
ABSTRACT
ATG3 (autophagy-related 3) is an E2-like enzyme essential for autophagy; however, it is unknown whether it has an autophagy-independent function. Here, we report that ATG3 is a relatively stable protein in unstressed cells, but it is degraded in response to DNA-damaging agents such as etoposide or cisplatin. With mass spectrometry and a mutagenesis assay, phosphorylation of tyrosine 203 of ATG3 was identified to be a critical modification for its degradation, which was further confirmed by manipulating ATG3Y203E (phosphorylation mimic) or ATG3Y203F (phosphorylation-incompetent) in Atg3 knockout MEFs. In addition, by using a generated phospho-specific antibody we showed that phosphorylation of Y203 significantly increased upon etoposide treatment. With a specific inhibitor or siRNA, PTK2 (protein tyrosine kinase 2) was confirmed to catalyze the phosphorylation of ATG3 at Y203. Furthermore, a newly identified function of ATG3 was recognized to be associated with the promotion of DNA damage-induced mitotic catastrophe, in which ATG3 interferes with the function of BAG3, a crucial protein in the mitotic process, by binding. Finally, PTK2 inhibition-induced sustained levels of ATG3 were able to sensitize cancer cells to DNA-damaging agents. Our findings strengthen the notion that targeting PTK2 in combination with DNA-damaging agents is a novel strategy for cancer therapy.
Introduction
Macroautophagy/autophagy is a tightly regulated pathway that can be stimulated by multiple forms of cellular stress.Citation1-5 During the autophagic process, cells form double-membraned compartments, termed phagophores, which sequester damaged organelles, proteins, or portions of the cytoplasm; maturation into an autophagosome is followed by subsequent delivery of the cargo to the lysosome.,Citation6,7 The molecular mechanisms of autophagosome formation are evolutionarily conserved and depend upon several autophagy-related (ATG) proteins.Citation8 For example, BECN1 (Beclin 1) can govern the autophagic process by regulating PIK3C3-dependent generation of phosphatidylinositol-3-phosphate and the subsequent recruitment of additional ATG proteins that orchestrate autophagosome formation.Citation9
Recently, a growing number of studies have shown that ATG proteins also play a role in autophagy-independent functions.Citation10-13 The ATG12–ATG5-ATG16L1 protein complex, whose well-known function is participating in autophagosome formation, has an autophagy-independent role in the antiviral activity triggered by IFNG/IFN-γ.Citation13 Another essential protein in autophagy induction, ATG7, is also reported to induce cell cycle arrest in response to metabolic stress.Citation11 In addition, BECN1 participates in the association of kinetochore proteins and mitosis independent of its function in autophagy.Citation12 Therefore, searching for autophagy-independent functions of ATG proteins seems to be a promising field of research.
As an E2-like enzyme essential for autophagy, ATG3 can catalyze the conjugation of MAP1LC3/ATG8 and phosphatidylethanolamine (PE).Citation14,15 In addition to regulating MAP1LC3–PE/MAP1LC3-II conjugation, ATG3 also forms a complex with ATG12 to regulate mitochondrial homeostasis and cell death mediated by mitochondrial pathways.Citation16-18 Because ATG3 is involved in many biological processes, mice deficient in the Atg3 gene die within the first d after birth with reduced amino acid levels.Citation19 However, it is unknown whether ATG3 is also involved in an autophagy-independent biologic function.
PTK2/FAK (protein tyrosine kinase 2) is a cytoplasmic protein tyrosine kinase that is overexpressed and activated in several advanced-stage solid cancers.Citation20 It can promote glucose consumption, lipogenesis, and glutamine dependency to promote cancer cell proliferation, motility, and survival.Citation21 Targeting PTK2 in endothelial cells is sufficient to induce tumor cell sensitization to DNA-damaging therapies by downregulating the NFKB/NF-κB pathway.Citation22 Small molecule PTK2 inhibitors (PTK2-Is) prevent tumor progression in mice and are being evaluated in clinical trials.Citation23-27 However, the greatest efficacy of PTK2-Is has been observed in combination with other tyrosine kinase inhibitorsCitation28,29 or cytotoxic drugs,Citation30,31 but the real mechanism has yet to be fully revealed.
In this study, we found that in response to cancer chemotherapeutic agent treatment, PTK2 induced ATG3 phosphorylation, which led to its significant degradation but was not associated with the induction of autophagy. In addition, PTK2 inhibition caused a sustained level of ATG3, leading to a significant decrease in cell viability. These results implicate ATG3 phosphorylation in the maintenance of cell viability in response to DNA damage and also support the notion that targeting PTK2 in combination with chemotherapy is a novel cancer therapeutic strategy.
Results
ATG3 is degraded during DNA damage treatment
ATG proteins have been reported to exert autophagy-independent functions. For example, ATG5 expression is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy.Citation32 Therefore, we examined several ATG protein levels in response to treatment with DNA-damaging drugs. Human colon cancer cell lines HCT116 and LoVo were treated with etoposide for 3 h or cisplatin for 6 h, washed, and incubated with fresh medium. As shown in , among the ATG proteins tested, only ATG3 protein levels were gradually decreased after etoposide or cisplatin treatment. To verify whether this phenomenon was cell type-dependent, the levels of ATG proteins were also measured in the cervical cancer cell line HeLa and osteosarcoma cell line U2OS after etoposide treatment. Consistent with the previous results, ATG3 was decreased at the protein level (Figure S1A-B). Furthermore, we used irradiation (IR) or camptothecin to treat HCT116 cells and found that this is a general phenomenon that occurs in response to DNA damage inducers (Figure S1C-D).
Figure 1. ATG3 is degraded in response to treatment with DNA-damaging drugs. (A) HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for the indicated time. Western blotting was performed to detect different ATG proteins. (B) HCT116 cells were treated with etoposide at various concentrations for 3 h and then incubated with fresh medium for 48 h. (C, D) Cisplatin (10 μM) (C) or etoposide (40 μM) (D) were introduced into HCT116 or LoVo cells, respectively. Cells were then treated as described in (A). (E, F) HCT116 cells were treated with etoposide (E) or cisplatin (F) as indicated, and then quantitative PCR (qPCR) was used to measure the mRNA levels of ATG3. The data are presented as the mean ± SD (n = 3). NS, no significance. (G) HCT116 cells were treated with etoposide for 3 h and then incubated with fresh medium for 48 h in the presence or absence of CHQ (10 μM) or MG132 (1 μM). Western blotting was performed to detect endogenous ATG3 protein levels. (H) HCT116 cells were transfected with HA-ubiquitin. Cells were treated 24 h after transfection with or without etoposide for 3 h and then incubated in fresh medium for 48 h. MG132 (1 μM) was added 12 h before collection. Cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-ATG3 or anti-HA antibody.
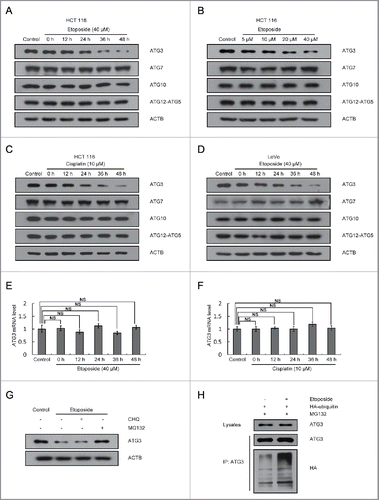
Because etoposide or cisplatin treatment had no effect on ATG3 mRNA levels (), protein degradation might be responsible for the decrease in ATG3 protein levels in response to DNA-damaging drug treatment. To determine the pathways involved in the ATG3 degradation, we pretreated HCT116 cells with a panel of inhibitors that included the proteasome inhibitor MG132 and the lysosome inhibitor chloroquine (CHQ). Treatment with MG132 significantly blocked the etoposide-induced reduction of ATG3 levels, but CHQ had no such effect on ATG3 degradation (), suggesting that ATG3 is degraded by the proteasomal pathway in response to etoposide treatment.
Next, to determine whether ubiquitination is required for ATG3 degradation, an HA-ubiquitin plasmid was transfected into HCT116 cells; ATG3 ubiquitination was detected by co-IP with an anti-ATG3 antibody followed by immunoblotting with an anti-HA antibody. As shown in , ATG3-conjugated ubiquitin after etoposide treatment was increased in comparison with that in untreated cells. These results demonstrate that the degradation of ATG3 occurs mainly through a ubiquitin-proteasome-dependent pathway.
PTK2 regulates ATG3 degradation
To test whether the post-translational modifications of ATG3 play a role in its degradation, a protein extract of HCT116 cells was immunoprecipitated with an anti-ATG3 antibody and probed with an anti-phosphorylated serine, an anti-phosphorylated threonine, an anti-phosphorylated tyrosine or an anti-acetylated lysine antibody. Acetylation of ATG3 at lysine and phosphorylation of ATG3 at serine or threonine did not show a significant change in HCT116 cells in response to etoposide treatment (Figure S2A-B). By contrast, a significant increase in phosphorylated ATG3 at a tyrosine was detected in etoposide-treated HCT116 cells (), suggesting that ATG3 tyrosine phosphorylation may be involved in regulating its stability.
Figure 2. Involvement of PTK2 kinase in ATG3 degradation. (A) MEF cells were treated with or without etoposide (20 μM) for 6 h, and then incubated with fresh medium. The cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-phospho-tyrosine antibody. (B) HCT116 cells were pre-incubated with PF-573228 (PTK2 inhibitor), SRC Inhibitor-1 (SRC inhibitor) or erlotinib (EGFR inhibitor) for 1 h and then treated with or without etoposide (40 μM). Three h later, the medium was changed, and cells were incubated in fresh medium in the presence or absence of kinase inhibitors for the indicated time. Western blotting was performed using anti-ATG3, anti-p-PTK2, anti-p-SRC or anti-p-EGFR antibodies. (C) HCT116 cells were transfected with a PTK2-specific siRNA or negative control. 48 h after transfection, the cells were treated with or without etoposide for 3 h and then incubated with fresh medium for 48 h. Endogenous ATG3 protein levels were measured by western blotting, and the efficiency of PTK2 siRNA was validated by monitoring total PTK2 and p-PTK2. The bands were quantified with ImageJ software. The numbers below the blot indicate the relative levels of p-PTK2 in each group.
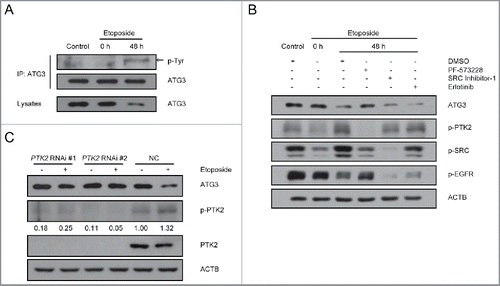
Next, we used several well-characterized tyrosine kinase inhibitors, SRC Inhibitor-1, an inhibitor of SRC; Erlotinib, an inhibitor of EGFR; and PF-573228, an inhibitor of PTK2, to investigate which cellular signaling pathway is involved in ATG3 degradation. HCT116 cells were pre-incubated with SRC Inhibitor-1, erlotinib or PF-573228 for 1 h followed by treatment with etoposide for another 3 h. Forty-eight h after etoposide treatment, blockage of PTK2 by PF-573228 completely inhibited etoposide-induced ATG3 degradation, whereas the others did not (). To further confirm the role of PTK2 in ATG3 degradation, HCT116 cells were transfected with either nonspecific or PTK2-specific siRNA, and ATG3 degradation was then detected. The efficiency of the RNAi against PTK2 was significant, and, as expected, upon etoposide treatment ATG3 was degraded in the cells transfected with the negative control siRNA. In contrast, ATG3 remained at higher levels in the cells transfected with the PTK2 siRNA (), suggesting that PTK2 plays a critical role in ATG3 degradation in response to DNA damage.
Y203 phosphorylation of ATG3 is required for its degradation
To test which phosphorylation site is critical for ATG3 degradation, we purified the ATG3 protein and performed an analysis with mass spectrometry (MS). As shown in Figure S3, a mass spectrometry analysis indicated that a highly conserved site, Y203, of ATG3 was phosphorylated after etoposide treatment. To further confirm this site, we generated an antibody for the Y203-phosphorylated peptide of ATG3 (). Using this antibody, we found that Y203-phosphorylated ATG3 was increased after etoposide treatment (). Moreover, etoposide-induced ATG3 phosphorylation at Y203 was downregulated by the PTK2 inhibitor PF-573228 ().
Figure 3. ATG3 degradation is dependent on its Y203 phosphorylation. (A) HCT116 cells were transfected with plasmids encoding FLAG-ATG3 WT or ATG3Y203F. The cell lysates were extracted 24 h after transfection for co-IP with an anti-FLAG antibody and probed with an anti-Y203-phosphorylated ATG3 antibody. (B) MEF cells were treated with or without etoposide (20 μM) for 6 h and then incubated with fresh medium. The cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-Y203-phosphorylated ATG3 antibody. The numbers below the blot indicate the relative levels of p-ATG3 in each group. (C) MEF cells were pre-incubated with PF-573228 for 1 h and then treated with or without etoposide (20 μM) for 6 h. After 48 h incubating in fresh medium, the cells were prepared for co-IP with an anti-ATG3 antibody and detected with an anti-Y203-phosphorylated ATG3 antibody. (D) ATG3-inducible MEF cells were treated with doxycycline (Doxy, 1 μg/ml) to induce WT, Y203E or Y203F ATG3 and then exposed to cycloheximide (CHX, 10 μM/ml). The protein level of ATG3 was measured by western blotting. (E) Graphs show the relative expression of WT or mutant ATG3 with the indicated treatments in (D). The data are presented as the mean ± SD (n = 3). (F) HCT116 cells were co-transfected with FLAG-ATG3 (WT, Y203E or Y203F) and HA-ubiquitin. The cells were treated with MG132 (2 μM) 24 h later for 8 h. Proteins were then immunoprecipitated from the cell extracts with anti-FLAG M2 beads, and the immune-complexes were probed with the anti-ATG3 or anti-HA antibody.
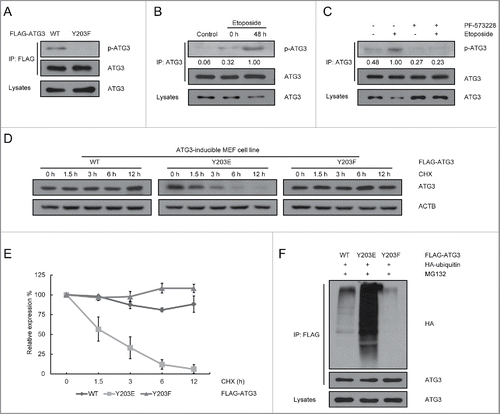
To further confirm the role of ATG3 phosphorylation at Y203 in its degradation, we generated ATG3 wild-type (ATG3-WT), ATG3Y203E (phosphorylation-mimic) and ATG3Y203F (phosphorylation-incompetent) inducible cell lines in Atg3 KO MEFs. As shown in and , ATG3 was a relatively stable protein, with a half-life in excess of 12 h. In contrast, the half-life of the phosphorylation-mimic protein ATG3Y203E was significantly shortened to 3 h. This reduction in the half-life of ATG3Y203E resulted from an increasing ubiquitination of ATG3. As shown in , ATG3Y203E-conjugated to ubiquitin was greatly increased compared with wild-type ATG3. The above results demonstrate that phosphorylation of ATG3 at Y203 is associated with its ubiquitination, which destabilizes the ATG3 protein under DNA damage conditions.
Y203 phosphorylation of ATG3 is dispensable for autophagy induction
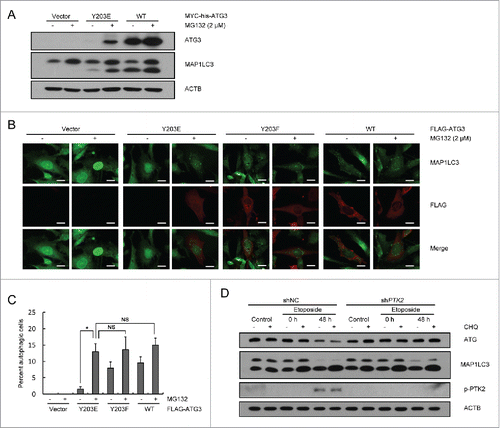
Because ATG3 is an autophagy essential gene, we next investigated the role of phosphorylation of ATG3 in autophagy induction. Autophagy in Atg3 knockout cells could not be rescued by expression of ATG3Y203E only, as the phosphorylation-mimic was unstable in cells. However, when Atg3 knockout cells transfected with the ATG3Y203E-encoding plasmid were incubated with MG132, autophagy was fully rescued (). These results suggested that Y203 phosphorylation of ATG3 is not critical for autophagy induction. Next, because ATG3 was degraded during the DNA damage process, we wanted to further explore whether there was any connection between ATG3 degradation and autophagy. A PTK2 RNAi plasmid was transfected into HCT116 cells to establish a stable PTK2 knockdown cell line; a nonspecific RNAi plasmid was used as a negative control. These cell lines were separately treated with etoposide for 3 h and then cultured in fresh medium for up to 48 h. Cell lysates were extracted for western blotting to detect changes in ATG3 and MAP1LC3, a marker of autophagy induction. As shown in , although ATG3 protein levels were rescued by PTK2 RNAi, no obvious difference of MAP1LC3-II accumulation was detected between the PTK2 stable-knockdown cell line and control cell line, suggesting that sustained levels of ATG3 caused by knocking down or inhibiting PTK2 are not related to autophagy induction.
Figure 4. Y203 phosphorylation of ATG3 is not critical for autophagy induction. (A) Atg3 KO MEF cells were transfected with plasmids encoding MYC-his-ATG3 WT or ATG3Y203E. MG132 (2 μM) was added into the medium 12 h after transfection for another 12 h to prevent ATG3Y203E degradation. Cell lysates were extracted to detect ATG3 and MAP1LC3 proteins. (B) Atg3 KO MEF cells were transfected with plasmids encoding FLAG-ATG3 WT, ATG3Y203E or ATG3Y203F. MG132 was added 12 h before collection. Immunofluorescence was performed after staining with anti-FLAG and anti-MAP1LC3 antibodies. Scale bars: 20 μm. (C) Quantification of the MAP1LC3 puncta-positive cells is shown in (B). The criterion for being counted was a cell with more than 10 puncta. The data are presented as the mean ± SD (n = 3). *p < 0.05; NS, no significance. (D) PTK2 stable-knockdown HCT116 cells and WT-HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for up to 48 h. CHQ (10 μM) was added 1 h before collection. Western blotting was performed to detect ATG3 and MAP1LC3 protein levels. An anti-p-PTK2 antibody was used to validate the efficiency of PTK2 siRNA.
ATG3Y203F promotes DNA damage-induced mitotic catastrophe by inhibiting BAG3
Previous studies have shown that some autophagy-related proteins can affect the DNA repair process.Citation32-36 Next, we tested whether sustained levels of ATG3 could regulate this process. Homologous recombination (HR) and nonhomologous end-joining (NHEJ) are 2 major pathways for the repair of DNA double-strand breaks.Citation37 Therefore, we used the direct-repeat (DR)-GFP reporter and the pEJ5-GFP reporter to monitor the HR and NHEJ pathways, respectively. As shown in Figure S4A-B, the efficiency of HR or NHEJ did not substantially change between the ATG3-WT cell line and ATG3Y203F cell line, suggesting that Y203 phosphorylation is not involved in the DNA repair process.
In addition to the involvement of ATG3 in autophagy, overexpressed ATG3 induces apoptosis in extracellular matrix (ECM)-attached colorectal cancer cells.Citation38 To investigate whether ATG3 degradation affects apoptosis, we used the ATG3-WT or ATG3Y203F-inducible cell lines for flow cytometry analysis. As shown in Figure S5, in response to etoposide treatment, there was no obvious difference in the apoptosis process between the ATG3-WT cell line and ATG3Y203F cell line.
As several reports have demonstrated that DNA-damaging agents are able to induce mitotic catastrophe,Citation32,39,40 we investigated the changes in nuclear morphology between ATG3-WT and ATG3Y203F cells following exposure to etoposide. In response to etoposide treatment, the nuclei became significantly larger, and some cells also contained micronuclei. Compared with the ATG3-WT cell line, the ATG3Y203F cells had more abnormal nuclei after etoposide treatment (). In addition, FACS analysis was used to detect the changes of DNA content in WT or Y203F cells treated with etoposide. Consistent with the above results, the hyperploid cell population (> 4N DNA cells) in Y203F cells was much higher than that in WT cells (). These results suggest that sustained ATG3 may be able to promote DNA damage-induced mitotic catastrophe.
Figure 5. ATG3Y203F is able to promote DNA damage-induced mitotic catastrophe. (A) ATG3-inducible MEF cells were treated with doxycycline (Doxy, 1 μg/ml) to induce WT or Y203F ATG3 and then exposed to etoposide. Then, the cells were stained with Giemsa. Scale bars: 20 μm. (B) Statistical analysis of the abnormal nuclei in (A). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (C) ATG3-inducible MEF cells were treated as described in (A), and immunostained with an antibody against TUBA (green). Nuclei were counterstained with DAPI (blue). Scale bars: 10 μm. (D) Statistical analysis of the abnormal nuclei in (C). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (E) ATG3-inducible MEF cells were treated as described in (A), and the cells were collected and analyzed by FACS. (F) HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for 48 h. The cell lysates were extracted for co-IP with an anti-ATG3 antibody and probed with an anti-BAG3 antibody.
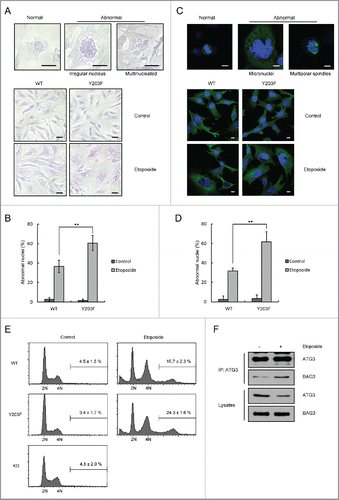
We next focused on the potential mechanisms of action for ATG3. It has been reported that ATG3 is able to interact with BAG3 (BCL2 associated athanogene 3),Citation41 which plays an important role in mitotic catastrophe.Citation42 As shown in , we found that the interaction between ATG3 and BAG3 was enhanced in response to etoposide treatment. These results suggest that ATG3 may promote DNA damage-induced mitotic catastrophe through its interaction with BAG3.
Sustained ATG3 is related to the inhibition of cell growth in response to DNA-damaging agents
Next, we generated an ATG3 knockout HCT116 cell line using the CRISPR/Cas9 system. Plasmids encoding ATG3-WT and ATG3Y203F were separately transfected into the ATG3 knockout HCT116 cell line (). Subsequently, we tested the colony formation of the ATG3-WT and ATG3Y203F cell lines under the treatment of etoposide, cisplatin or IR. Surprisingly, we found that the ATG3Y203F cell line exhibited reduced survival rates relative to the ATG3-WT cell line in response to DNA-damaging drugs ( and Figure S6A-B). In addition, we also tested colony formation in ATG7 knockout cells and obtained similar results (Figure S7A-B), suggesting that autophagy is independent of the function of ATG3 that mediates inhibition of cell growth in response to DNA damage. Furthermore, we used NOD-SCID mice to find whether sustained levels of ATG3 can sensitize cancer cells to treatment with DNA-damaging drugs ex vivo. Plasmids encoding ATG3-WT and ATG3Y203F were separately transfected into ATG3 knockout HCT116 cells, and then the cells were injected subcutaneously into NOD-SCID mice. Consistent with the colony formation data, the ATG3Y203F tumors showed less growth relative to the ATG3-WT tumors in response to etoposide treatment (). These results suggested that sustained levels of ATG3 may be beneficial for the inhibition of cell growth both in vitro and in vivo when cells are undergoing DNA damage. Accordingly, we predicted that PTK2 inhibition, which can maintain the stability of ATG3, would be able to sensitize cancer cells to DNA-damaging reagents.
Figure 6. Sustained ATG3 is related to tumor cell growth inhibition in response to DNA-damaging agents. (A) Cas9 ATG3 knockout cells were transfected with plasmids encoding FLAG-ATG3 WT ATG3Y203F. Western blotting was performed to detect the ATG3 and MAP1LC3 protein levels. (B) Cells overexpressing FLAG-ATG3-WT or FLAG-ATG3Y203F from (A) were treated with or without etoposide (40 μM) for 3 h or cisplatin (10 μM) for 6 h. Then, the cells were washed with fresh medium and allowed to grow for approximately 2 wk. The cells were stained with methylene blue before counting. (C) Statistical analysis of the surviving fraction in (B). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (D) Cells overexpressing FLAG-ATG3-WT or FLAG-ATGY203F cells were injected into the NOD-SCID mice, and the tumors were detected 3 weeks later (4 mice per group). (E) Tumors were weighed and analyzed. *p < 0.05. (F) HCT116 cells were pre-incubated with PF-573228, SRC Inhibitor-1 or erlotinib for 1 h and then treated with or without etoposide (40 μM) for 3 h. Cell proliferation was tested with the WST-1 assay, and the data are presented as the mean ± SD. Student t test was performed by comparing drug-treated cells versus untreated cells. *p < 0.05; **p < 0.01. (G) HCT116 cells were pre-incubated with PF-573228, SRC Inhibitor-1 or erlotinib for 1 h and then treated with or without etoposide or cisplatin. Then, the cells were treated as described in (B). (H) Statistical analysis of the surviving fraction in (G). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (I) Western blotting was performed to detect the endogenous PTK2 level in stable PTK2 knockdown and negative control cell lines. (J) PTK2 stable-knockdown HCT116 cells and WT-HCT116 cells were treated with or without etoposide or cisplatin. Then, the drug was removed and the colonies were counted after 2 wk. (K) Quantification of the surviving fraction in (J). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01.
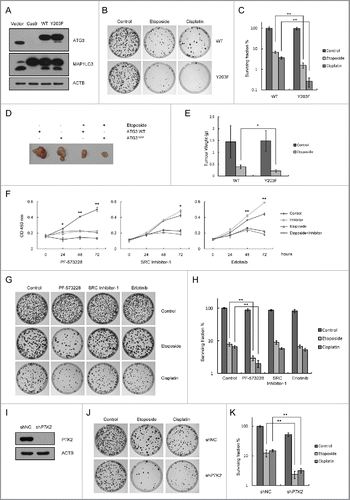
HCT116 cells were treated with etoposide alone or in combination with different tyrosine kinase inhibitors to observe changes in cell proliferation assayed with the WST-1 reagent. Consistent with our hypothesis, the PTK2 inhibitor PF-573228, but not the SRC nor EGFR inhibitors, was able to enhance etoposide-induced inhibition of cell proliferation (). For the long-term cell survival assay, we measured the colony formation of HCT116 cells under the treatment of DNA-damaging drugs combined with different tyrosine kinase inhibitors. After etoposide or cisplatin treatment, the colony formation rate was dramatically decreased in the PTK2 inhibitor PF-573228-treated cells but not in the SRC Inhibitor-1- or Erlotinib-treated cells (). The nature of PTK2 inhibition combined with chemotherapeutic agent-induced cell growth inhibition was further explored by knocking down PTK2 in HCT116 cells. As we expected, both etoposide and cisplatin decreased the efficiency of colony formation in PTK2 knockdown cells (), suggesting that the efficiency of DNA-damaging drugs is enhanced by PTK2 inhibition.
Discussion
In this study, we identified an autophagy-independent function of ATG3 during the DNA damage process. After treatment with DNA-damaging agents, PTK2 phosphorylates ATG3 at Y203, through which the degradation of ATG3 is promoted via the ubiquitin-proteasome-dependent pathway. Inhibition of PTK2 combined with DNA-damaging drug treatment causes sustained levels of ATG3, leading to a significant decrease in cell proliferation in a mitotic catastrophe-dependent manner.
Previous studies have shown that several autophagy-related proteins can be degraded through the proteasomal pathway.Citation43-48 For example, ubiquitin E3 ligase RNF5 interacts with ATG4B and controls ATG4B stability through proteasome-dependent degradation.Citation44 In addition, free ATG12 is highly unstable and rapidly degraded by the proteasomal pathway.Citation45 These results suggest that autophagy-related regulators may be regulated by the ubiquitin-proteasome system. Although ATG3 is very stable in normal conditions ()Citation49 our data showed that ATG3 can also be ubiquitinated and degraded under the conditions of DNA damage (). Consistently, it has already been reported that there are several potential ubiquitination sites on ATG3.Citation50,51 Therefore, further studies need to be performed to determine whether these ubiquitination sites are also responsible for ATG3 degradation in response to DNA-damaging agents.
As a protein-conjugating enzyme for MAP1LC3 lipidation, the biological function of ATG3 is mainly focused on autophagy.Citation14,15 The fact that autophagy protects cells from genotoxic stress and maintains genome integrity has already been established.Citation33,52-56 Autophagy, however, is unlikely itself to be responsible for the DNA damage-induced tumor cell growth inhibition. It has been reported that very low levels of ATG5 expression are sufficient for autophagy induction.Citation57 Similar to ATG5, we also found that although most ATG3 was degraded after etoposide treatment, autophagy could still be induced by the small amount of nondegraded ATG3 (). These results are consistent with previous reports that show incomplete deletion of Atg3 in macrophages can still allow MAP1LC3 lipidation.Citation58 In addition, we found that sustained levels of ATG3 induced by PTK2 inhibition can still sensitize cancer cells to DNA-damaging reagents in autophagy-deficient cells (Figure S7A-B), suggesting that autophagy is not crucial for ATG3 mediated inhibition of cell growth in response to DNA damage. Thus, we speculate that activity of ATG3 other than autophagy induction, leads to its crosstalk with the DNA damage response pathway. Recent evidence has shown that overexpressed ATG3 triggers apoptosis in the ECM-attached colorectal cancer cells.Citation38 However, in our system, we found that there was no difference between ATG3-WT and ATG3Y203F in the apoptosis process after DNA-damaging drug treatment (Figure S5). Instead, we found sustained levels of ATG3 were able to enhance DNA damage-induced mitotic catastrophe (), and this might be a new function of ATG3 independent of autophagy and apoptosis.
Furthermore, we also revealed that ATG3 may take part in the mitotic process by binding BAG3. As a member of the BAG family of co-chaperones that interacts with HSPA (heat shock protein family A [Hsp70] member), BAG3 can regulate many important biologic processes such as development, apoptosis and autophagy.Citation59 Recently, a novel HSPB8-dependent mitotic function of BAG3, which can guide spindle orientation and proper chromosome segregation, has been uncovered.Citation42 In addition, a proteomic study of BAG3 showed that ATG3 is a newly identified binding partner.Citation41 In this study, we observed an increase in levels of interaction between BAG3 and ATG3 in response to DNA-damaging drug treatment (). Accordingly, we postulate that ATG3 can inhibit the mitotic function of BAG3 after DNA-damaging drug treatment, which may be mediated by interfering with the formation of the BAG3 protein complex. When degradation of ATG3 was blocked after DNA-damaging drug treatment, the increased levels of ATG3 could sequester more BAG3, which may improve the suppression of BAG3 activity, leading to enhanced DNA damage-induced mitotic catastrophe.
Accumulating evidence suggests that PTK2 can promote cancer cell proliferation, motility, and survival.Citation21 These multiple functions of PTK2 work through kinase-dependent or kinase-independent mechanisms. As a protein tyrosine kinase, PTK2 can activate the phosphoinositide 3-kinase-AKT1 pathway by binding with the PIK3R/p85 subunit of phosphoinositide 3-kinaseCitation60 to induce survival signals and prevent cancer cell death.Citation31,61,62 Targeting of PTK2 in endothelial cells is sufficient to induce tumor cell sensitization to DNA-damaging therapies by downregulating the NFKB pathway.Citation22 Additionally, a recent study showed that some ATG proteins can colocalize with PTK2 and PXN.Citation63 Consistently, our data showed that in response to treatment with DNA-damaging agents, PTK2-induced ATG3 phosphorylation led to its degradation. Furthermore, PTK2 inhibition combined with DNA-damaging drugs induced sustained levels of ATG3, leading to a significant decrease in cell viability.
Previous studies have shown that PTK2 is overexpressed and activated in several advanced-stage solid cancers.Citation20,64,65 As a promising target for cancer therapy, some specific small molecule inhibitors (PTK2-Is) are being evaluated in clinical trials.Citation23-27 In addition, several studies have shown that combination regimens may be more effective for cancer therapy.Citation27,66,67 For example, PTK2 inhibition combined with irradiation can improve the radio-sensitivity of radio-resistant cancer stem cells via the WNT signaling pathway.Citation68 PTK2 knockout in endothelial cells can enhance the effects of DNA-damaging cancer therapy.Citation22 Y15, a small molecule PTK2 inhibitor, can inhibit cell growth in vitro and in vivo and enhance the efficacy of chemotherapy.Citation27 Consistent with previous reports, we demonstrated that sustained levels of ATG3 induced by PTK2 inhibition can sensitize cancer cells to DNA-damaging reagents both in vitro and in vivo. We also found that among several tyrosine kinase inhibitors, the PTK2 inhibitor had a much stronger effect on cancer cell colony formation when combined with DNA-damaging agents in this study. Although the specific mechanism of the tumor suppression function of ATG3 is not clear, targeting PTK2 combined with chemotherapeutic agents may be a useful strategy for the development of novel cancer therapies in the future.
In conclusion, our results showed that ATG3 is phosphorylated and degraded in response to cancer chemotherapeutic agents in a PTK2-dependent manner. Sustained levels of ATG3 after a combination regimen result in a significant decrease in cell viability in cancer cells through a mitotic catastrophe-dependent manner. Further studies exploring the mechanism of PTK2 inhibitors in combinatorial therapies will help us promote the rational use of chemotherapy drugs.
Methods
Reagents
Etoposide (E1383), cisplatin (P4394), chloroquine (C6628), PF-573228 (PZ0117), SRC Inhibitor-1 (S2075), doxycycline (D9891) and Anti-FLAG M2 beads (A2220) were purchased from Sigma-Aldrich; erlotinib (S1023) was purchased from Selleck Chemicals; MG132 (474790) was purchased from Merck; and Tet System Approved fetal bovine serum (631106) was purchased from Clontech.
Antibodies
Antibodies to ATG3 (M133–3), ATG10 (M151–3) and TUBA (PM054) were from MBL; ATG5 (2630), phospho-tyrosine (9416), acetylated-lysine (9441), phospho-PTK2 (Tyr576/577; 3281), phospho-SRC (Tyr416; 6943), and MAP1LC3 (2775) were purchased from Cell Signaling Technology; ATG7 (sc-33211), ACTB (sc-7210), and HA-probe (sc-7392) were purchased from Santa Cruz Biotechnology Inc.; phospho-serine (ab9332) and phospho-EGFR (Tyr1068; 1138–1) were purchased from Abcam; PTK2 (BS3583) was purchased from Bioworld Technology, Inc.; and BAG3 (10599–1-AP) was purchased from Proteintech Group Inc.
Plasmids
Based on the obtained DNA sequence of the murine Atg3 homolog, we amplified an open-reading frame of the murine Atg3 cDNA by high-fidelity PCR, cloned the fragment into the p3XFLAG-CMV-10 (Sigma-Aldrich, E4401), pcDNA3.1-MYC-his (Invitrogen, V855–20) or pTRIPZ (Thermo Fisher Scientific, RHS4750) vectors, and designated the resultant plasmids as the FLAG-ATG3, MYC-his-ATG3 and pTRIPZ-ATG3 plasmids. ATG3Y203E- and ATG3Y203F-encoding plasmids were generated with a site-directed mutagenesis kit (Agilent Technologies, 200523).
Cell culture and transfection
HCT116 (CCL−247) and LoVo (CCL−229) cell lines were purchased from the American Type Culture Collection and grown in McCoy's 5A medium (Macgene, CM10050) or DMEM (Macgene, CM15019) supplemented with 10% fetal bovine serum in a 37°C incubator with a humidified, 5% CO2 atmosphere. Atg3 knockout MEF cells were kindly provided by Dr. M. Komatsu (Niigata University) and grown in DMEM supplemented with 10% fetal bovine serum and 1X non-essential amino-acid (Macgene, CC25025) in 5% CO2 at 37°C. DR-GFP-U2OS and EJ5-GFP-HEK293 stable cell lines were from Dr. Xingzhi Xu (Shenzhen University Health Science Center), and grown in DMEM. For transfecting cancer cell lines, Lipofectamine 2000 (Invitrogen, 11668–027) was used following the manufacturer's instructions, while the Neon transfection system (Invitrogen, MPK5000) was used for transfection of MEF cells.
Establishment of ATG3-inducible MEF cell lines
The ATG3-inducible cell line was generated using the pTRIPZ-ATG3 WT, Y203E and Y203F plasmids. Plasmids were transfected into Atg3 knockout MEF cells, and the stable cell line was established by selection with 4 μg/ml puromycin (Sigma-Aldrich, P8833).
Generation of Cas9 ATG3 knockout and PTK2 knockdown cell lines
The Cas9 ATG3 knockout cell lines were generated using CRISPR–Cas9 methods in HCT116 cells. We used the SpCas9–2A-Puro vector purchased from Addgene (#48139; deposited by Feng Zhang).Citation69 The ATG3 gRNA was designed by online software (http://crispr.mit.edu), and the gRNA sequence of ATG3 was 5′-GTGAAGGCATACCTACCAAC-3′. The plasmids were transfected into HCT116 cells and selected with 2.5 μg/ml puromycin. The stable PTK2 knockdown cell lines were established by a PTK2-specific RNAi plasmid. The nonspecific or shPTK2 plasmids were transfected into HCT116 cells and selected using 500 μg/ml hygromycin B (Roche, 10843555001).
Immunoblot analysis
Equal amounts of proteins were size-fractionated by 7.5–15% SDS-PAGE. For immunoprecipitation, cells were harvested and then lysed in a Nonidet P40 buffer (1% Nonidet P40 [Amersco, E109], 150 mM NaCl, 50 mM Tris at pH 7.5, and 5 mM EDTA) supplemented with complete protease inhibitor cocktail (Roche, 5892791001) and phosphatase inhibitor cocktail (Applygen, P1260). Whole-cell lysate proteins were used for immunoprecipitation with the indicated antibodies. Generally, 2 μg of antibody was added to 1 ml of cell lysate, which was incubated at 4°C for 8 to 12 h. After the addition of protein G-agarose beads (GE Healthcare, 17–0618–01), the incubation was continued for 2 h. Immunoprecipitates were extensively washed with lysis buffer and eluted with SDS loading buffer by boiling for 5 min. All bands of blots were scanned with a phosphorimager, and the relative intensity of each band was normalized to each band of ACTB. The data collected came from at least 3 independent experiments.
Real-time PCR analysis of mRNA
Total RNA was isolated with TRIzol reagent (Invitrogen, 15596026). The cDNA was synthesized from 2 μg of RNA by using the Quantscript RT Kit (TianGen, KR103). The primer sequences of ATG3 and ACTB for RT-PCR were as follows: ATG3, GATGGCGGATGGGTAGATACA, TCTTCACATAGTGCTGAGCAATC; ACTB, CCAACCGCGAGAAGATGA, CCAGAGGCGTACAGGGATAG.
Immunofluorescence analysis
Cells were cultured on confocal dishes to approximately 60% confluence. After transfection and treatment, cells were fixed with 4% paraformaldehyde and permeabilized with methanol. The dishes were incubated with blocking solution (0.8% BSA [Amresco, 0332] in phosphate-buffered saline [Macgene, CC008]) and exposed overnight to a primary antibody (1:100 dilution for all antibodies) at 4°C. After being washed 3 times with blocking solution, the dishes were exposed to a secondary antibody (1:100 dilution) and conjugated to FITC-TRITC (fluorescein isothiocyanate-tetramethyl rhodamine isothiocyanate; ZSGB-Bio, ZF-0312). The cells were observed and documented under a confocal microscope (Olympus BX-51, America Inc.).
Colony formation assay
Cells were plated and exposed to 40 μM etoposide for 3 h. Then, etoposide was removed by washing 3 times. Cells were trypsinized and plated into 60-mm plates. After 2 wk, methanol fixation and staining with methylene blue was undertaken to identify visible colonies. Plating efficiencies were calculated as follows: number of colonies formed/number of cells plated. Surviving fractions were calculated as follows: number of colonies formed/number of cells plated (treated) x plating efficiency (untreated).
WST-1 assay
Equal numbers of cells (approximately 5000/well) were seeded into a 96-well plate 24 h before experimentation. Cells were treated with etoposide separately or combined with different tyrosine kinase inhibitors. After treatment, WST-1 (Dojindo, S311) was added into the 96-well plate. The absorbance of each sample was read at 450 nm.
DR-GFP assay and EJ5-GFP assay
DR-U2OS and EJ5-HEK293 cells were plated in 6-well plates and transfected with plasmids encoding FLAG-ATG3 WT or ATG3Y203F. The cells were then transfected with the pCBASce plasmid (encoding I-SceI enzyme; Addgene, #26477; deposited by Maria Jasin). After 48 h, the percentage of GFP+ cells per well was determined by FACS.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download Zip (7.8 MB)Acknowledgment
We thank Dr. Masaaki Komatsu for providing Atg3 knockout MEF cells.
Funding
This study was supported by National Natural Science Foundation of China (31570812, 81222028, 81321003, 81472581, 81530074, 81672712,91319302 and 31261140372); Discipline Construction Funding of Shenzhen (2016), and grants (JCYJ20160427104855100) from Shenzhen Municipal Commission of Science and Technology Innovation.
Related Research Data
References
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nature reviews Molecular cell biology 2015; 16:461-72; PMID:26177004; http://dx.doi.org/10.1038/nrm4024
- Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature 2015; 517:302-10; PMID:25592535; http://dx.doi.org/10.1038/nature14190
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature cell biology 2010; 12:665-75; PMID:20543840; http://dx.doi.org/10.1038/ncb2069
- Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J, Fu W, Wei FZ, Wang L, Xie D, et al. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell research 2013; 23:491-507; PMID:23277279; http://dx.doi.org/10.1038/cr.2013.2
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Navone F, Genevini P, Borgese N. Autophagy and Neurodegeneration: Insights from a Cultured Cell Model of ALS. Cells 2015; 4:354-86; PMID:26287246; http://dx.doi.org/10.3390/cells4030354
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, Vandenabeele P. Beclin1: a role in membrane dynamics and beyond. Autophagy 2012; 8:6-17; PMID:22170155; http://dx.doi.org/10.4161/auto.8.1.16645
- Bestebroer J, V'Kovski P, Mauthe M, Reggiori F. Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 2013; 14:1029-41; PMID:23837619; http://dx.doi.org/10.1111/tra.12091
- Lee J, Kim HR, Quinley C, Kim J, Gonzalez-Navajas J, Xavier R, Raz E. Autophagy suppresses interleukin-1beta (IL-1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem 2012; 287:4033-40; PMID:22167182; http://dx.doi.org/10.1074/jbc.M111.280065
- Fremont S, Gerard A, Galloux M, Janvier K, Karess RE, Berlioz-Torrent C. Beclin-1 is required for chromosome congression and proper outer kinetochore assembly. EMBO Rep 2013; 14:364-72; PMID:23478334; http://dx.doi.org/10.1038/embor.2013.23
- Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell host & microbe 2012; 11:397-409; http://dx.doi.org/10.1016/j.chom.2012.03.002
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488-92; PMID:11100732; http://dx.doi.org/10.1038/35044114
- Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem 2002; 277:13739-44; PMID:11825910; http://dx.doi.org/10.1074/jbc.M200385200
- Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142:590-600; PMID:20723759; http://dx.doi.org/10.1016/j.cell.2010.07.018
- Metlagel Z, Otomo C, Takaesu G, Otomo T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proceedings of the National Academy of Sciences of the United States of America 2013; 110:18844-9; PMID:24191030; http://dx.doi.org/10.1073/pnas.1314755110
- Moloughney JG, Monken CE, Tao H, Zhang H, Thomas JD, Lattime EC, Jin S. Vaccinia virus leads to ATG12-ATG3 conjugation and deficiency in autophagosome formation. Autophagy 2011; 7:1434-47; PMID:22024753; http://dx.doi.org/10.4161/auto.7.12.17793
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Molecular biology of the cell 2008; 19:4762-75; PMID:18768753; http://dx.doi.org/10.1091/mbc.E08-03-0309
- Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer 2014; 14:598-610; PMID:25098269; http://dx.doi.org/10.1038/nrc3792
- Zhang J, Hochwald SN. The role of FAK in tumor metabolism and therapy. Pharmacol Ther 2014; 142:154-63; PMID:24333503; http://dx.doi.org/10.1016/j.pharmthera.2013.12.003
- Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP, Alexopoulou A, Elia G, et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014; 514:112-6; PMID:25079333; http://dx.doi.org/10.1038/nature13541
- Schwock J, Dhani N, Hedley DW. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin Ther Targets 2010; 14:77-94; PMID:20001212; http://dx.doi.org/10.1517/14728220903460340
- Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, Fingert H, Pierce KJ, Xu H, Roberts WG, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol 2012; 30:1527-33; PMID:22454420; http://dx.doi.org/10.1200/JCO.2011.38.9346
- Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacology & therapeutics 2015; 146:132-49; http://dx.doi.org/10.1016/j.pharmthera.2014.10.001
- Shanthi E, Krishna MH, Arunesh GM, Venkateswara Reddy K, Sooriya Kumar J, Viswanadhan VN. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: a patent review. Expert opinion on therapeutic patents 2014; 24:1077-100; PMID:25113248; http://dx.doi.org/10.1517/13543776.2014.948845
- Heffler M, Golubovskaya VM, Dunn KM, Cance W. Focal adhesion kinase autophosphorylation inhibition decreases colon cancer cell growth and enhances the efficacy of chemotherapy. Cancer biology & therapy 2013; 14:761-72; http://dx.doi.org/10.4161/cbt.25185
- Lu H, Wang L, Gao W, Meng J, Dai B, Wu S, Minna J, Roth JA, Hofstetter WL, Swisher SG, et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Molecular cancer therapeutics 2013; 12:2864-73; PMID:24130049; http://dx.doi.org/10.1158/1535-7163.MCT-13-0233
- Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, Tuthill T, Andresen CJ. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer biology & therapy 2009; 8:856-65; http://dx.doi.org/10.4161/cbt.8.9.8246
- Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer research 2007; 67:10976-83; PMID:18006843; http://dx.doi.org/10.1158/0008-5472.CAN-07-2667
- Kang Y, Hu W, Ivan C, Dalton HJ, Miyake T, Pecot CV, Zand B, Liu T, Huang J, Jennings NB, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. Journal of the National Cancer Institute 2013; 105:1485-95; PMID:24062525; http://dx.doi.org/10.1093/jnci/djt210
- Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nature communications 2013; 4:2130. PMID:23945651; http://dx.doi.org/10.1038/ncomms3130
- Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K, Li X, Wang L, Wang J, Zhang H, et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Molecular cell 2016; 63:34-48; PMID:27345151; http://dx.doi.org/10.1016/j.molcel.2016.05.027
- Liu C, He W, Jin M, Li H, Xu H, Liu H, Yang K, Zhang T, Wu G, Ren J. Blockage of Autophagy in C6 Glioma Cells Enhanced Radiosensitivity Possibly by Attenuating DNA-PK-Dependent DSB Due to Limited Ku Nuclear Translocation and DNA Binding. Current molecular medicine 2015; 15:663-73; PMID:26321753; http://dx.doi.org/10.2174/1566524015666150831141112
- Zhang D, Tang B, Xie X, Xiao YF, Yang SM, Zhang JW. The interplay between DNA repair and autophagy in cancer therapy. Cancer biology & therapy 2015; 16:1005-13; http://dx.doi.org/10.1080/15384047.2015.1046022
- Mo N, Lu YK, Xie WM, Liu Y, Zhou WX, Wang HX, Nong L, Jia YX, Tan AH, Chen Y, et al. Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol Rep 2014; 32:1905-12; PMID:25175062
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723
- Yoo BH, Zagryazhskaya A, Li Y, Koomson A, Khan IA, Sasazuki T, Shirasawa S, K VR. Upregulation of ATG3 contributes to autophagy induced by the detachment of intestinal epithelial cells from the extracellular matrix, but promotes autophagy-independent apoptosis of the attached cells. Autophagy 2015; 11:1230-46; PMID:26061804; http://dx.doi.org/10.1080/15548627.2015.1056968
- Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S, Kim WH, Yoon G, Choi KS. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005; 24:4765-77; PMID:15870702; http://dx.doi.org/10.1038/sj.onc.1208627
- Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proceedings of the National Academy of Sciences of the United States of America 2005; 102:1065-70; PMID:15650047; http://dx.doi.org/10.1073/pnas.0409130102
- Chen Y, Yang LN, Cheng L, Tu S, Guo SJ, Le HY, Xiong Q, Mo R, Li CY, Jeong JS, et al. Bcl2-associated athanogene 3 interactome analysis reveals a new role in modulating proteasome activity. Molecular & cellular proteomics : MCP 2013; 12:2804-19; http://dx.doi.org/10.1074/mcp.M112.025882
- Fuchs M, Luthold C, Guilbert SM, Varlet AA, Lambert H, Jette A, Elowe S, Landry J, Lavoie JN. A Role for the Chaperone Complex BAG3-HSPB8 in Actin Dynamics, Spindle Orientation and Proper Chromosome Segregation during Mitosis. PLoS genetics 2015; 11:e1005582. PMID:26496431; http://dx.doi.org/10.1371/journal.pgen.1005582
- Jiao H, Su GQ, Dong W, Zhang L, Xie W, Yao LM, Chen P, Wang ZX, Liou YC, You H. Chaperone-like protein p32 regulates ULK1 stability and autophagy. Cell death and differentiation 2015.
- Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, Niesman IR, Yang HJ, Lopez-Otin C, Yang WY, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS genetics 2012; 8:e1003007. PMID:23093945; http://dx.doi.org/10.1371/journal.pgen.1003007
- Haller M, Hock AK, Giampazolias E, Oberst A, Green DR, Debnath J, Ryan KM, Vousden KH, Tait SW. Ubiquitination and proteasomal degradation of ATG12 regulates its proapoptotic activity. Autophagy 2014; 10:2269-78; PMID:25629932; http://dx.doi.org/10.4161/15548627.2014.981914
- Platta HW, Abrahamsen H, Thoresen SB, Stenmark H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. The Biochemical journal 2012; 441:399-406; PMID:21936852; http://dx.doi.org/10.1042/BJ20111424
- Zhang T, Dong K, Liang W, Xu D, Xia H, Geng J, Najafov A, Liu M, Li Y, Han X, et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. eLife 2015; 4:e06734. PMID:25821988
- Li J, Qi W, Chen G, Feng D, Liu J, Ma B, Zhou C, Mu C, Zhang W, Chen Q, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 2015; 11:1216-29; PMID:26018823; http://dx.doi.org/10.1080/15548627.2015.1017180
- Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proceedings of the National Academy of Sciences of the United States of America 2012; 109:13325-30; PMID:22847423; http://dx.doi.org/10.1073/pnas.1120193109
- Wagner SA, Beli P, Weinert BT, Scholz C, Kelstrup CD, Young C, Nielsen ML, Olsen JV, Brakebusch C, Choudhary C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Molecular & cellular proteomics : MCP 2012; 11:1578-85; http://dx.doi.org/10.1074/mcp.M112.017905
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Molecular cell 2011; 44:325-40; PMID:21906983; http://dx.doi.org/10.1016/j.molcel.2011.08.025
- Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harbor symposia on quantitative biology 2011; 76:397-402; PMID:21813637; http://dx.doi.org/10.1101/sqb.2011.76.011023
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65-75; PMID:24679527; http://dx.doi.org/10.1016/j.cell.2014.02.049
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer 2013; 13:714-26; PMID:24060863; http://dx.doi.org/10.1038/nrc3599
- Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell death and differentiation 2013; 20:1444-54; PMID:23933813; http://dx.doi.org/10.1038/cdd.2013.103
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS letters 2006; 580:2623-9; PMID:16647067; http://dx.doi.org/10.1016/j.febslet.2006.04.008
- Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, Fujita N, Saitoh T, Akira S, Yoshimori T, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 2014; 40:924-35; PMID:24931121; http://dx.doi.org/10.1016/j.immuni.2014.05.006
- Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis 2011; 2:e141. PMID:21472004; http://dx.doi.org/10.1038/cddis.2011.24
- Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem 1999; 274:12361-6; PMID:10212207; http://dx.doi.org/10.1074/jbc.274.18.12361
- Lane D, Goncharenko-Khaider N, Rancourt C, Piche A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene 2010; 29:3519-31; PMID:20400979; http://dx.doi.org/10.1038/onc.2010.107
- Zheng Y, Gierut J, Wang Z, Miao J, Asara JM, Tyner AL. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene 2013; 32:4304-12; PMID:23027128; http://dx.doi.org/10.1038/onc.2012.427
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nature cell biology 2012; 14:51-60; http://dx.doi.org/10.1038/ncb2386
- Canel M, Secades P, Rodrigo JP, Cabanillas R, Herrero A, Suarez C, Chiara MD. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clinical cancer research : an official journal of the American Association for Cancer Research 2006; 12:3272-9; PMID:16740747; http://dx.doi.org/10.1158/1078-0432.CCR-05-1583
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer metastasis reviews 2009; 28:35-49; PMID:19169797; http://dx.doi.org/10.1007/s10555-008-9165-4
- Thakur R, Trivedi R, Rastogi N, Singh M, Mishra DP. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Scientific reports 2015; 5:10194. PMID:25973915; http://dx.doi.org/10.1038/srep10194
- Golubovskaya VM, Huang G, Ho B, Yemma M, Morrison CD, Lee J, Eliceiri BP, Cance WG. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Molecular cancer therapeutics 2013; 12:162-72; PMID:23243059; http://dx.doi.org/10.1158/1535-7163.MCT-12-0701
- Williams KE, Bundred NJ, Landberg G, Clarke RB, Farnie G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem cells 2015; 33:327-41; PMID:25187396; http://dx.doi.org/10.1002/stem.1843
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols 2013; 8:2281-308; PMID:24157548; http://dx.doi.org/10.1038/nprot.2013.143