
ABSTRACT
The MAP1LC3/LC3 family plays an essential role in autophagosomal biogenesis and transport. In this report, we show that the HECT family E3 ubiquitin ligase NEDD4 interacts with LC3 and is involved in autophagosomal biogenesis. NEDD4 binds to LC3 through a conserved WXXL LC3-binding motif in a region between the C2 and the WW2 domains. Knockdown of NEDD4 impaired starvation- or rapamycin-induced activation of autophagy and autophagosomal biogenesis and caused aggregates of the LC3 puncta colocalized with endoplasmic reticulum membrane markers. Electron microscopy observed gigantic deformed mitochondria in NEDD4 knockdown cells, suggesting that NEDD4 might function in mitophagy. Furthermore, SQSTM1 is ubiquitinated by NEDD4 while LC3 functions as an activator of NEDD4 ligase activity. Taken together, our studies define an important role of NEDD4 in regulation of autophagy.
Introduction
Autophagy is a lysosome-mediated cellular degradation process for recycling and removal of misfolded proteins, insoluble aggregates (aggresomes), damaged organelles and external pathogens.Citation1-3 Autophagy is activated by many cellular signals, mainly nutritional and oxidative stress signals.Citation4,5 The effect of autophagy on cell survival or death is variable,Citation6,7 probably due to circumstances in which autophagy is activated. In general autophagy is an important cellular process for relieving cellular stress conditions, such as nutritional starvation, endoplasmic reticulum (ER) stress and mitochondrial damage. In cancer cells, autophagy has been observed required for cell survival.Citation8,9 However, constitutive activation of autophagy by continuous stress may result in cell death. Autophagy has been classified as one of the 3 cell death forms, along with apoptosis and necrosis.Citation10
MAP1LC3 (microtubule associated protein 1 light chain 3), abbreviated as LC3 henceforth, is a mammalian family of orthologs of the yeast autophagic protein Atg8. The LC3 family is essential in autophagy involved in biogenesis and transport of autophagosomes by functioning in elongation of the phagophore double-layer membrane and in the recruitment of proteins for autophagic processes.Citation11-13 LC3 is structurally similar to ubiquitin and can be biochemically modified by cleavage and lipidation with conjugation to phosphatidylethanolamine (PE), which resembles a ubiquitination process.Citation13-15 This PE–conjugated LC3, named as LC3-II, is the functional form on phagophores. LC3-II has been used as a marker of autophagosomes and formation or turnover rate of LC3-II used as an index of autophagic activity.Citation16,17
Although LC3 plays an essential role in autophagy, the molecular mechanism underlying the effect of LC3 on autophagy is largely unknown. One important function of LC3 is recruiting autophagic proteins to autophagosomes for assembly of functional autophagosomal complexes by interacting with the LIR (LC3-interacting region) domain-containing proteins.Citation18,19 Currently, more than 100 proteins have been identified containing the LIR domain.Citation19 One relevant example is the core autophagic protein SQSTM1/p62. SQSTM1 interacts with LC3 and E3 ubiquitin ligases, such as KEAP1 and TRAF6,Citation20-23 and mediates ubiquitination-dependent selective autophagy.Citation24,25 However, the ubiquitination signaling in autophagic processes remains poorly characterized.
NEDD4/NEDD4–1 is a member of the HECT ubiquitin E3 ligase family. Early studies found that NEDD4 ubiquitinates epithelial sodium channel ENaC for proteasomal degradation.Citation26,27 The defect in this regulation was found in patients with the Liddle syndrome, a hypertension disease.Citation28 In yeast, the NEDD4 ortholog Rsp5 regulates the cargo sorting process of late endosomes and multivesicular bodies (MVBs) by ubiquitination of the cargos.Citation29 NEDD4 also plays an important role in the retroviral budding process, which resembles the sorting process of MVBs, by ubiquitination of Gag polyproteins.Citation30,31 In mammalian cells, NEDD4 is involved in regulation of intracellular trafficking of receptor tyrosine kinases by ubiquitination of either endocytic or vesicle sorting proteins, such as CBL, EPS15, TSG101, HGS/HRS, SCAMPs, and TNK2/ACK1,Citation32-37 or the respective receptors.Citation38 Furthermore, NEDD4 interacts and ubiquitinates the tumor suppressor PTEN and facilitates its degradation,Citation39 suggesting that NEDD4 may function as an oncogenic protein and promote tumorigenesis.
The involvement of NEDD4 in autophagy was first found in a study on the characterization of the cellular autophagy network that shows interaction of LC3 with NEDD4.Citation40 Consistent with this, a recent report observes that knockdown of NEDD4 in cancer cells inhibits autophagy.Citation41 In this report, we found that NEDD4 interacts with LC3 through an LIR domain. Functional analysis by knockdown or mutation of NEDD4 indicates that NEDD4 is required for autophagy and involved in biogenesis of autophagosomes. Ubiquitination or the in vitro ligase activity assay suggests that LC3 is an activator, not a substrate, of NEDD4. Our studies have demonstrated that NEDD4 plays an essential role in autophagy.
Results
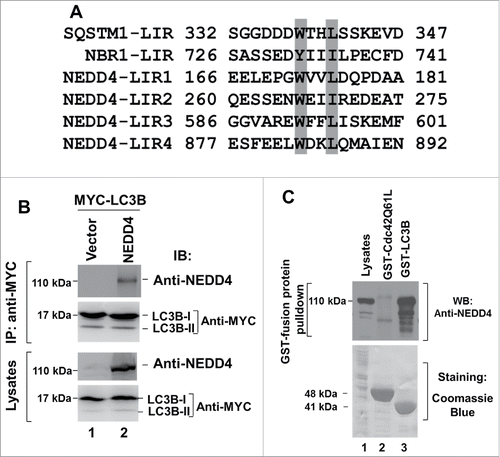
NEDD4 is an LC3-interactive protein
Ubiquitination is an important biochemical process in selective autophagy.Citation24,25 To determine whether the HECT family E3 ubiquitin ligase NEDD4 is involved in regulation of autophagy, we investigated interaction of NEDD4 with autophagic proteins. It is known that LC3 binds to the WXXL motif in the LC3-interaction region (LIR) of proteins.Citation18 Therefore, we searched for the WXXL motif in human NEDD4 peptide sequence and found 4 putative LIRs (), suggesting that NEDD4 may directly interact with LC3 to regulate autophagy. To verify this possibility, we coexpressed NEDD4 with MYC-tagged MAP1LC3B (abbreviated as LC3B henceforth), a common isoform of LC3, in HEK293 cells and performed coimmunoprecipitation assay. As shown in , NEDD4 was coimmunoprecipitated with MYC-tagged LC3B (lane 2). To confirm the binding, we used the bead-bound GST-LC3B to pull down NEDD4 from the HEK293 cell lysates. As shown in , GST-LC3B precipitated significant amount of NEDD4 (lane 3), while GST-CDC42-Q61L, which is a GST-fused GTPase defective mutant of the small G-protein CDC42 and does not interact with NEDD4, used as a control for GST-LC3B, did not precipitate NEDD4 from the cell lysates (lane 2). These results demonstrate that NEDD4 is an LC3-binding protein.
Figure 1. NEDD4 is an LC3-binding protein. (A) Alignment of the 4 putative LIRs of NEDD4 with the identified LIR in SQSTM1 and NBR1. (B) NEDD4 is coimmunoprecipitated with LC3B in HEK293 cells coexpressing NEDD4 and LC3B. (C) Endogenous NEDD4 was precipitated using GST-LC3B-conjugated beads in A549 cells.
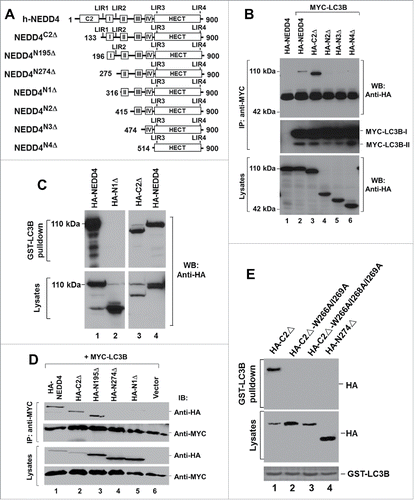
NEDD4 interacts with LC3 through a conserved LIR domain
We first used the truncation mutants of NEDD4, as shown in , to narrow the LC3-binding region. We examined the binding of the NEDD4 truncation mutants to LC3 by coimmunoprecipitation assay and found that NEDD4C2Δ, but not NEDD4N2Δ, was coimmunoprecipitated with LC3B (), indicating that the LC3-binding region is located between amino acids 133 and 415 that contains the putative LIR1 and LIR2 (). We confirmed that the LC3-binding region is within amino acids 133 and 316 by the GST-LC3B affinity precipitation assay (). In addition, we observed that NEDD4C2Δ had a higher binding affinity to LC3B than wild type NEDD4 (lanes 2 and 3, ). This increase in the binding affinity might be due to elimination of the intramolecular interaction between the C2 and the HECT domains upon deletion of the C2 domain, as shown in our previous studies.Citation42 To determine which of the putative LIR1 and LIR2 is responsible for the binding, we further tested the truncation mutants missing the N-terminal 195 amino acids and the N-terminal 274 amino acids (). As shown in , deletion of the N-terminal 195 amino acids did not significantly affect the binding to LC3B (lane 3); however, deletion of the N-terminal 274 amino acids diminished the binding, indicating the binding site is localized between amino acids 196 and 275, which contains the putative LIR2. Thus, NEDD4 may bind to LC3 through the putative LIR2. To confirm that the WXXL motif 266(WEII)269 in the LIR2 is the site for binding to LC3, we mutated W266, I268 and I269 into alanine in NEDD4C2Δ and tested the binding of these mutants to LC3 using an GST-LC3B affinity precipitation assay. As shown in , while NEDD4C2Δ, as a positive control, was precipitated by GST-LC3B, none of the point mutants of the LIR2 WXXL motif was coprecipitated with GST-LC3B. These results demonstrate that the LIR2 motif 266(WEII)269 is the LC3 binding site.
Figure 2. NEDD4 interacts with LC3 through a conserved LIR domain. (A) Truncation constructs of human NEDD4. C2, the C2 domain; I, II, III and IV stand for 4 WW domains; HECT, the HECT domain; the numbers labeled in NEDD4 structure sketches indicate the amino acid residue positions. Positions of the 4 putative LIRs are also marked on the structure sketches. (BE) The LIR2 of NEDD4 is the LC3 binding site. In (B) and (D), interaction of NEDD4 or the truncation mutants with LC3B was determined by coimmunoprecipitation assay; in (C), interaction of NEDD4 or the truncation mutants with LC3B was determined by GST-LC3B pulldown assay; in (E), mutation of the LC3-binding motif WEII in the LIR2 impaired binding of NEDD4 to LC3B.
Knockdown of NEDD4 reduced the level of LC3 and increased the level of SQSTM1
To determine the cellular function of the interaction of NEDD4 with LC3, we made a stable NEDD4 knockdown cell line in lung cancer A549 cells using lentiviral vector-loaded NEDD4 shRNA, and examined the effect of the knockdown on protein levels of key autophagic proteins. As shown in , NEDD4 shRNA eliminated more than 90% of NEDD4 in the cell line as determined by immunoblotting. Depletion of NEDD4 dramatically reduced the LC3 protein level and elevated the SQSTM1 protein level. Furthermore, depletion of NEDD4 also caused a decrease in BECN1/Beclin1, an important autophagic protein in autophagosomal biogenesis,Citation43,44 but did not affect protein level of PDCD6IP/ALIX, a protein involved in endosomal and exosomal trafficking.Citation45,46 This result suggests that NEDD4 might play a role in autophagy.
Figure 3. Knockdown of NEDD4 reduced LC3 protein levels and increased SQSTM1 protein levels. (A) The effect of NEDD4 knockdown on autophagic protein levels in lung cancer A549 cells. (B) The low molecular weight NEDD4 (NEDD4 [LM]) is a degradation product of full-length NEDD4 (NEDD4 [HM]). The HA-tagged NEDD4 was ectopically expressed in HEK293 cells and detected by immunoblotting with either anti-NEDD4 (the left panel) or anti-HA (the right panel). (C) Re-expression of NEDD4 in the NEDD4 shRNA cell line rescued the protein level of LC3 and SQSTM1. **P<0.01; ***P<0.001.
![Figure 3. Knockdown of NEDD4 reduced LC3 protein levels and increased SQSTM1 protein levels. (A) The effect of NEDD4 knockdown on autophagic protein levels in lung cancer A549 cells. (B) The low molecular weight NEDD4 (NEDD4 [LM]) is a degradation product of full-length NEDD4 (NEDD4 [HM]). The HA-tagged NEDD4 was ectopically expressed in HEK293 cells and detected by immunoblotting with either anti-NEDD4 (the left panel) or anti-HA (the right panel). (C) Re-expression of NEDD4 in the NEDD4 shRNA cell line rescued the protein level of LC3 and SQSTM1. **P<0.01; ***P<0.001.](/cms/asset/8a7079c4-de8a-4c49-b073-234b2c7be0a3/kaup_a_1268301_f0003_b.gif)
We noticed that 2 forms of NEDD4, the high molecular mass (HM; 110 kDa) and the low molecular mass (LM; 90 kDa), were depleted by NEDD4 shRNA (the top panel, ). The 110 kDa (HM) band is the full length of NEDD4 isoform 1 that is usually referred to as NEDD4. To determine whether the low molecular weight NEDD4 is a degradation product or an isoform, we examined if ectopically expressed HA-tagged NEDD4 in HEK293 is able to produce a 90-kDa degradation product. As shown in , blotting the HA-NEDD4-expressed lysates with anti-NEDD4 detected 2 bands, one is 110 kDa and the other is 90 kDa, matching the knockdown bands in A549 cells, while blotting with anti-HA detected only one band at 110 kDa. In addition, re-expression of NEDD4 in the shNEDD4 cells also recovered the 90-kD band (). These data suggest that the 90-kDa NEDD4 band is likely a degradation product of the full-length NEDD4, and the degradation site is localized at the N terminus (which eliminated the HA-tag).
To demonstrate the effect of shNEDD4 on protein levels of LC3 and SQSTM1 is truly caused by depletion of NEDD4, not the off-target effect of the shRNA, we re-expressed NEDD4 in the shNEDD4 cells. As shown in , re-expression of NEDD4 completely abolished the effects of shNEDD4 on protein levels of LC3 and SQSTM1, confirming that the shNEDD4-induced changes in autophagic protein levels is truly caused by knockdown of NEDD4.
Knockdown of NEDD4 caused defective autophagy
Next we examined the effect of NEDD4 knockdown on autophagic activity in lung cancer A549 cells by detection of degradation of SQSTM1 and elevation of LC3-II, the 2 indicators of autophagy activation,Citation17 in response to autophagy stimuli. As shown in , treatment with either the starvation medium (glucose and amino acid depleted medium) or rapamycin, 2 known autophagy activators, for 2 and 4 h in the vector control cell line induced a significant degradation of SQSTM1 and an elevation of LC3-II, indicating that autophagy is activated. However, knockdown of NEDD4 in the shNEDD4 cells eliminated both the starvation medium- and rapamycin-induced activation of autophagy (downregulation of SQSTM1 and elevation of LC3-II), indicating that depletion of NEDD4 causes defect in autophagy.
Figure 4. Knockdown of NEDD4 caused defective autophagy. The vector control and the NEDD4 shRNA cell lines were treated with the amino acid-depleted RPMI-1640 (starvation) or 1 μM rapamycin for 0, 2 and 4 h (panels A and B), or 1 μM rapamycin for 0 and 2 h (panels C and D). The cells were lysed by SDS-PAGE sample buffer and the proteins of interest were detected by immunoblotting. The amount of SQSTM1 and LC3-II was quantified using a Gel-Logic 100 Imaging System, from 3 independent experiments. Panels (A) and (B), lung cancer A549 cells; panel (C), hepatocellular carcinoma HepG2 cells; panel (D), neuroblastoma BE(2)-C cells. *P < 0.05; **P < 0.01; ***P < 0.001.
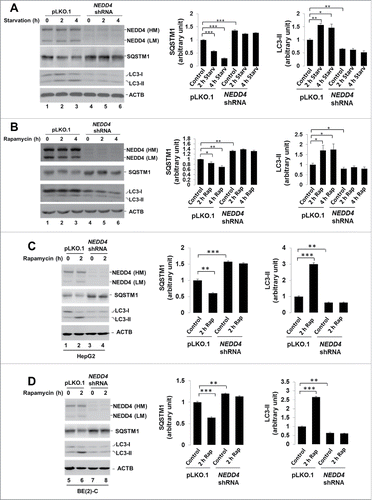
We further examined the effect of NEDD4 on autophagy in other cancer cell lines. As shown in , knockdown of NEDD4 in hepatocellular carcinoma HepG2 cells or neuroblastoma BE(2)-C cells caused the same effect on autophagic protein levels and rapamycin-induced autophagic response as in A549 cells, suggesting that NEDD4 may play a ubiquitous role in autophagy of cancer cells.
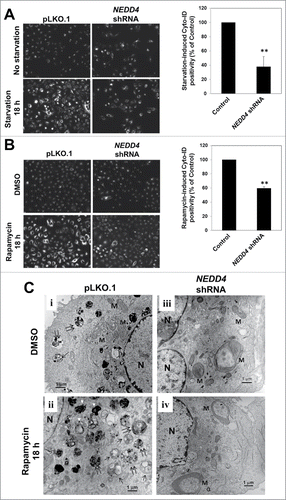
Knockdown of NEDD4 reduced formation of autophagosomes
To confirm the role of NEDD4 in autophagy, we used the Cyto-ID autophagy assay kit to detect formation of autophagosomes upon treatment with the starvation medium or rapamycin in both the vector control and the NEDD4 knockdown cells. As shown in , in the vector control cells, treatment with starvation or rapamycin for 18 h enhanced formation of autophagosomes, indicating activation of autophagy. However, knockdown of NEDD4 significantly inhibited the starvation- and rapamycin-induced formation of autophagosomes by about 60% and 40% respectively. These results suggest a role of NEDD4 in biogenesis of autophagosomes.
Figure 5. Knockdown of NEDD4 reduced formation of autophagosomes. (A and B), Autophagosomes in A549 cells containing either the vector control or the NEDD4 shRNA were labeled live with the Cyto-ID autophagy detection kit, followed by treatment with the amino acid-depleted RPMI-1640 (starvation) or 1 μM rapamycin for 0 and18 h. The labeled fluorescence (autophagosomes) was visualized under an inverted Nikon fluorescent microscope, and quantified by a fluorescence cytometer. The data in the graphs are from 3 independent replicate samples measured with a fluorescent cytometer. (C) Electron microscopy observation of ultra-structure of the cells. Panels i and ii, the vector control cell line; iii and iv, the NEDD4 shRNA cell line. The cells in panels i and iii were treated with DMSO (solvent control) and in panels ii and iv treated with rapamycin (1 μM) for 18 h. M, mitochondria; N, nuclei. Single arrows indicate autophagosomes; double arrows for autolysosomes; arrowheads for lamella bodies. **P<0.01.
We further determined the effect of NEDD4 knockdown on autophagy using electron microscopy. As shown in , in the vector control cell line, rapamycin treatment significantly enhanced formation of autophagosomes and autolysosomes (panel ii), while in the NEDD4 knockdown cell line, very few autophagosomes and autolysosomes were seen in either solvent (DMSO) or rapamycin-treated cells (panels iii and iv). Interestingly, knockdown of NEDD4 caused formation of gigantic mitochondria (panels iii and iv), indicating a severe defect in mitophagy. This defect in mitophagy was seen in both DMSO- and rapamycin-treated NEDD4 knockdown cells, thus is associated only with NEDD4, not rapamycin. Taken together, these data suggest that NEDD4 may function in both selective and canonical autophagic processes.
Depletion of NEDD4 induced aggregation of LC3 puncta in cells
We further determined the effect of NEDD4 knockdown on cellular localization and morphology of autophagosomes by visualizing GFP-LC3B or immuno-stained SQSTM1 fluorescence. The GFP-LC3B fluorescence was monitored in live cells with or without 18 h treatment of rapamycin (). Without treatment of rapamycin, GFP-LC3B in the vector control cells was displayed as diffused or tiny fluorescent puncta, while in the NEDD4 knockdown cells displayed as randomly distributed large fluorescent puncta. The number and size of GFP-LC3B puncta per cell were measured and quantified using ImageJ software (). A total 1713 GFP-LC3B puncta in 40 vector control cells, 3735 GFP-LC3B puncta in 31 rapamycin-treated vector control cells, 1384 GFP-LC3B puncta in 30 shNEDD4 cells, and 1023 GFP-LC3B puncta in 26 rapamycin-treated shNEDD4 cells were counted and measured for statistical analysis. While the average number of the GFP-LC3B puncta per cell had no significant difference between the vector control and the NEDD4 knockdown cell lines, there was a huge increase in size of the GFP-LC3B fluorescent puncta in the NEDD4 knockdown cell line (). Upon treatment with rapamycin, a significant increase in number of GFP-LC3B puncta (autophagosomes) was observed in control cells (the left bottom panel, , and quantification data in ), indicating a rapamycin-induced activation of autophagy. These rapamycin-induced GFP-LC3B puncta were relatively homogenous in size and evenly distributed around nuclei (). In the NEDD4 knockdown cell line, treatment with rapamycin had no significant change in number of the GFP-LC3B puncta (autophagosomes) (), but induced aggregation of GFP-LC3B puncta (), thus further enhanced the size of the puncta (). Furthermore, the GFP-LC3B puncta in the rapamycin-treated NEDD4 knockdown cells were heterogenous in size and had completely different cellular localization from that in the control cells (the right bottom panel, ). These results indicate that knockdown of NEDD4 impairs process of autophagosomes and caused aberrant distribution and aggregation of the LC3-positive phagophores and/or autophagosomes.
Figure 6. Depletion of NEDD4 induced aggregation of the LC3B-positive puncta in cells. (A) lentiviral vector-loaded GFP-LC3B was stably expressed in A549 cells containing the vector control or NEDD4 shRNA. Knockdown of NEDD4 by shNEDD4 is shown in the right panel. NEDD4 (LM), low molecular weight NEDD4; NEDD4 (HM), high molecular weight NEDD4. The cells were treated with 1 μM rapamycin for 18 h to activate autophagy. The GFP-LC3B-positive autophagosomes were visualized live under an inverted Nikon fluorescent microscope. (B) Quantification of numbers and sizes of the GFP-LC3B fluorescent puncta in the fluorescent microscopic images from the experiment. A total of 1713 GFP-LC3B puncta in 40 vector control cells, 3735 GFP-LC3B puncta in 31 rapamycin-treated vector control cells, 1384 GFP-LC3B puncta in 30 shNEDD4 cells, and 1023 GFP-LC3B puncta in 26 rapamycin-treated shNEDD4 cells were counted and measured with ImageJ program for statistical analysis. The statistical analysis was performed based on the numbers and average sizes of the puncta in each of the cells. (C) Experimental procedures were the same as that in (A) except the GFP-LC3B-expressed cells were fixed and stained with anti-SQSTM1 and DAPI. Bar, 10 μm. *P<0.05.
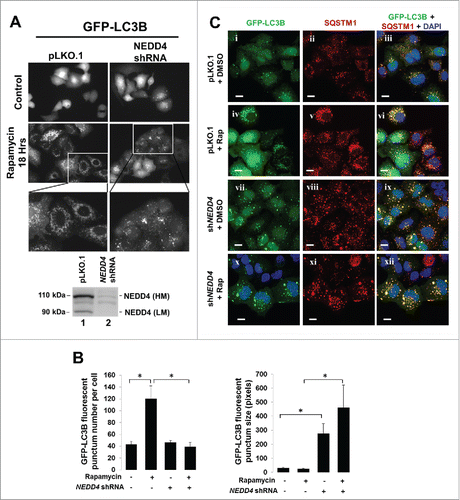
To further determine the effect of NEDD4 on autophagic function of LC3, we examined colocalization of GFP-LC3B with SQSTM1 in both the vector control and the NEDD4 knockdown cell lines by immunofluorescent staining. Consistent with the live observation, depicted in in , knockdown of NEDD4 significantly enhanced size of GFP-LC3B puncta, but did not interrupt colocalization of LC3B with SQSTM1 in both control and rapamycin-treated cells (), suggesting that NEDD4 is not required for interaction of LC3 with SQSTM1.
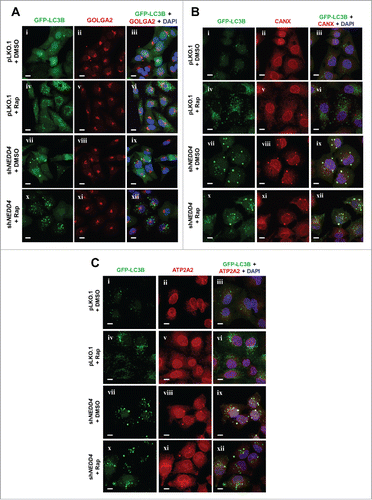
The NEDD4 knockdown-induced aggregated GFP-LC3B puncta were colocalized with endoplasmic reticulum (ER) membrane markers
We further examined the effect of NEDD4 on localization of GFP-LC3B on Golgi and ER, the 2 organelles proposed for biogenesis of autophagosomes,Citation47,48 in A549 cells, by immunofluorescent staining. GOLGA2/GM130 and CANX (calnexin), the 2 known Golgi and ER membrane markers, respectively,Citation49,50 were stained along with GFP-LC3B. As shown in , the same as observed in , a portion of GFP-LC3B puncta was aberrantly enlarged in the NEDD4 knockdown cell line, but not in the vector control cell line. Furthermore, the enlarged GFP-LC3B puncta in the NEDD4 knockdown cell line was colocalized with the ER membrane marker CANX even upon treatment with rapamycin (), but not with the Golgi marker GOLGA2 (), suggesting that GFP-LC3B puncta are retained in the CANX-positive membrane vesicles upon NEDD4 knockdown, which might be a biogenic site of autophagosomes.Citation48 To further confirm that GFP-LC3B puncta are retained in the ER membrane vesicles upon depletion of NEDD4, we examined colocalization of GFP-LC3B with a second ER membrane marker ATP2A2/SERCA2 in A549 cells.Citation51 As shown in , the enlarged GFP-LC3B puncta induced by knockdown of NEDD4 are colocalized with ATP2A2 in both control and rapamycin-treated samples, which is consistent with the results in . These results suggest that the NEDD4 depletion-caused defect in autophagy might occur in the early stage of autophagosomal biogenic process that is required for maturing autophagosomes.
Figure 7. The NEDD4 knockdown-induced aggregates of the GFP-LC3B puncta were colocalized with the endoplasmic reticulum (ER) membrane markers CANX and ATP2A2. (A-C), Lentiviral vector-loaded GFP-LC3B was stably transfected and expressed in cells containing either the vector control or NEDD4 shRNA and the cells were treated with DMSO (solvent control) or 1 μM rapamycin for 18 h. The cells were fixed and stained with anti-GOLGA2 (Golgi marker) (A), anti-CANX (ER membrane marker) (B), anti-ATP2A2 (ER membrane marker) (C) and DAPI. Bar, 10 μm.
LC3 is not a ubiquitination substrate of NEDD4
Because NEDD4 is an E3 ubiquitin ligase and interacts with LC3, we examined whether LC3 is a ubiquitination substrate of NEDD4 by the GST-UBA pulldown and immunoprecipitation assays. The GST-UBA affinity precipitation assay has been successfully used for isolation of ubiquitinated proteins in our previous studies.Citation37 As shown in and , no ubiquitinated LC3B was detected when LC3B was coexpressed with NEDD4 in HEK293 cells either by GST-UBA pulldown assay () or by immunoprecipitation assay (), despite significant amount of NEDD4 and LC3B proteins were expressed in the cells. These results suggest that LC3 may not be a ubiquitination substrate of NEDD4.
Figure 8. SQSTM1, but not LC3, is a ubiquitination substrate of NEDD4. (A) and (B), NEDD4 and Myc-tagged LC3B were coexpressed in HEK293 cells. Ubiquitinated proteins were precipitated by GST-UBA and immunoblotted with an anti-LC3 (in (A)), or Myc-tagged LC3B was immunoprecipitated by anti-MYC antibody and immunoblotted with an anti-ubiquitin antibody (B). (C) SQSTM1 was coexpressed with NEDD4 or its ligase-dead mutant NEDD4C867A (NEDD4-LD) and Myc-tagged LC3B in HEK293 cells. LC3B was immunoprecipitated with anti-MYC, and the coimmunoprecipitated SQSTM1 was detected with an anti-SQSTM1 antibody. HE, high exposure; LE, low exposure. The shifted SQSTM1 bands are framed by a white line. (D) SQSTM1 was coexpressed with HA-NEDD4 in HEK293 cells. Ubiquitinated proteins were precipitated by GST-UBA and immunoblotted with an anti-SQSTM1.
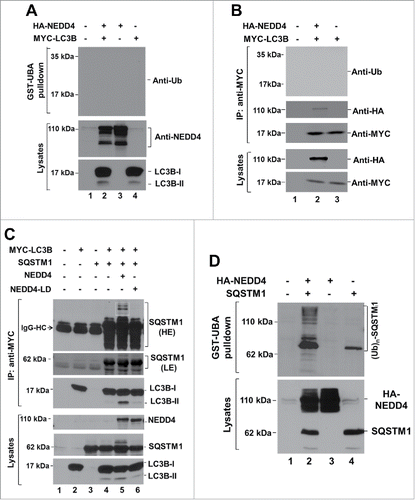
Surprisingly, when we examined the effect of NEDD4 on interaction of LC3 with SQSTM1 upon coexpression of NEDD4 or its ligase-dead mutant NEDD4C867A (labeled as NEDD4-LD in figures) with SQSTM1 and LC3B, we observed that SQSTM1 coexpressed with NEDD4, but not NEDD4-LD, had a gel-shift (circled by the frame in the top panel of ). This suggests that SQSTM1 might be a ubiquitination substrate of NEDD4. In addition, LC3B either immunoprecipitated or in the lysates had no any gel-shift band (), indicating that LC3B was not ubiquitinated by coexpression with NEDD4, which is consistent with the conclusion that LC3 is not a ubiquitination substrate of NEDD4 made from .
To confirm that SQSTM1 is ubiquitinated by NEDD4, we performed the GST-UBA affinity precipitation assay to detect the ubiquitination level of SQSTM1 upon coexpression with NEDD4 in HEK293 cells as we did for LC3 in . As shown in , unlike LC3, SQSTM1 had a robust polyubiquitination when coexpressed with NEDD4, indicating that SQSTM1 is a ubiquitination substrate of NEDD4 in cells.
LC3 functions as an activator of NEDD4
As suggested by that SQSTM1 is a ubiquitination substrate of NEDD4, we speculate that the interaction of LC3 with NEDD4 might be involved in regulation of the NEDD4-mediated ubiquitination of SQSTM1. To test this hypothesis, we employed 2 N-terminal truncation mutants NEDD4C2Δ and NEDDN274Δ (). These 2 truncation mutants, lacking the C2 domain-mediated autoinhibition,Citation42 thus are active ubiquitin ligases. We know that NEDD4N274Δ does not have the LIR2 and is defective in binding to LC3 in absence of SQSTM1 (). To confirm that NEDD4N274Δ is still an LC3-binding defective mutant when coexpressed with SQSTM1, we coexpressed NEDD4C2Δ and NEDD4N274Δ with SQSTM1 and examined the binding of these 2 NEDD4 mutants using a GST-LC3B affinity precipitation assay. As shown in , when coexpressed with SQSTM1, NEDD4C2Δ still binds to LC3B while NEDD4N274Δ does not, indicating that the binding is independent of SQSTM1. We further examined ubiquitination of SQSTM1 upon coexpression with wild-type NEDD4 and the 2 N-terminal truncation mutants. SQSTM1 was heavily ubiquitinated when coexpressed with NEDD4 or NEDD4C2Δ (). However, when coexpressed with NEDD4N274Δ, the ubiquitination of SQSTM1 was significantly reduced (). Quantification of the ubiquitination indicates that coexpression with the LC3-binding defective mutant NEDD4N274Δ had only about 30% of the ubiquination level in SQSTM1, compared with the level when coexpressed with wild-type NEDD4 or NEDD4C2Δ (). This raises a possibility that LC3 functions as an activator of the NEDD4 ligase activity for effectively ubiquitinating SQSTM1.
Figure 9. LC3 functions as an activator of NEDD4. (A) HA-tagged NEDD4C2Δ or LC3-binding defective truncation mutant NEDD4N274Δ was transfected or cotransfected with GFP-SQSTM1 into HEK293 cells. The HA-tagged NEDD4 mutants or/and GFP-SQSTM1 were precipitated with GST-LC3B conjugated beads and detected by immunoblotting with anti-HA and anti-SQSTM1 antibodies. (B and C), GFP-SQSTM1 was transfected or cotransfected with HA-tagged NEDD4, or the NEDD4 truncation mutants into HEK293 cells. GFP-SQSTM1 was immunoprecipitated with an anti-SQSTM1 antibody and the ubiquitinated SQSTM1 was detected by immunoblotting with an anti-ubiquitin antibody. In (C), quantification of the SQSTM1 ubiquitination by NEDD4 and its truncation mutants from 3 independent experiments. (D and E) activation of the NEDD4 E3 ubiquitin ligase activity by LC3 determined by an in vitro E3 ubiquitin ligase activity assay. NEDD4 or its LC3-binding defective mutant N274Δ was transfected in HEK293 cells, immunoprecipitated with an anti-NEDD4 antibody and used for the in vitro E3 ubiquitin ligase activity assay. The indicated amount of purified LC3B protein was directly added into the ubiquitin ligase assay mix before the reaction was initiated. The polyubiquitin product was detected by immunoblotting with an anti-ubiquitin antibody. In (E), amount of the polyubiquitin product was quantified by Multi Gauge V3.0 (Fujifilm) from 3 independent repeats of the in vitro E3 ubiquitin ligase assay. The ligase activity was calculated as the ratio of amount of polyubiquitin product to amount of NEDD4 or the truncation mutant protein. **P < 0.01; ***P < 0.001.
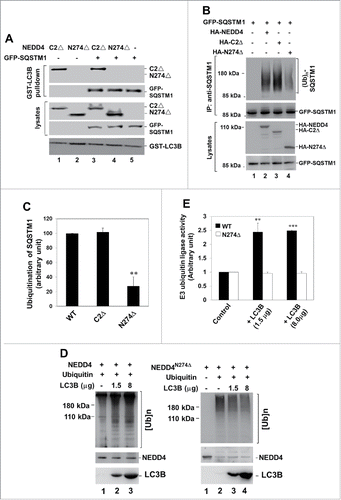
To test whether LC3 functions as an activator of NEDD4, we performed an in vitro ubiquitin ligase activity assay of NEDD4 in presence or absence of LC3, in which the amount of polyubiquitin formed from monoubiquitin conjugations is used for evaluating the E3 ligase activity of NEDD4 and the activation effect of LC3.Citation42 As shown in , when purified LC3B protein was added to the reaction mixture in the in vitro NEDD4 ligase activity assay, the polyubiquitin products were dramatically increased, indicating a significant activation of the ligase activity by LC3B. However, when LC3B was added in the NEDD4274Δ (the LC3-binding defective) ligase activity assay mixture, no activation of the ligase was observed (the right panel in ). Notice that NEDD4274Δ is an active E3 ubiquitin ligase (lane 2, right panel in ), just not activated by LC3. In addition, the NEDD4274Δ protein level in the negative control sample (lane 1, right panel in ) was higher than in the samples with polyubiquitination (lane 2 to 4, right panel in ), probably due to the auto-ubiquitination-caused gel shift that reduced the level of unubiquitinated NEDD4274Δ. Taken together, the results indicate that LC3 is an activator of NEDD4 and suggest that LC3 binding might be an activation process for NEDD4 in ubiquitination of SQSTM1.
Discussion
In this report, we showed that human NEDD4, an E3 ubiquitin ligase, is a regulatory protein of autophagy via a direct interaction with the autophagic protein LC3. Knockdown of NEDD4 resulted in severe defect in autophagy as evaluated by the effects on accumulating SQSTM1 protein levels, diminishing rapamycin- or starvation-induced changes in LC3-II and SQSTM1, impairing formation of autophagosomes, inducing deformation and aggregation of the LC3-positive phagophores and autophagosomes and interrupting mitophagy. In addition, the NEDD4-depeletion-induced aggregates of the LC3 puncta seem to be retained in the ER membrane marker-positive vesicles. Finally, we observed that SQSTM1, but not LC3, is ubiquitinated by NEDD4, and that LC3 activates the E3 ligase activity of NEDD4 in vitro. These results indicate that NEDD4 could be a pivotal E3 ubiquitin ligase regulating autophagy through interacting with LC3 and ubiquitination of SQSTM1.
We identified the LIR2 in NEDD4 as the LC3 binding site. Alignment of the LIR2 with the sequences of the NEDD4 E3 ubiquitin ligase family members found that the LC3-binding site LIR2 is conserved in NEDD4 and NEDD4L (also named as NEDD4–2) across mammalian species, but not in other members of the NEDD4 E3 ubiquitin ligases, such as WWP1/2, ITCH, SMURF1/2 and HECW1/2, suggesting that the role in regulation of autophagy might be conserved only in NEDD4 and NEDD4L E3 ubiquitin ligases. Our preliminary studies observed that NEDD4L, similar to NEDD4, binds to LC3 (data not shown), suggesting that NEDD4L might also play a role in regulating autophagy. Consistent with this, a recent research report found that ER stress upregulates NEDD4L that is required for activation of autophagy.Citation52 We will investigate how NEDD4L regulates autophagy in cancer cells and determine the difference between NEDD4 and NEDD4L in interacting with LC3, ubiquitinating SQSTM1 and regulating autophagy in future studies.
It has been shown that NEDD4 interacts with an autophagic protein BECN1, a PtdIns3K interactive protein that functions in biogenesis of phagophores, and ubiquitinates BECN1 in HeLa cells.Citation53 Ubiquitination of BECN1 by NEDD4 has been proposed as a function for proteasomal degradation of BECN1.Citation53 Because BECN1 is an important protein required for autophagy, this previous study suggests a negative role of NEDD4 in autophagy. However, our study has demonstrated a positive role of NEDD4 in autophagy. In addition, we observed that knockdown of NEDD4 caused a decrease in BECN1 protein level in A549 cells (), suggesting that NEDD4 may upregulate or impede degradation of BECN1 in A549 cells. This discrepancy in the effect of NEDD4 on BECN1 degradation between the studies might be due to differences in the context of autophagy between HeLa and A549 cell lines used in the experiments. Further studies should be pursued for clarifying the role of NEDD4 in ubiquitination and degradation of BECN1.
Many ubiquitination substrates of NEDD4 have been identified by an in vitro substrate screening assay.Citation38 Most of these substrates containing the PPXY motif that interacts with the WW domains of NEDD4 and are involved in receptor endocytic trafficking, vesicle secretion or viral budding processes. Regulation of assembly of the Endosomal Sorting Complex Required for Transport (ESCRT) in the membrane vesicle trafficking by ubiquitination may be a major functional role of NEDD4, through interaction with CBL, EPS15, TSG101, HGS/HRS, SCAMPs, and TNK2/ACK1.Citation32-37 The results in this report clearly show that NEDD4 interact with LC3 and plays an essential role in autophagy. It has been observed that the autophagic process shares the endosomal vesicle trafficking machinery.Citation54 However, the connection of the NEDD4-involved endosomal vesicle trafficking to autophagy has not been established. To further define the role of NEDD4-mediated ubiquitination in cellular catabolism, it is necessary to investigate the cross-talk between autophagy and endosomal trafficking regulated by NEDD4, LC3 and ESCRTs, elucidate the mechanism underlying the effect of NEDD4 on endosomal vesicle trafficking and autophagic process, and understand coordination between the endosome-dependent and the autophagosome-dependent lysosomal degradation processes.
How does NEDD4 regulate autophagy? Based on the data in this report, we hypothesize that NEDD4 forms a functional complex with LC3 to promote biogenesis of autophagosomes. Here we propose a model for the mechanism by which NEDD4 regulates autophagy: upon stimulation by an autophagic activation signal (nutritional stress or rapamycin), LC3-II interacts with NEDD4 and recruits NEDD4 to the phagophore assembly site (PAS), releases the autoinhibition of NEDD4 by disrupting the intramolecular interaction between the C2 and the HECT domain,Citation42 and activates the ligase activity of NEDD4. Activated NEDD4 subsequently ubiquitinates autophagic proteins such as SQSTM1and BECN1. The ubiquitinated autophagic proteins then recruit downstream effectors to assemble autophagic complexes for biogenesis of autophagosomes. Thus, when NEDD4 is defective or downregulated, the autophagic process will be halted at the phagophore stage and the autophagic protein LC3-II or SQSTM1 will be retained and accumulated in the PAS to form aggregates, presumably at the initiation site derived from ER membrane, as shown in . In addition, the electron microscopic study observed that knockdown of NEDD4 caused dramatic enlargement of mitochondria (), suggesting that NEDD4 may play a pivotal role in mitophagy. We speculate that ubiquitination of SQSTM1 by NEDD4 might be a key process in mitophagy. Previous studies have shown that SQSTM1 is a central player in selective autophagy by interacting with ubiquitinated cargos or binding proteins, such as KEAP1 and CHDH (choline dehydrogenase), involved in mitophagy.Citation20,21,24,55,56 NEDD4-mediated ubiquitination may modulate molecular structure of SQSTM1 and lead to an increase in binding affinity to the ubiquitinated cargos or the binding proteins, thus to activate mitophagic process. Currently, we know little about NEDD4-mediated ubiquitination of SQSTM1 and the ubiquitination for function of SQSTM1 in autophhagy. In future studies, we will further characterize the NEDD4-mediated ubiquitination of SQSTM1 and determine how the ubiquitination regulates autophagy through effecting the molecular structure and function of SQSTM1 and communication of SQSTM1 with the downstream binding proteins.
Materials and methods
Materials
Antibodies: anti-SQSTM1 (D3; SC-28359) and anti- PDCD6IP/ALIX (SC-53540) were purchased from Santa Cruz Biotechnology; anti-NEDD4 from Millipore (07–049); anti-LC3 from Abgent (AP1802a); anti-BECN1 from Epitomics (2026–1); anti-CANX and anti-GOLGA2/GM130 from ECM Biosciences (OK7670); anti-ATP2A2/SERCA2 from Novus Biologicals (NB300–529); anti-GFP (MMS-118R), anti-MYC (MMS-150R) and anti-HA (MMS-101R) from Covance; Anti-ACTB from Sigma-Aldrich (A5441). Purified MAP1LC3B/LC3B protein was purchased from MyBioSource (MBS203837). The DNA mutagenesis kit (QuikChange® Site-Directed Mutagenesis Kit) was purchased from Strategene (200518). The Cyto-ID autophagy detection kit (ENZ-51031-K200) was from Enzo Life Sciences. The NEDD4 shRNA (5′-AUUUGAACCGUAUAGUUCAGC-3′) in the lentiviral expression vector pLKO.1 was purchased from Open Biosystems (RHS4533-EG4734). The Human NEDD4 cDNA used in experiments is the transcript variant 1 (Accession# NM_006145). The lung cancer cell line A549, the hepatocellular carcinoma cell line HepG2 and the neuroblastoma cell line BE(2)-C cell line were purchased from ATCC. The amino acid-depleted RPMI-1640 culture medium (for starvation treatment; R9010–01) was purchased from US Biological Life Science.
Cell culture and transfection
HEK293T, HEK293A, A549, HepG2 and BE(2)-C cells were maintained in Dulbecco's modified Eagle's medium (Gibco, 11965092) with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin and streptomycin at 37°C with 5% CO2. For transfection, HEK293 or HEK293A cells were seeded one d before the transfection. The transfection procedures were the same as described previously.Citation57
Construction of plasmids and mutagenesis
The human SQSTM1 or MAP1LC3B cDNA was subcloned into the mammalian expression vectors pcDNA3-HA, pcDNA3-MYC, lentiviral GFP vector pLVTHM-GFP (a gift from Dr. Jihe Zhao at University of Central Florida) or GST fusion vector pGEX4T3 (GE Health Care Life Sciences, 28–9545–52). Human NEDD4 or the mutant cDNA was subcloned into the lentiviral expression vector pFUW (a gift from Dr. Jihe Zhao at University of Central Florida) for establishing stable cell lines or re-expressing in the NEDD4-depleted A549 cells, and into the mammalian expression vector pcDNA3-HA for transient transfection in HEK293 cells. Point mutations and truncations of NEDD4 were created using a mutagenesis kit from Strategene. For re-expression of NEDD4 in the shNEDD4 cells, 3 synonymous point mutations in the region of NEDD4 cDNA corresponding to the sequence of NEDD4 shRNA (AUUUGAACCGUAUAGUUCAGC) were created for overcoming the knockdown effect of shNEDD4.
Preparation of cell lysates, immunoprecipitation, immunoblot and GST-fusion protein affinity precipitation assay
Cells were rinsed once with ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and lysed in ice-cold mammalian lysis buffer (40 mM HEPES [Sigma-Aldrich, H3374], pH 7.4,100 mM NaCl, 1% Triton X-100 [Sigma-Aldrich, T8787], 25 mM glycerol phosphate, 1 mM sodiumorthovanadate, 1 mM EDTA, 10 µg/ml aprotinin [Sigma-Aldrich, A1153], and 10 µg/ml leupeptin [Sigma-Aldrich, L9783]) or RIPA buffer (40 mM HEPES, pH 7.4, 1% Triton X-100, 0.5% Na-deoxylcholate [Sigma-Aldrich, D6750], 0.1% SDS [Sigma-Aldrich, L3771], 100 mM NaCl, 1 mM EDTA, 25 mM μ-glycerolphosphate, 1 mM Na-orthovanadate, 10 μg/ml leupeptin and aprotinin) as indicated. The cell lysates were cleared by centrifugation at 15,000 xg for 15 min. In immunoprecipitation, primary antibodies were added to the lysates and incubated with rotation at 4°C for 30 min, followed by adding 20 μl of the protein A-Sepharose (Sigma-Aldrich, P3391) bead slurry (1:1) into the lysates and incubating with rotation for an additional 3 h. The immunoprecipitates were washed 3 times with lysis buffer. The cell lysates or immunoprecipitated proteins were denatured by addition of SDS-PAGE sample buffer and boiled for 5 min, resolved by 8%–14% SDS-PAGE. The proteins in the gel were transferred to PVDF membranes (EMD Millipore, IPFL00010). The immunoblot was performed as described previously, using chemo-luminescence.Citation57
GST fusion protein expression, purification and affinity precipitation assay were performed as previously described.Citation37,57
Immunofluorescent staining
The cells were cultured in glass-bottom culture dishes with coverslips (MatTek, P35GCOL-0–10-C) to 50 to 80% confluence. After the culture medium was aspirated, the cells were rinsed with PBS twice, fixed with 3.7% paraformaldehyde at 25°C for 10 min, and permeabilized with 0.2% Triton X-100 in PBS at 25°C for 10 min. After washing with PBS, the cells were incubated with primary antibody at 8°C overnight. The cells were washed with PBS 3 times and incubated with secondary antibody conjugated with a fluorescent dye (Molecular Probes, T-6390 or T-6391) at 37°C for 1 to 2 h. After washing with PBS 3 times, fluorescent staining of the cells was visualized under a Zeiss LSM710 confocal microscope (Peabody, MA 01960) or a Nikon Eclipse TE2000 inverted fluorescent microscope (Melville, NY 11747).
Quantification of fluorescent punctate numbers and sizes in the fluorescent microscopic images
The analysis and quantification of fluorescent images were performed using the ImageJ program. The threshold in detection of the fluorescence was set to cover all the visible fluorescent puncta. Numbers of fluorescent puncta were counted from 2 randomly selected fluorescence microscopy fields (25 to 47 cells). The size of the fluorescent puncta in each cell was measured and averaged. The statistical analysis was performed based on the numbers and average sizes of puncta from each of the cells.
Virus packaging and transduction
The viral packaging was performed as described previously.Citation58 Briefly, the lentiviral plasmids were cotransfected with psPAX2 (Addgene, 12260, deposited by Didier Trono) and pMD2.G (Addgene, 12259, deposited by Didier Trono) packaging plasmids into actively growing HEK293T cells using Lipofectamine 2000 transfection reagent. Viral particle-containing culture medium was collected every 24 h, 3 times. The medium was cleared by centrifugation at 1000 xg for 5 min, and used for infecting target cells in the presence of 6 μg/ml polybrene (Sigma-Aldrich, H9268). The infected cells were selected with puromycin.
Analysis of autophagy
The autophagy was activated by treatment of cells with the amino acid-depleted RPMI-1640 (starvation) or the MTOR inhibitor rapamycin (LC Laboratory, R5000) for the indicated time. The LC3- or SQSTM1-positive autophagosomes were visualized by either immunofluorescent staining or GFP-tag under a Zeiss LSM710 confocal fluorescence microscope or a Nikon Eclipse TE2000 inverted fluorescence microscope. To determine live autophagic processes, the Cyto-ID autophagy detection kit was used to label autophagosomes in living cells as instructed by the manufacturer. The labeled autophagosomes were visualized under a Nikon Eclipse TE2000 inverted fluorescent microscope and the Cyto-ID positivity was quantified using a Beckman Coulter Cytomics FC 500 cytometer (Danvers, MA 01923)by measuring the fluorescent density from 104 cells in each sample.
Electron microscopy
A549 cells were trypsinized, resuspended in PBS and spun down (100xg, 5 min) in 1.5-ml centrifuge tubes. The PBS was removed and the cell pellet was fixed in 3% glutaraldehyde overnight at 4°C. Following fixation in the tube the pellets were gently removed and placed in a specimen jar with 3% glutaraldehyde and fixed for an additional 24 h. The cell pellet was then processed for standard electron microscopy as follows: 3×10-min washes in 1 M sodium cacodylate buffer; fixation in 1% osmium tetroxide for 1 h; a 10 min rinse in 1 M sodium cacodylate buffer; fixation in 6% uranyl magnesium acetate for 1 h and then dehydrated through a series of ethanol washes beginning at 50% and ending with 2 washes in 100%. Following dehydration, the cells were washed in propylene oxide for 10 min and then fixed in a 1:1 mixture of propylene oxide and Epon plastic (Electron Microscopy Sciences, EMS14120) for 90 min. This solution was removed and replaced with a 3:1 mixture of Epon to propylene oxide for 90 min. The cells were then embedded in a mold of 100% plastic, placed in a 70 degree oven and allowed to harden overnight. The block was trimmed down using a razor blade and thick sections were obtained using a Sorvall RMC MT600-XL ultramicrotome (Liverpool, NY13090). These sections were stained with toulidine blue for direct observation. Thin sections were then obtained using a diamond knife and these sections were placed on hex copper grids (Electron Microscopy Sciences, H200-Cu). Following a drying period of 2 h these grids were stained with Reynolds lead citrate for 10 min, washed, and then allowed to dry. Transmission Electron Microscopy was performed on these grids, using a JEOL 1200 EX Electron microscope (Peabody, MA 01960). Reagents for preparation and staining were obtained from Electron Microscopy Sciences.
Detection of ubiquitinated proteins and the in vitro E3 ubiquitin ligase activity assay
Detection of ubiquitinated proteins was performed using both GST-UBA affinity precipitation and immunoprecipitation assays as described previously.Citation37 Briefly, cells were lysed with RIPA buffer (40 mM HEPES, pH 7.4, 1% Triton X-100, 0.5% Na-deoxylcholate, 0.1% SDS, 100 mM NaCl, 1 mM EDTA, 25 mM β-glycerolphosphate, 1 mM Na-orthovanadate, 10 μg/ml leupeptin and aprotinin), and the ubiquitinated proteins were detected either by immunoprecipitation with the primary antibody followed by immunoblotting with an anti-ubiquitin antibody (BioLegend, 646302), or by affinity precipitation with GST-UBA-conjugated glutathione beads followed by immunoblotting with an anti-SQSTM1 antibody.
The in vitro E3 ubiquitin ligase assay of NEDD4 was performed as previously described.Citation42 Briefly, NEDD4 was overexpressed in HEK293T cell and isolated by immunoprecipitation. The immunoprecipitated NEDD4 was then incubated with or without purified LC3B on ice for 30 min. The ubiquitin ligation reaction was initiated by addition of monoubiquitin to the reaction mixture at 25°C. The reaction was performed at 25°C for 30 min and terminated by addition of SDS-PAGE sample buffer. The polyubiquitin was separated by SDS-PAGE and detected by immunoblotting with an anti-ubiquitin antibody. The polyubiquitin product conjugated from monoubiquitin was used for evaluating the ligase activity of NEDD4.
Statistical analysis
The Student t test was used in statistical analysis of experimental data. A P value less than 0.05 was considered as statistically significant.
Abbreviations
| ACTB | = | actin β |
| ATP2A2/SERCA2 | = | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 |
| E3 | = | ubiquitin protein ligase |
| GFP | = | green fluorescent protein |
| GOLGA2/GM130 | = | golgin A2 |
| HECT | = | homologous to the E6-AP carboxyl terminus |
| HGS/HRS | = | hepatocyte growth factor-regulated tyrosine kinase substrate |
| LC3B | = | MAP1LC3B |
| LIR | = | LC3-interacting region |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| NEDD4 | = | neural precursor cell expressed; developmentally downregulated 4 |
| PDCD6IP/ALIX | = | programmed cell death 6 interacting protein |
| SQSTM1 | = | sequestosome 1 |
| TNK2/ACK1 | = | tyrosine kinase nonreceptor 2. |
Disclosure of potential conflicts of interests
No potential conflicts of interest were disclosed.
Acknowledgments
We want to thank Dr. Jihe Zhao at University of Central Florida for sending the lentiviral expression vectors and the packaging plasmids.
Funding
This work is supported by National Natural Science Foundation of China (NSFC) (No. 81372208 to Q. L. and No. 81472558 to W.Y.).
References
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-1721; PMID:11099404; http://dx.doi.org/10.1126/science.290.5497.1717
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 2(12 Suppl):1542-1552; http://dx.doi.org/10.1038/sj.cdd.4401765
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4(2):151-75; PMID:18188003; http://dx.doi.org/10.4161/auto.5338
- Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr 1987; 7:539-64; PMID:3300746; http://dx.doi.org/10.1146/annurev.nu.07.070187.002543
- Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. Autophagy-physiology and pathophysiology. Histochem Cell Biol 2008; 129(4):407-20; PMID:18320203; http://dx.doi.org/10.1007/s00418-008-0406-y
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-995; PMID:15528435; http://dx.doi.org/10.1126/science.1099993
- Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both?. Cancer Res 2006; 66:9349-9351; PMID:17018585; http://dx.doi.org/10.1158/0008-5472.CAN-06-1597
- Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell 2013; 155(6):1216-9; PMID:24315093; http://dx.doi.org/10.1016/j.cell.2013.11.019
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 2004; 16(6):663-9; PMID:15530778; http://dx.doi.org/10.1016/j.ceb.2004.09.011
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19(21):5720-8; PMID:11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Lang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J 1998; 17(13):3597-607; PMID:9649430; http://dx.doi.org/10.1093/emboj/17.13.3597
- Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 2011; 12(7):226; PMID:21867568; http://dx.doi.org/10.1186/gb-2011-12-7-226
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36(12):2503-18; PMID:15325588; http://dx.doi.org/10.1016/j.biocel.2004.05.009
- Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem 2013; 55:39-50; PMID:24070470; http://dx.doi.org/10.1042/bse0550039
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15(3):1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8(4):445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013; 126(Pt 15):3237-47.
- Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci 2014; 127(Pt 1):3-9; PMID:24345374; http://dx.doi.org/10.1242/jcs.140426
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12(3):213-23; PMID:20173742
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30(13):3275-85; PMID:20421418; http://dx.doi.org/10.1128/MCB.00248-10
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 2013; 51(3):283-96; PMID:23911927; http://dx.doi.org/10.1016/j.molcel.2013.06.020
- Isakson P, Lystad AH, Breen K, Koster G, Stenmark H, Simonsen A. TRAF6 mediates ubiquitination of KIF23/MKLP1 and is required for midbody ring degradation by selective autophagy. Autophagy 2013; 9(12):1955-64; PMID:24128730; http://dx.doi.org/10.4161/auto.26085
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34(3):259-69; PMID:19450525; http://dx.doi.org/10.1016/j.molcel.2009.04.026
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7(3):279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J 1996; 15(10):2371-80; PMID:8665844
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 1997; 16(21):6325-36; PMID:9351815; http://dx.doi.org/10.1093/emboj/16.21.6325
- Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 2007; 8(9):1246-64; PMID:17605762; http://dx.doi.org/10.1111/j.1600-0854.2007.00602.x
- Katzmann DJ, Sarkar S, Chu T, Audhya A, Emr SD. Multivesicular body sorting: ubiquitin ligase Rsp5 is required for the modification and sorting of carboxypeptidase S. Mol Biol Cell 2004; 15(2):468-80; PMID:14657247; http://dx.doi.org/10.1091/mbc.E03-07-0473
- Blot V, Perugi F, Gay B, Prévost MC, Briant L, Tangy F, Abriel H, Staub O, Dokhélar MC, Pique C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J Cell Sci 2004; 117(Pt 11):2357-67; PMID:15126635; http://dx.doi.org/10.1242/jcs.01095
- Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci U S A 2008; 105(10):3974-9; PMID:18305167; http://dx.doi.org/10.1073/pnas.0710629105
- Magnifico A, Ettenberg S, Yang C, Mariano J, Tiwari S, Fang S, Lipkowitz S, Weissman AM. WW domain HECT E3s target Cbl RING finger E3s for proteasomal degradation. J Biol Chem 2003; 278:43169-77; PMID:12907674; http://dx.doi.org/10.1074/jbc.M308009200
- Katz M, Shtiegman K, Tal-Or P, Yakir L, Mosesson Y, Harari D, Machluf Y, Asao H, Jovin T, Sugamura K, Yarden Y. Ligand-independent degradation of epidermal growth factor receptor involves receptor ubiquitylation and Hgs, an adaptor whose ubiquitin-interacting motif targets ubiquitylation by Nedd4. Traffic 2002; 3(10):740-51; PMID:12230472; http://dx.doi.org/10.1034/j.1600-0854.2002.31006.x
- Woelk T, Oldrini B, Maspero E, Confalonieri S, Cavallaro E, Di Fiore PP, Polo S. Molecular mechanisms of coupled monoubiquitination. Nat Cell Biol 2006; 8:1246-54; PMID:17013377; http://dx.doi.org/10.1038/ncb1484
- Segura-Morales C, Pescia C, Chatellard-Causse C, Sadoul R, Bertrand E, Basyuk E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J Biol Chem 2005; 280(29):27004-12; PMID:15908698; http://dx.doi.org/10.1074/jbc.M413735200
- Aoh QL, Castle AM, Hubbard CH, Katsumata O, Castle JD. SCAMP3 negatively regulates epidermal growth factor receptor degradation and promotes receptor recycling. Mol Biol Cell 20(6):1816-32; PMID:19158374; http://dx.doi.org/10.1091/mbc.E08-09-0894
- Lin Q, Wang J, Childress C, Sudol M, Carey DJ, Yang W. HECT E3 ubiquitin ligase NEDD4-1 ubiquitinates ACK and regulates EGF-induced degradation of EGFR and ACK. Mol Cell Biol 2010; 30:1541-1554; PMID:20086093; http://dx.doi.org/10.1128/MCB.00013-10
- Persaud A, Alberts P, Amsen EM, Xiong X, Wasmuth J, Saadon Z, Fladd C, Parkinson J, Rotin D. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol Syst Biol 2009; 5:333; PMID:19953087; http://dx.doi.org/10.1038/msb.2009.85
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007; 128:129-39; PMID:17218260; http://dx.doi.org/10.1016/j.cell.2006.11.039
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466(7302):68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Li Y, Zhang L, Zhou J, Luo S, Huang R, Zhao C, Diao A. Nedd4 E3 ubiquitin ligase promotes cell proliferation and autophagy. Cell Prolif 2015; 48(3):338-47; PMID:25809873; http://dx.doi.org/10.1111/cpr.12184
- Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem 2010; 285(16):12279-12288; PMID:20172859; http://dx.doi.org/10.1074/jbc.M109.086405
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447(7148):1121-5; PMID:17589504
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10(7):776-87; PMID:18552835; http://dx.doi.org/10.1038/ncb1740
- Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Fauré J, Blanc NS, Matile S, Dubochet J, Sadoul R, Parton RG, Vilbois F, Gruenberg J. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 2004; 303(5657):531-4; PMID:14739459; http://dx.doi.org/10.1126/science.1092425
- Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P, David G. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol 2012; 14(7):677-85; PMID:22660413; http://dx.doi.org/10.1038/ncb2502
- Geng J, Klionsky DJ. The Golgi as a potential membrane source for autophagy. Autophagy 2010; 6(7):950-1; PMID:20729630; http://dx.doi.org/10.4161/auto.6.7.13009
- Hamasaki M, Yoshimori T. Where do they come from? Insights into autophagosome formation. FEBS Lett 2010; 584(7):1296-301; PMID:20188731; http://dx.doi.org/10.1016/j.febslet.2010.02.061
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 1995; 131(6 Pt 2):1715-26; PMID:8557739; http://dx.doi.org/10.1083/jcb.131.6.1715
- Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ 2nd, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem 1991; 266(29):19599-610; PMID:1918067
- Padar S, van Breemen C, Thomas DW, Uchizono JA, Livesey JC, Rahimian R. Differential regulation of calcium homeostasis in adenocarcinoma cell line A549 and its Taxol-resistant subclone. Br J Pharmacol 2004; 142(2):305-16; PMID:15066902; http://dx.doi.org/10.1038/sj.bjp.0705755
- Wang H, Sun RQ, Camera D, Zeng XY, Jo E, Chan SM, Herbert TP, Molero JC, Ye JM. Endoplasmic reticulum stress up-regulates Nedd4-2 to induce autophagy. FASEB J 2016; 30(7):2549-56; PMID:27022162; http://dx.doi.org/10.1096/fj.201500119
- Platta HW, Abrahamsen H, Thoresen SB, Stenmark H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem J 2012; 441(1):399-406; PMID:21936852; http://dx.doi.org/10.1042/BJ20111424
- Metcalf D, Isaacs AM. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem Soc Trans 2010; 38(6):1469-73; PMID:21118109; http://dx.doi.org/10.1042/BST0381469
- Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8(13):1986-90; PMID:19502794; http://dx.doi.org/10.4161/cc.8.13.8892
- Park S, Choi SG, Yoo SM, Son JH, Jung YK. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014; 10(11):1906-20; PMID:25483962; http://dx.doi.org/10.4161/auto.32177
- Shen F, Lin Q, Gu Y, Childress C, Yang W. Activated Cdc42-associated Kinase 1 (ACK1) Is a Component of EGF Receptor Signaling Complex and Regulates EGF Receptor Degradation. Mol Biol Cell 2007; 18(3):732-42; PMID:17182860; http://dx.doi.org/10.1091/mbc.E06-02-0142
- Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH, Shabahang M, Yang W. Geranylgeranylation signals to the Hippo pathway for breast cancer cell proliferation and migration. Oncogene 2015; 34(24):3095-106; PMID:25109332; http://dx.doi.org/10.1038/onc.2014.251