incubation ((L) and M). Orthogonal projection of stack image and 3D reconstruction show PLGA-aNP surrounded by LAMP1-GFP signal ((M)and N). Scale bar applies to all pictures: 10 µm. (N) Spacing in the frame: 1 µm.
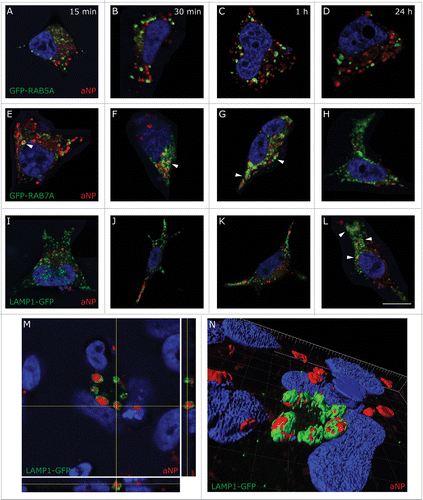
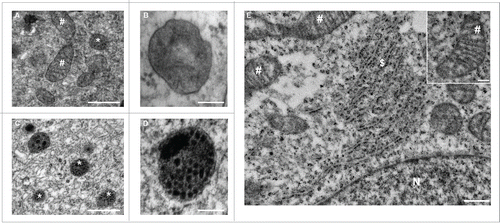
Figure 2. Ultrastructural examination of cells incubated in the absence or presence of PLGA-aNP. Ultrastructural examination of M17 cells, untreated ((A) and B) or incubated with PLGA-aNP for 24 h ((C) to E). High magnification picture of a lysosomal-related vesicle containing PLGA-aNP (D). Other organelles did not contain PLGA-aNP after 24 h incubation (E). Inset is a high magnification picture of mitochondria. #, mitochondria; *, autophagic vesicles; $, endoplasmic reticulum; N, nucleus. Scale bars: 500 nm (A,C,E); 200 nm (E, inset); 100 nm (B,D).
aNP rescue lysosomal pH after lysosomal inhibition
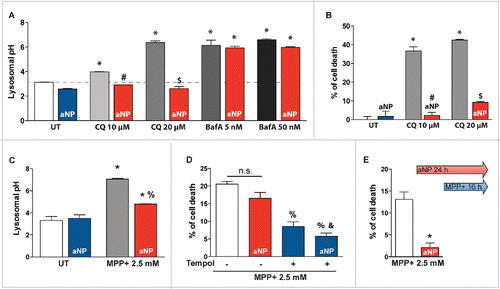
We further investigated the effects of PLGA-aNP on lysosomal pH and cell viability in the presence or absence of 2 different autophagy inhibitors, namely chloroquine (CQ), and bafilomycin A1 (BafA) at concentrations reported in the literature. First, PLGA-aNP treatment per se neither lowered lysosomal pH () nor induced cell death (). Treatment of cells with the lysomotropic agent CQ alkalinizes lysosomes (), which in turn causes lysosomal membrane permeabilization (LMP) leading to apoptosis.Citation21 When we incubated PLGA-aNP concomitantly with CQ at 2 different doses (10 and 20 µM) for 24 h, lysosomal pH was restored to a basal level, associated with reduced cell death (). Conversely, PLGA-aNP were not able to restore lysosomal pH after 1 h cotreatment with the lysosomal V-type ATPase inhibitor BafA at 5 nM and 50 nM (). This effect is at least in part due to a decrease of lysosomal addressing of PLGA-aNP following BafA treatment (Fig. S3).
Figure 3. Acidic nanoparticles rescued lysosomal pH after lysosomal inhibitor treatment and PD-related toxin exposure. (A) Lysosomal pH values as measured ratiometrically using LysoSensor Yellow/Blue DND-160 in untreated cells, 10 and 20 µM chloroquine (CQ) or 5 and 50 nM bafilomycin A1 (BafA)-treated cells, with or without PLGA-aNP incubated concurrently for 24 h or 1 h respectively. (B) Cell death in CQ-treated cells incubated with or without PLGA-aNP. (C) Lysosomal pH values in untreated (UT) and MPP+-intoxicated M17 cells, in the absence or presence of PLGA-aNP treatment. (D) Cell death in MPP+-treated M17 cells after incubation with PLGA-aNP for 24 h, in the presence or the absence of Tempol (500 µM). (E) Cell death measured after PLGA-aNP incubation for 24 h prior to 16 h of MPP+ treatment. In all panels, n=3 to 5 per experimental group. *, P<0.05 compared with control untreated cells; #, P<0.05 compared with CQ (10 µM)-treated cells; $, P<0.05 compared with CQ (20 µM)-treated cells; %, P<0.05 compared with MPP+-treated cells; and &, P<0.05 compared with MPP+-intoxicated cells treated with PLGA-aNP.
aNP prevent lysosomal alkalization after MPP+ treatment without cell death attenuation
Mitochondrial parkinsonian neurotoxin 1-methyl-4-phenylpyridinium ion (MPP+), the active form of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), reproduces several PD-related cellular alterations, such as inhibition of mitochondrial complex I, BAX activation, increased reactive oxygen species (ROS) production, the latter responsible for LMP, leading to disruption of lysosomal structure integrity and contributing to subsequent cell death.Citation22,23 Cotreatment of PLGA-aNP and MPP+(2.5 mM for 24 h) successfully reduced lysosomal pH (), an effect that was not sufficient to reduce cell death (). We posited that the absence of protection versus MPP+-induced cell death was due to the strong ROS production following MPP+ intoxication.Citation4,23,24 Supporting the latter hypothesis, we show that a combined treatment with PLGA-aNP and the free radical scavenger superoxide dismutase mimetic compound tempol (500 µM for 24 h) did not further attenuate MPP+-induced cell death compared to the administration of either compound separately (). Interestingly, 8 h of PLGA-aNP incubation prior to MPP+ addition was then capable of markedly protecting from MPP+ toxicity (), indicating that pretreatment with PLGA-aNP is able to protect cells through lysosomes against mitochondrial-derived ROS insults. Our results indicate that aNP are able to prevent loss of lysosomal pH after MPP+-induced lysosomal deficiency in this experimental system.
aNP restores lysosomal impairment in lysosomal-related genetic models
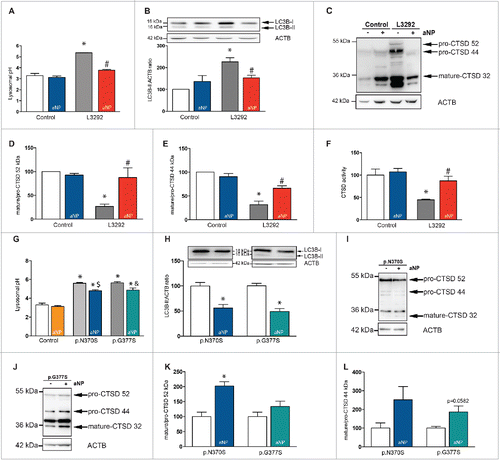
After having demonstrated in different drug-treated cells the ability of aNP to directly acidify lysosomes, we next explored whether PLGA-aNP may rescue lysosomal function in an in vitro genetic model of PD. PD patient-derived fibroblast harboring ATP13A2 mutations (heterozygous for 1306+5GA and 3057delC), named L3292, and control human fibroblasts from our fibroblast libraryCitation1 were used. Treatment with PLGA-aNP restored a normal lysosomal pH (), significantly increased clearance of AP, as indicated by an attenuation of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β)-II accumulations by immunoblot (). CTSD (cathepsin D), the predominant lysosomal protease,Citation25 is processed from a proCTSD 52-kDa form to a proCTSD 44-kDa intermediate and finally to a 32-kDa mature form in the lysosomes. While proforms were barely detectable in control fibroblasts, L3292 fibroblasts exhibited an increase in total amount of CTSD and enrichment in immature forms. We observed a decrease of proforms (proCTSD 52 kDa and proCTSD 44 kDa) in favor of the mature form in PLGA-aNP-treated L3292 fibroblasts compared to untreated L3292 fibroblasts, as indicated by an increased mature/immature ratio (). Functional assay of CTSD activity in lysosomal fractions from PLGA-aNP-treated L3292 fibroblasts confirmed restoration of proteolytic activity of this lysosomal enzyme compared to untreated L3292 fibroblasts ().
Figure 4. Acidic nanoparticle treatment restored impaired lysosomal function in ATP13A2 mutant fibroblasts and partially in GBA mutant fibroblasts. (A) Lysosomal pH values in control and mutant ATP13A2 L3292 fibroblasts, in the absence or presence of PLGA-aNP treatment. (B) LC3B immunoblot levels in mutant ATP13A2 L3292 fibroblasts in the absence or presence of PLGA-aNP. ((C) to E). CTSD immunoblot levels in mutant ATP13A2 L3292 fibroblasts in the absence or presence of PLGA-aNP treatment. (F) In vitro assay of CTSD enzyme activity in lysosomal fractions of control and mutant ATP13A2 fibroblasts with or without PLGA-aNP treatment. (G) Lysosomal pH values in control and mutant GBA p.N370S and p.G377S fibroblasts, in the absence or presence of PLGA-aNP treatment. (H) LC3B immunoblot levels in mutant GBA fibroblasts with or without PLGA-aNP treatment. ((I) to L) CTSD immunoblot levels in mutant GBA fibroblasts in the absence or presence of PLGA-aNP treatment. In all panels, n=3 to 5 per experimental group. *, P<0.05 compared with control untreated cells; #, P<0.05 compared with L3292 untreated cells. $, P<0.05 compared with untreated p.N370S mutant fibroblasts. and &, P<0.05 compared with untreated p.G377S mutant fibroblasts.
To corroborate the potential of PLGA-aNP to rescue lysosomal-mediated degradation in dopaminergic cell lines, we used the previously described BE-M17 cells stably depleted of ATP13A2 (shATP13A2(403-1)) and their respective control (shScr-1).Citation1 ATP13A2-knockdown cells recapitulate similar lysosomal alterations observed in fibroblasts from PD patients.Citation1 In agreement with the data collected in fibroblasts derived from patients with PD, PLGA-aNP treatment was able to restore normal lysosomal pH (Fig. S4A), increase clearance of AP (Fig. S4B), rescue lysosomal proteolytic activity through improvement of both maturation and activity of CTSD (Fig. S4C to F), decrease macroautophagy-associated accumulation of substrates such as SQSTM1/p62 (Fig. S4G) and was associated to a clear attenuation of cell death (Fig. S4H). Overall, these results demonstrate the feasibility of restoring lysosomal function in vitro in both ATP13A2-knockdown cells and ATP13A2 mutant fibroblasts.
To extend our reported results, we used another pathological model involving the second lysosomal-related gene linked to PD. On the one hand, homozygous mutations in the GBA gene encoding for GBA protein cause Gaucher disease (GD), which is the most frequent lysosomal storage disorder (LSD).Citation26 On the other hand, heterozygous mutations have been reported to be an important genetic risk factor for PD.Citation9 GBA produce glucose and ceramide from the glycolipid glucocerebroside inside lysosomes, which in turn results in glucocerebroside accumulation in GD.Citation27
While the underlying mechanism linking GBA mutations to parkinsonism remains unknown, mutations in GBA gene have been shown to alter endoplasmic reticulum and compromised proteolysis of long-lived proteins, such as the PD-linked SNCA/α-synuclein.Citation28,29 Here, we used fibroblasts from PD patients with 2 different GBA point mutations: p.N370S and p.G377S. When GBA-mutant cells were incubated with PLGA-aNP, the abnormal lysosomal pH of these fibroblasts was slightly decreased (). A previous study has reported a lower amount of CTSD in Lewy body dementia patients with GBA mutations.Citation28 In our experimental models, PLGA-aNP were able to increase both clearance of AP () and CTSD maturation process (). However, the trend for the decrease in CTSD immature forms varies between the 2 GBA mutant fibroblasts (), suggesting that part of the effect of mutant GBA is due to a gain of toxic function.Citation30 Our results indicate that PLGA-aNP are nevertheless capable of restoring, at least in part, the pathological changes afforded by the mutations.
To determine the broad applicability of such a strategy, we further explored the effects of PLGA-aNP in a non-brain-related disorder. To this purpose, we used a fibroblast model of X-linked myopathy with excessive autophagy (XMEA) (Fig. S5). This childhood disease is characterized by autophagic vacuolation and atrophy of skeletal muscles.Citation31 A recent study reports that XMEA is caused by mutations of the VMA21 gene, which reduce the amount of the protein, an essential assembly chaperone of the vacuolar-type ATPase (V-ATPase), and reduce V-ATPase activity to 10 to 30% of normal.Citation32 Decreased V-ATPase activity, in turn, raises lysosomal pH. Remarkably, treatment with PLGA-aNP restored a normal lysosomal pH (Fig. S5), suggesting that such innovative strategy could be applied to other lysosomal-related diseases.
aNP are detected in dopaminergic neurons after intracerebral injections in mice and attenuate nigrostriatal dopaminergic neurodegeneration in MPTP-treated mice
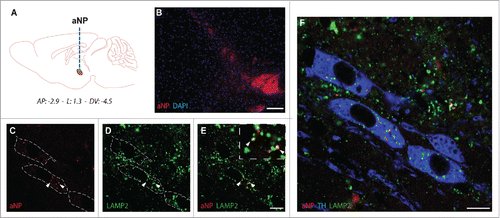
Finally, we explored the translational potential of such innovative strategy. PD is classically characterized by the degeneration of dopaminergic neurons of the substantia nigra pars compacta (SNpc) responsible for most of the motor symptomatology in PD.Citation33,34 To demonstrate the feasibility and therapeutic potential of this strategy, we next assessed whether PLGA-aNP may be used in the brain. To this purpose, PLGA-aNP were administered stereotaxically into the SNpc of wild-type mice (). Seven d after injection, PLGA-aNP were detected around the injection site without evident cytotoxicity (). PLGA-aNP localized inside lysosomes (as evidence by colocalization with LAMP2) () of tyrosine hydroxylase-positive cells, a marker for dopaminergic neurons ().
Figure 5. Acidic nanoparticles are detected in lysosomes of dopaminergic neurons after intracerebral injections in mice. (A) Schematic diagram indicating the site of nigral stereotactic LB inoculations in mice. (B) Representative images of PLGA-aNP (in red; nucleus in blue) 5 d after the injection in SNpc. ((C) to F) Colocalization of PLGA-aNP (in red) with LAMP2 (in green) in TH-immunopositive neurons (in blue) in midbrain sections of PLGA-aNP injected mice. Arrowheads highlight colocalization of PLGA-aNP and LAMP2. Scale bars: 100 µm (B); 20 µm ((C)and D).
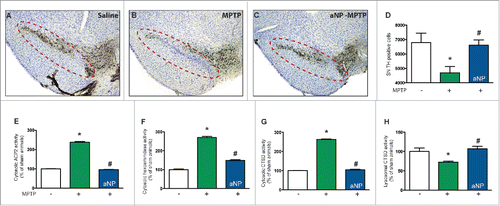
To determine the relevance of our in vitro results to an in vivo situation, we next assessed the effects of PLGA-aNP in MPTP-treated mice.Citation35 PLGA-aNP were administered prior to MPTP intoxication. Mice received one intraperitoneal injection of 30 mg/kg/d MPTP for 2 consecutive d and were terminated 5 d after the last MPTP injection. Similar to the in vitro results reported above, preadministration of PLGA-aNP in MPTP-treated mice was associated with a significant attenuation of dopaminergic neurodegeneration as determined by stereological neuronal cell counts (). Previous reports have shown in the same experimental model of PD that inhibition of MPTP-induced LMP was beneficial.Citation22,23 Consequently, we assessed whether the observed survival of dopaminergic neurons occurred through a decrease of MPTP-induced LMP. To this purpose, we measured the activity of 3 lysosomal enzymes (i.e. ACP2 [acid phosphatase 2, lysosomal], HEX/β-hexosaminidase and CTSD) in lysosome-free cytosolic fractions from ventral midbrain of MPTP- or saline-injected animals. Preadministration of PLGA-aNP efficiently reduced LMP in MPTP-injected mice (). A role for lysosome in protecting against neurodegeneration is supported by the observation that PLGA-aNP restored the CTSD activity in lysosomes of MPTP-injected mice (). These results demonstrate the feasibility and therapeutic potential of biotechnologically targeting lysosome in vivo and in an in vivo situation related to PD.
Figure 6. Acidic nanoparticles protect dopaminergic neurons from MPTP-induced cell death through rescue of lysosomal function after intracerebral injections in mice. ((A) to C) Representative photomicrographs of TH-immunostained SNpc from saline-and MPTP-treated mice, injected with PLGA-aNP, or controls. Dotted lines circle the SNpc containing dopaminergic neurons. (D) Stereological cell counts of SNpc TH-immunoreactive neurons from saline- or MPTP-intoxicated mice, intracerebrally injected with PLGA-aNP, at 5 d post-MPTP (n=5 for vehicle-injected animals, n=6 for MPTP-treated mice, n=7 for MPTP + aNP-treated animals). ((E) to G) Enzymatic activities of lysosomal enzymes: ACP2/acid phosphatase isoform 2, HEX/β-hexosaminidase and CTSD in cytosolic, lysosomal-free cytosolic fractions from the ventral midbrain of saline, MPTP-treated mice with or without PLGA-aNP. (H) Enzymatic activity of CTSD in lysosomal fraction from the ventral midbrain of saline, MPTP-treated mice treated with PLGA-aNP, or controls. *, P<0.05 compared with vehicle-injected mice; #, P<0.05 compared with MPTP-intoxicated mice.
Discussion
Lysosomal impairment is increasingly regarded as a major pathogenic event in neurodegenerative diseases, including PD.Citation11 The lysosome is now recognized as a multifactorial organelle involved in cellular clearance, signaling and energy metabolism.Citation36 However, none of the ALPs-based strategies used so far directly targeted the lysosome, but instead aimed at enhancing the whole autophagy machinery.Citation37
Here we show that PLGA-aNP are internalized into mammalian cells and trafficked to lysosomes. Furthermore, we reveal the ability of acidic PLGA-aNP to reacidify defective lysosomes to basal level and restore lysosomal deleterious effects caused by PD-linked mutations related to ALPs in 3 different pathological PD models. We here demonstrate that PLGA-aNP may be successfully delivered to dopaminergic neurons after intracerebral injection and attenuate MPTP-induced degeneration of the dopaminergic neurons in mice through alleviation of LMP, supporting the translating opportunity of such biotechnology-based strategy.
Furthermore, in order to go beyond PD (thus central nervous system-centered) and extend our innovative approach to other lysosomal and non-brain related disorders, we demonstrate that PLGA-aNP can efficiently counteract lysosomal acidification impairment induced by V-ATPase activity deficiency in XMEA disorder, suggesting that such innovative strategy could also be applied to other diseases.
Interestingly, PLGA-aNP were not able to restore lysosomal pH after BafA treatment but efficiently after CQ. In this regard, several studies shows that BafA treatment induce the blockade of endosomal carrier vesicle formation Citation38 as well as transport from late endosomes to lysosomes.Citation39 These effects might explain the absence of PLGA-aNP efficacy after BafA treatment as we also observed a decreased addressing of PLGA-aNP following BafA-treatment (). However, we cannot exclude that the specific inhibition of the proton-ATPase by BafA might inhibit the change of surface charge from anionic to cationic that happens in the acidic environment of secondary endosomesCitation19 and subsequently prevent PLGA-aNP from exerting their beneficial effect (). This suggests that PLGA-aNP would need at least a partially functional V-ATPase as PLGA-aNP still exert their effect in XMEA-mutant fibroblasts with a range of ATPase activity reduction between 11% and 13% of normal.
Figure 7. Proposed PLGA-aNP mechanism. PLGA-aNP are endocytosed by a nonspecific event and then traffic from early endosomes to late endosomes and ultimately are delivered to lysosomes where they can exert their effect. A key step in PLGA-aNP effect is the protonation in the late endosome (aNP+). Once in the lysosome, PLGA-aNP can stabilize lysosomal pH (pHL) and maintain lysosomal membrane integrity. MPTP and its active ion MPP+ induce lysosomal membrane permeabilization (LMP), which is blocked by PLGA-aNP. The lysosomotropic drug chloroquine (CQ) induces lysosomal alkalinisation and LMP that are both inhibited by PLGA-aNP. Only bafilomycin A1 (BafA) blocks the effect of PLGA-aNP. We showed that it is, at least partially, due to the inhibition of trafficking of PLGA-aNP but might also involve the inhibition of PLGA-aNP protonation due to the inhibition of lysosomal V-ATPase proton pump (in blue). PLGA-aNP might also be degraded and their derivative (lactic and glycolic acids) might support the mitochondrial membrane potential (ΔΨm).
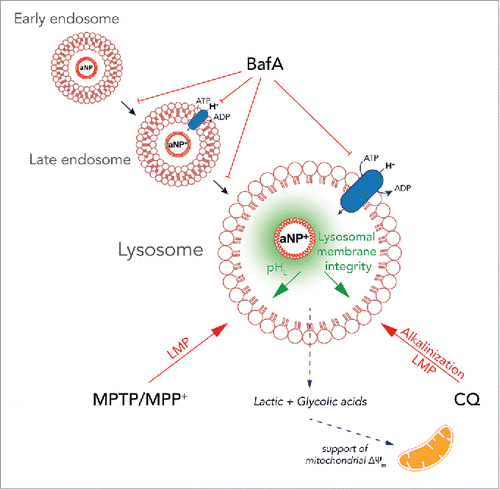
Using the mitochondrial toxin MPP+, we showed that lysosomal alkalinization associated with LMP, could be reversed by a combined treatment with PLGA-aNP and a ROS scavenger. In this paradigm, PLGA-aNP alone were not able to decrease cell death when administered concomitantly, although rescuing lysosomal pH. Thus, most of the toxicity is imputable to oxidative stress as previously described.Citation22,24 Interestingly, a pretreatment with PLGA-aNP decrease cell death induced by MPP+ in vitro and decrease both cell death and MPTP-induced LMP in vivo. While we cannot fully explain this effect, previous reports have shown that PLGA-aNP exert an excellent biocompatibility and are easily metabolized by the Krebs cycle.Citation40 Moreover, degradation products of PLGA (i.e., lactic and glycolic acids) might lower lysosomal pH through their chemical properties. Interestingly, these degradation products have also been shown to support mitochondrial membrane potential as well as neuronal survival in several models related to mitochondrial insults and PD.Citation41 We cannot exclude that PLGA-aNP, after exerting their lysosomal effect, could also have a mitochondrial effect (). Further studies are needed to fully understand this biphasic effect.
Regarding the discrepancy in PLGA-aNP effectiveness between ATP13A2 and GBA mutant fibroblasts, one possible explanation lies in the nature of these 2 genes and chosen mutations. PLGA-aNP were able to rescue lysosomal pH in both ATP13A2 mutated fibroblasts and ATP13A2-knockdown cells. The combination of these 2 models suggests that the lysosomal dysfunction related to ATP13A2 might be due to a loss of function, which can be rescued by PLGA-aNP. In GBA mutant fibroblasts, PLGA-aNP did not fully restore lysosomal pH. Some authors have suggested that mutant GBA induces lysosomal dysfunction through a gain of toxic function,Citation30 inhibiting the effect of PLGA-aNP and explaining the weak efficacy of PLGA-aNP in this model. Moreover, while a causal relationship has been clearly established between ATP13A2 mutations and juvenile forms of PD, mutations in the GBA gene remain only a genetic risk factor.Citation11
So far, the proposed therapeutic strategies aiming at restoring lysosomal function are based upon pharmacological compoundsCitation4 or viral-mediated overexpression of autophagy components.Citation12-14 With regard to drug development, whole autophagy process enhancement should be carefully considered as autophagy can determine cell fate as it has been shown to have either survival or death promoting roles.Citation42 In respect to this concept, we here show that solely restoration of lysosomal physiology without impacting the autophagy flux can be a successful strategy. Mounting evidence indicates that lysosomal impairment is a common feature between idiopathic and genetic forms of PD. Thus, restoration of lysosomal physiology might be used not only in hereditary forms of PD but also in the sporadic condition. Although we focused on PD, we also demonstrate that PLGA-aNP exert their effect in other pathological contexts, such as a genetic model of lysosomal-related myopathy. This paves the way for other diseases such as Alzheimer disease in which defect in lysosomal acidification through mutations in PSEN1 (presenilin 1) has been describedCitation43 without neglecting the wide range of LSD.Citation44 Finally, and more importantly, we validated for the first time that solely restoration of lysosomal physiology, using a lysosome-targeted strategy and without cellular machinery intervention, might be of therapeutic interest for pathologies associated with lysosomal impairment.Citation45
Material and methods
Cell lines and transfections
Cultured fibroblasts from one PD patient harboring the ATP13A2 mutation (L3292), and from control individuals come from our fibroblast library.Citation1 L3292 is a compound heterozygous fibroblast for 3057delC and 1306+5G®A.Citation10 Cultured fibroblasts from 2 PD patients with GBA mutations (p.N370S and p.G377S) were obtained from Coriell Cell Repositories (NJ, USA). XMEA fibroblasts (c*6A to G mutation) were obtained generously from Dr. Berge Minassian's laboratory (Division of Neurology, Department of Pediatrics, The Hospital for Sick Children, Toronto, Canada). Control, as well as mutant ATP13A2, GBA and XMEA fibroblasts were maintained at 37°C in 5% CO2 in DMEM medium (Sigma-Aldrich, D5671), supplemented with 10% fetal calf serum (Life Technologies, 26140-0179) and 1% penicillin/streptomycin (Sigma-Aldrich, P0781). Human neuroblastoma cell lines BE(2)-M17 were obtained from ATCC (CRL-2267) and grown in OPTIMEM (Life Technologies, 31985-047) plus 10% fetal bovine serum (Sigma-Aldrich, D0899). ATP13A2 stable knockdown BE-(2)-M17 (shATP13A2(403-1)) human dopaminergic neuroblastoma cells were maintained at 37°C in 5% CO2 in OPTIMEM supplemented with 10% fetal calf serum, 1% penicillin/streptomycin and 2 µg/ml puromycin (Sigma-Aldrich, P8833). Transient transfections with LAMP1-GFP or GFP-targeted to mitochondria (mts-GFP) were performed with Lipofectamine 2000 (Life Technologies, 11668019) following the manufacturer's recommendations. The LAMP1-GFP plasmid was obtained from Dr. Jennifer Lippincott-Schwartz (National Institutes of Health, Bethesda, MD). We thank Muriel Priault (CNRS UMR 5095, Bordeaux, France) for RAB5A and RAB7A constructs. CellLight reagents for labeling of endoplasmic reticulum or Golgi apparatus (Life Technologies, C10590 and C10592 respectively) were used according manufacturer's recommendations. For drug treatments, cells were grown at 70% to 80% confluence and treated for either 1 or 24 h, as indicated in the text. Cells were treated with 10 μM or 20 μM chloroquine (Sigma-Aldrich, C6628), or either 5 nM or 50 nM bafilomycin A1 (Sigma-Aldrich, B1793). BE(2)-M17 cells were treated with 2.5 mM MPP+ (Sigma-Aldrich, D048) in the presence or the absence of 500 μM tempol (Sigma-Aldrich, 176141).
aNP preparation
Resomer® RG 503H PLGA (Sigma-Aldrich, 719870) was used with a lactide-glycoside ratio of 50:50, Molecular weight 24000 to 38000. A stock solution of polymer was prepared by dissolving 31 mg of PLGA in 3.1 mL of THF (Sigma-Aldrich, 401757). 200 µL of this stock solution are added to 20 mL of deionized water under sonication. The suspension of PLGA-aNP was then slowly concentrated to provide a sample with a typical concentration of 0.18 mg/mL. PLGA-aNP containing Nile red fluorophore (Sigma-Aldrich, 19123) were formulated similarly, except that 0.9 mg of Nile red was introduced in the polymer stock solution. For all experiments, PLGA-aNP solutions were used as freshly prepared.
aNP characterization
Transmission electron microscopy was carried out using a HITACHI H7650 electron microscope (HITACHI Ltd., Tokyo, Japan). The copper grid coated with a carbon membrane was pretreated using the Glow discharge technique to yield positively charged hydrophilic carbon surface to allow stronger interaction between the sample and the grid itself and thus easier imaging. One droplet of the PLGA-aNP aqueous suspension was deposited on the grid and the excess liquid was dried off with a paper. A staining procedure using uranyl acetate was used to enhance the contrast.
Cell viability assay and lysosomal pH measurement
Cell viability was estimated by an MTT assay (ATCC, 30-1010K) following the manufacturer's recommendations. Quantification of lysosomal pH was determined using dextran conjugates LysoSensor Yellow/Blue DND-160 (Life Technologies, L-22460), and was performed as previously described.Citation1 Briefly, control and mutant ATP13A2 cells were grown in their respective media. Cells were then trypsinized, harvested (1×106 cells/ml) and loaded with 1mg/ml of LysoSensor-dextran for 1 h at 37°C with 5% CO2. The cells were then washed 3X in HBSS (Gibco, 14060) and aliquoted at 100 μl into a black 96-well microplate. pH calibration was performed as previously described.Citation1 Wild-type and mutant cells were treated with 10 μM monensin (Sigma-Aldrich, M5273) and 10 μM nigericin (Sigma-Aldrich, N7143) in MES buffer (5 mM NaCl, 115 mM KCl, 1.3 mM MgSO4, 25 mM MES), with the pH adjusted to a range from 3.5-7.0. The samples were read in a FLUOstar Optima fluorimeter (BMG Labtech, Champigny sur Marne, France) with excitation at 355 nm. The ratio of emission 440/535 nm was then calculated for each sample. The pH values were determined from the linear standard curve generated via the pH calibration samples.
CTSD activity assay
CTSD activity was measured in cell lysates using a fluorometric CTSD activity assay kit (Abcam, ab65302) in accordance with the manufacturer's instructions. Fluorescence was measured on a FLUOstar Optima microplate analyzer (BMG Labtech, Champigny sur Marne, France).
HEX/β-hexosaminidase and ACP2/acid phosphatase enzymatic assays
The integrity of the lysosomal membrane in cultured cells was monitored by analyzing the presence of 2 lysosomal enzymes, namely, ACP2 (acid phosphatase 2, lysosomal) and HEX/β-hexosaminidase, in the cytosol. Fresh ventral midbrain samples, from saline- or MPTP-intoxicated mice, intracerebrally injected with PLGA-aNP, or controls, at 5 d post-MPTP (n=5 for each group) were dissected. The tissue was homogenized immediately in buffered sucrose (Sigma-Aldrich, S9378; 4 mM HEPES, 150 mM sucrose, pH 7.4) and following differential centrifugation the cytosolic fraction (supernatant fraction after 100,000 × g centrifugation for 30 min) and the light mitochondrial-lysosomal enriched fraction were obtained. Phosphatase activity was assayed with the help of the acid phosphatase assay kit (Sigma-Aldrich, CS0740), according to the manufacturer's instructions, as well as the β-hexosaminidase assay kit (Sigma-Aldrich, CS0780). These experiments were performed by measuring the ectopic activities of 2 lysosomal enzymes, HEX/β-hexosaminidase and ACP2 (acid phosphatase 2, lysosomal) in cytosolic lysosomal-free fractions.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde. LAMP2 antibody (mouse monoclonal; Santa Cruz Biotechnology, H4B4, sc-18822) was used at 1:600. Cell nuclei were visualized using Hoechst (Life Technologies, 33342) at 10 μM for 10 min. Quantification of the number of PLGA-aNP was performed by taking random pictures and manually counting the number of red round-shaped objects and nuclei in the field. The ratio of counted objects on counted nuclei was the plot. Fluorescence was analyzed using a Zeiss Axio Imager.M2 fluorescence microscope (Oberkochen, Germany) or a confocal Leica microscope DM 6000 TCS SP8 X (Wetzlar, Germany). 3D reconstruction images were modeled as mixed rendering using Bitplane Imaris software (Zurich, Switzerland). Orthogonal views were generated with ImageJ.Citation46
Flow cytometry
Following treatments, cells were harvested in ice-cold phosphate-buffered saline (Gibco, 70011044). Fluorescence measurements and analyses were performed using a BD LSRFortessa cytometer (Franklin Lakes, New Jersey, USA).
Electron microscopy
For EM, cells were fixed with a solution of 0.1 M phosphate buffer (PB) pH 7.4 containing 1.6% glutaraldehyde, at 4°C for 45 min. Then, cells were rinsed in 0.1 M PB and postfixed in osmium tetroxide with 1% uranyl acetate in 70% ethanol. Cultures were embedded in resin (Fluka, Durcupan ACM) on a capsule filled with resin and polymerized at 58°C for 48 h. Finally, coverslips were removed by heat shock in liquid nitrogen and boiling water. Ultrathin sections were cut, collected on pioloform-coated single-slop copper grids (Electron Microscopy Sciences, FCF2010-Cu), stained with lead acetate and examined with a HITACHI-H7650 (HITACHI Ltd., Tokyo, Japan) electron microscope.
Cell lysates and western blot analysis
Cells were washed with cold phosphate-buffered saline at 4°C and lysed in a buffer containing 25 mM Tris HCl, pH 6.8, 1% SDS (Sigma-Aldrich, L3771), 250 mM DTT, glycerol 7.5%, 0.05% bromophenol blue. For immunoblotting, 40 µg of protein were loaded per lane and separated by SDS-PAGE (18% for LC3B; 12% for SQSTM1), transferred to nitrocellulose membranes, and immunoblotted with LC3B (rabbit polyclonal; Novus Biologicals, NB100-2220), SQSTM1 (guinea-pig polyclonal; Progen, GP62-C), CTSD (mouse monoclonal; Sigma-Aldrich, C0715). ACTB/β-actin (mouse monoclonal; Sigma-Aldrich, A5441) was used to control equal loading.
Animals and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-intoxicated mice
Experiments were performed in accordance with the European Union directive of September 22, 2010 (2010/63/EU) on the protection of animals used for scientific purposes. The Institutional Animal Care and Use Committee of Bordeaux (CE50) approved the experiments under the license number 5012099-A. For PLGA-aNP detection in the SNc, 2 wild-type C57BL/6 mice (2 mo old) received 1 µl of PLGA-aNP by stereotactic delivery to the substantia nigra (anteroposterior: −2.9; lateral: 1.3; dorsoventral: 4.5) at a flow rate of 0.4 µl/min. For proof-of-concept neuroprotective activity, 5 to 7 mice were used in each group. Thirteen 8- to 10-wk-old male C57BL/6 mice received one intraperitoneal injection of MPTP-HCl per day (30 mg/kg/day of free base; Sigma-Aldrich, M0896) for 2 consecutive d.Citation35,47 Control mice received saline injections only. Mice were killed 5 d after the last MPTP administration. Animals were deeply anesthetized with chloral hydrate (VWR, MOLEM70482875), 150 mg/kg (i.p.). Brains were collected fresh, frozen in isopentane and immediately stored at −80°C until further post-mortem processing.
Immunohistochemistry and stereological counting
For stereological counting of dopaminergic neurons and immunofluorescence, mice were terminated and brains were collected, immediately immersed in cold 4% paraformaldehyde for 24 h, then cryoprotected with 20% sucrose (Fluka, 84100) and finally frozen by immersion in isopentane at −50°C.Citation48 Brains were cut at 50-µm thick sections and SN was collected.
For colocalization studies of PLGA-aNP with lysosomes in vivo, TH (tyrosine hydroxylase; Abcam, ab6211) and LAMP2 (Abcam, ab25339) immunohistochemistry was performed as previously described.Citation4 Donkey anti-mouse IgG conjugated to Alexa Fluor 488 (Abcam, ab150109) and goat anti-rabbit Atto647N (Sigma, 40839) were used as secondary antibody. Cell nuclei were visualized using Hoechst (Life Technologies, 33342) at 1:5,000 for 30 sec. Fluorescence was analyzed using a Leica fluorescence microscope with Morpho Strider (Explora Nova, La Rochelle, France) or Leica confocal fluorescence microscope (SPE). All images were then analyzed with ImageJ.Citation46
For stereological counting, TH immunochemistry was performed with the aforementioned protocol and revealed by an anti-mouse peroxidase EnVision system (Dako, K400311) followed by VECTOR Peroxidase (Vector Laboratories, SK4100) incubation. Boundaries of the SN were chosen by examining the size and shape of the different TH-immunopositive neuronal groups, cellular relationships to axonal projections and nearby fiber bundles. Unbiased stereological counting of TH-positive neurons was performed using Exploranova Mercator (Explora Nova, La Rochelle, France) as previously described.Citation48,49 Ten sections per animal were used and optical dissectors were distributed using a systemic sampling scheme. Dissectors (50 µm × 40 µm) were separated from each other by 150 µm (x) and 120 µm (y). In these dissectors, the nuclei of neurons were counted into focus.
Statistical analysis
All data are representative of at least 3 independent experiments. Statistical significance of the data was evaluated after the calculation of a one-way or 2-way analyses of variance (ANOVA) followed by Tukey's multiple comparison test using GraphPad Prism. Values of P<0.05 were considered statistically significant. All values are expressed as the mean ± standard error of the mean.
Abbreviations
| 3D | = | 3-dimensional |
| ACP2 | = | acid phosphatase 2, lysosomal |
| ALPs | = | autophagy-lysosomal pathways |
| AP | = | autophagosome |
| BafA | = | bafilomycin A1 |
| CQ | = | chloroquine |
| CTSD | = | cathepsin D |
| EM | = | electron microscopy |
| GBA/glucocerebrosidase | = | glucosidase, β, acid |
| GD | = | Gaucher disease |
| GFP | = | green fluorescent protein |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3 β |
| LMP | = | lysosomal membrane permeabilization; |
| LSD | = | lysosomal storage disorder |
| MPP+ | = | 1-methyl-4-phenylpyridinium ion |
| MPTP | = | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mts-GFP | = | GFP-targeted to mitochondria |
| PD | = | Parkinson disease |
| PLGA-aNP | = | poly(DL-lactide-co-glycolide) acidic nanoparticles |
| ROS | = | reactive oxygen species |
| SNpc | = | substantia nigra pars compacta |
| V-ATPase | = | vacuolar-type ATPase |
| XMEA | = | X-linked myopathy with excessive autophagy. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Files
Download Zip (7.1 MB)Acknowledgments
We thank Marie-Laure Thiolat and Evelyne Doudnikoff for their technical assistance. The microscopy was done in the Bordeaux Imaging Center, a service unit of the CNRS-INSERM and Bordeaux University, member of the national infrastructure France BioImaging. The help of Sebastien Marais, Fabrice Cordelieres and Sabrina Lacomme is acknowledged. Flow cytometry experiments were done in the Cytometry core facility, a service unit of the SFR TransBioMed. The help of Vincent Pitard and Santiago Gonzalez is acknowledged. The University of Bordeaux and the Centre National de la Recherche Scientifique provided infrastructural support.
Funding
This work was supported by Marie Curie Reintegration Grant FP7-PEOPLE-2009-ERG256303 from the European Commission and PEPS-Idex 1024R-3219 (B.D.), Fondation pour la Recherche Médicale (B.D.), Fondation de France (E.B.) Agence Nationale de la Recherche ANR-12-BSV4-0001-01 (E.B.) and LABEX BRAIN ANR-10-LABX-43. M.B. has been supported by a MESR fellowship and the France Parkinson Foundation. M.B-D gratefully acknowledges Conseil Régional d'Aquitaine for financial support (Chaire d'Accueil grant). J.D. received a fellowship from Conseil Régional d'Aquitaine.
References
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 2012; 109:9611-6; PMID:22647602; http://dx.doi.org/10.1073/pnas.1112368109
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 2012; 32:4240-6; PMID:22442086; http://dx.doi.org/10.1523/JNEUROSCI.5575-11.2012
- Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson disease. EMBO Mol Med 2012; 4:380-95; PMID:22407749; http://dx.doi.org/10.1002/emmm.201200215
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson disease. J Neurosci 2010; 30:12535-44; PMID:20844148; http://dx.doi.org/10.1523/JNEUROSCI.1920-10.2010
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson disease: relationship to α-synuclein inclusions. Neurobiol Dis 2009; 35:385-98; PMID:19505575; http://dx.doi.org/10.1016/j.nbd.2009.05.023
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson disease. Histol Histopathol 1997; 12:25-31; PMID:9046040
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 2010; 67:1464-72; PMID:20697033; http://dx.doi.org/10.1001/archneurol.2010.198
- Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson disease. Physiol Rev 2011; 91:1161-218; PMID:22013209; http://dx.doi.org/10.1152/physrev.00022.2010
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med 2009; 361:1651-61; PMID:19846850; http://dx.doi.org/10.1056/NEJMoa0901281
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006; 38:1184-91; PMID:16964263; http://dx.doi.org/10.1038/ng1884
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. Lysosomal impairment in Parkinson disease. Mov Disord 2013; 28:725-32; PMID:23580333; http://dx.doi.org/10.1002/mds.25462
- Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013; 136:2130-46; PMID:23757764; http://dx.doi.org/10.1093/brain/awt131
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110:E1817-26; PMID:23610405; http://dx.doi.org/10.1073/pnas.1305623110
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson and Lewy body diseases. J Neurosci 2009; 29:13578-88; PMID:19864570; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009
- Diebold Y, Calonge M. Applications of nanoparticles in ophthalmology. Prog Retin Eye Res 2010; 29:596-609; PMID:20826225; http://dx.doi.org/10.1016/j.preteyeres.2010.08.002
- Hasadsri L, Kreuter J, Hattori H, Iwasaki T, George JM. Functional protein delivery into neurons using polymeric nanoparticles. J Biol Chem 2009; 284:6972-81; PMID:19129199; http://dx.doi.org/10.1074/jbc.M805956200
- Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release 2010; 145:182-95; PMID:20226220; http://dx.doi.org/10.1016/j.jconrel.2010.01.036
- Baltazar GC, Guha S, Lu W, Lim J, Boesze-Battaglia K, Laties AM, Tyagi P, Kompella UB, Mitchell CH. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PloS One 2012; 7:e49635; PMID:23272048; http://dx.doi.org/10.1371/journal.pone.0049635
- Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J 2002; 16:1217-26; PMID:12153989; http://dx.doi.org/10.1096/fj.02-0088com
- Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(D,L-lactide-co-glycolide) nanoparticles. Int J Pharm 2003; 262:1-11; PMID:12927382; http://dx.doi.org/10.1016/S0378-5173(03)00295-3
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27:6434-51; PMID:18955971; http://dx.doi.org/10.1038/onc.2008.310
- Vila M, Bove J, Dehay B, Rodriguez-Muela N, Boya P. Lysosomal membrane permeabilization in Parkinson disease. Autophagy 2011; 7:98-100; PMID:21045565; http://dx.doi.org/10.4161/auto.7.1.13933
- Bove J, Martinez-Vicente M, Dehay B, Perier C, Recasens A, Bombrun A, Antonsson B, Vila M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 2014; 10:889-900; PMID:24686337; http://dx.doi.org/10.4161/auto.28286
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson disease. Proc Natl Acad Sci U S A 2003; 100:6145-50; PMID:12721370; http://dx.doi.org/10.1073/pnas.0937239100
- Dean RT. Lysosomes and protein degradation. Ciba Found Symp 1979:139-49; PMID:399886
- Ginns EI, Brady RO, Pirruccello S, Moore C, Sorrell S, Furbish FS, Murray GJ, Tager J, Barranger JA. Mutations of glucocerebrosidase: discrimination of neurologic and non-neurologic phenotypes of Gaucher disease. Proc Natl Acad Sci U S A 1982; 79:5607-10; PMID:6957882; http://dx.doi.org/10.1073/pnas.79.18.5607
- Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014; 137:1304-22; PMID:24531622; http://dx.doi.org/10.1093/brain/awu002
- Kurzawa-Akanbi M, Hanson PS, Blain PG, Lett DJ, McKeith IG, Chinnery PF, Morris CM. Glucocerebrosidase mutations alter the endoplasmic reticulum and lysosomes in Lewy body disease. J Neurochem 2012; 123:298-309; PMID:22803570; http://dx.doi.org/10.1111/j.1471-4159.2012.07879.x
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146:37-52; PMID:21700325; http://dx.doi.org/10.1016/j.cell.2011.06.001
- Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol 2011; 69:940-53; PMID:21472771; http://dx.doi.org/10.1002/ana.22400
- Kalimo H, Savontaus ML, Lang H, Paljarvi L, Sonninen V, Dean PB, Katevuo K, Salminen A. X-linked myopathy with excessive autophagy: a new hereditary muscle disease. Ann Neurol 1988; 23:258-65; PMID:2897824; http://dx.doi.org/10.1002/ana.410230308
- Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZP, et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol 2013; 125:439-57; PMID:23315026; http://dx.doi.org/10.1007/s00401-012-1073-6
- Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, Singleton A, Olanow CW, Merchant KM, Bezard E, et al. Targeting α-synuclein for treatment of Parkinson disease: mechanistic and therapeutic considerations. Lancet Neurol 2015; 14(8):855-66
- Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, et al. Priorities in Parkinson disease research. Nat Rev Drug Discov 2011; 10:377-93; PMID:21532567; http://dx.doi.org/10.1038/nrd3430
- Bezard E, Imbert C, Gross CE. Experimental models of Parkinson disease: from the static to the dynamic. Rev Neurosci 1998; 9:71-90; PMID:9711900; http://dx.doi.org/10.1515/REVNEURO.1998.9.2.71
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/10.1038/emboj.2012.32
- Bourdenx M, Bezard E, Dehay B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front Neuroanat 2014; 8:83; PMID:25177278; http://dx.doi.org/10.3389/fnana.2014.00083
- Clague MJ, Urbe S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem 1994; 269:21-4; PMID:8276796
- van Weert AW, Dunn KW, Geuze HJ, Maxfield FR, Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol 1995; 130:821-34; PMID:7642700; http://dx.doi.org/10.1083/jcb.130.4.821
- Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012; 161:505-22; PMID:22353619; http://dx.doi.org/10.1016/j.jconrel.2012.01.043
- Toyoda Y, Erkut C, Pan-Montojo F, Boland S, Stewart MP, Muller DJ, Wurst W, Hyman AA, Kurzchalia TV. Products of the Parkinson disease-related glyoxalase DJ-1, D-lactate and glycolate, support mitochondrial membrane potential and neuronal survival. Biol Open 2014; 3:777-84; PMID:25063200; http://dx.doi.org/10.1242/bio.20149399
- Eskelinen EL. Doctor Jekyll and Mister Hyde: autophagy can promote both cell survival and cell death. Cell Death Differ 2005; 12 Suppl 2:1468-72; PMID:16247492; http://dx.doi.org/10.1038/sj.cdd.4401721
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146-58; PMID:20541250; http://dx.doi.org/10.1016/j.cell.2010.05.008
- Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal 2012; 17:766-74; PMID:22098160; http://dx.doi.org/10.1089/ars.2011.4405
- Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2010; 2:189-204
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/10.1038/nmeth.2089
- Sterky FH, Hoffman AF, Milenkovic D, Bao B, Paganelli A, Edgar D, Wibom R, Lupica CR, Olson L, Larsson NG. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum Mol Genet 2012; 21:1078-89; PMID:22090423; http://dx.doi.org/10.1093/hmg/ddr537
- Bastide MF, Dovero S, Charron G, Porras G, Gross CE, Fernagut PO, Bézard E. Immediate-early gene expression in structures outside the basal ganglia is associated to l-DOPA-induced dyskinesia. Neurobiol Dis 2014; 62:179-92; PMID:24103779; http://dx.doi.org/10.1016/j.nbd.2013.09.020
- Engeln M, Bastide MF, Toulme E, Dehay B, Bourdenx M, Doudnikoff E, Li Q, Gross CE, Boué-Grabot E, Pisani A, et al. Selective Inactivation of Striatal FosB/DeltaFosB-Expressing Neurons Alleviates L-Dopa-Induced Dyskinesia. Biol Psychiatry 2014; 79(5):338-340; PMID:25146322.