
ABSTRACT
Autophagy is a cellular process in which the cell degrades and recycles its own constituents. Given the crucial role of autophagy in physiology, deregulation of autophagic machinery is associated with various diseases. Hence, a thorough understanding of autophagy regulatory mechanisms is crucially important for the elaboration of efficient treatments for different diseases. Recently, ion channels, mediating ion fluxes across cellular membranes, have emerged as important regulators of both basal and induced autophagy. However, the mechanisms by which specific ion channels regulate autophagy are still poorly understood, thus underscoring the need for further research in this field. Here we discuss the involvement of major types of ion channels in autophagy regulation.
Autophagy
Autophagy is a cellular process in which the cell degrades and recycles its own constituents.Citation1 Autophagy is generally considered to play predominantly a cell-prosurvival role. Indeed, under basal conditions, autophagy participates in maintaining normal cellular homeostasis by elimination of misfolded proteins, protein aggregates and damaged organelles, whereas in stress autophagy mobilizes various energy sources, provides nutrients and removes potentially dangerous elements.Citation2,3 Autophagy has been implicated in a number of physiological processes, including embryonic development, cell differentiation, oocyte fertilization, innate and adaptive immunity, senescence, life-span extension and others (for review see ref. Citation1, 2). Therefore, it is not surprising that deregulation of autophagy has been linked to many diseases, such as infectious, metabolic, cardiovascular, pulmonary, and neurodegenerative diseases as well as cancer.Citation4 Accordingly, a thorough understanding of autophagy regulatory mechanisms is crucially important for the elaboration of efficient treatments for different diseases.
Normally, autophagy occurs under basal conditions and it is stimulated in response to various types of cellular stress, including endoplasmic reticulum (ER) stress, oxidative stress, nutrient and growth factor starvation, hypoxia, mitochondrial damage, and others.Citation5 Various types of autophagy have been described, including macro- and microautophagy (which can be selective or nonselective) as well as chaperone-mediated autophagy (CMA; which selectively targets a specific subset of cytosolic proteins).Citation1,6,7 During macroautophagy, hereafter called autophagy, cytoplasmic components (autophagic substrates) are sequestered within double-membrane structures (termed autophagosomes) which eventually fuse with lysosomes, resulting in substrate degradation.Citation1,8
The process of autophagosome formation in mammals is mediated by at least 18 autophagy-related (ATG) gene products and is triggered by changes in growth conditions and energy supply which are sensed by central regulators MTOR (mechanistic target of rapamycin) complex 1 (MTORC1) and 5′ adenosine monophosphate-activated protein kinase (AMPK).Citation9–Citation12 In fact, MTORC1 and AMPK control the activity of the ULK (unc-51 like autophagy activating kinase) protein complex (comprising ULK1/2, RB1CC1/FIP200 [RB1 inducible coiled-coil 1] protein and ATG13), which is essential for the initiation of autophagosome formation.Citation13 Under fed conditions, active MTORC1 binds and inactivates the ULK complex, thereby inhibiting autophagosome formation. Upon amino acid starvation, MTORC1 is inactivated and dissociates from the ULK complex, resulting in ULK1/2 activation and autophagy induction.Citation14 Along with regulation by MTORC1, the ULK complex is also regulated by a cellular key energy sensor, AMPK. Under glucose starvation, AMPK promotes autophagy by directly activating ULK1 through phosphorylation of Ser317 and Ser777.Citation13 In addition, AMPK inactivates MTORC1 (through the activation of MTORC1 inhibitor TSC2 [TSC complex subunit 2]) and in this way further favors ULK complex activation.Citation15 The activated ULK complex translocates to the autophagosome initiation site (the phagophore), where it enhances the activity of another recruited protein complex containing PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), along with regulatory subunits BECN1 (Beclin 1), ATG14 and PIK3R4/VPS15 (phosphoinositide-3-kinase regulatory subunit 4).Citation16 Activation of the PIK3C3/VPS34-containing complex results in a production of phosphatidylinositol-3-phosphate (PtdIns3P) allowing binding of autophagy-specific PtdIns3P effectors (such as ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1), and WIPI (WD repeat domain, phosphoinositide interacting) proteins to the forming autophagosomal membrane and subsequent recruitment of the ATG12–ATG5-ATG16L1 protein complex, which is important for membrane expansion and closure.Citation12,17-19 The ATG12–ATG5-ATG16L1 complex functions as an E3-like ligase to mediate the lipidation of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) and its family members including GABARAPL2/GATE16 (GABA type A receptor associated protein like 2) and GABARAP (GABA type A receptor-associated protein).Citation20,21 This allows LC3 and its family members to associate with the phagophore membrane and promote membrane expansion and closure as well as fusion of the autophagosome with a lysosome.Citation22 Moreover, LC3 also functions to recruit selective cargo to the phagophore via interaction with cargo receptors.Citation23 Of note, LC3 family members remain associated with autophagosomes until their degradation in lysosomes; therefore, the detection of these proteins by immunoblotting or immunofluorescence is now the most widely used technique for monitoring autophagy.Citation24 For a more detailed description of the molecular mechanisms underlying the execution of autophagy we refer the reader to the comprehensive reviews available.Citation1,2,8–12,17
Ion channels
Accumulated evidence indicates a close interplay between signaling pathways regulating autophagy and those regulating cell cycle, cell growth, cell survival, and death.Citation5 Furthermore, numerous studies have demonstrated the implication of ion channels in all the abovementioned processes.Citation25–Citation27
Ion channels are integral pore-forming membrane proteins that mediate passive influx/efflux of essential signaling ions into/from the cell or intracellular organelles thereby controlling cytoplasmic/intraorganellar ion concentrations, membrane potential and cell volume. Ion channels contribute to virtually all basic cellular processes and are critically involved in such fundamental physiological processes as muscle contraction, synaptic transmission, action potential propagation and secretion. Ion channels control our sleep-wake cycle, vision, heartbeat, movements, thinking and memory and it is not surprising that ion channel dysfunctions have been linked to a number of diseases.Citation28 At present, ion channels are regarded as promising drug targets for the development of novel therapeutic agents.Citation29 The crucial importance of ion channel research is also demonstrated by 3 Nobel Prizes (in physiology or medicine in 1963 and 1991, as well as in chemistry in 2003) for the discoveries concerning the structure and function of ion channels.
A striking molecular variety of ion channels exists. They are classified by ion selectivity (sodium channels, potassium channels, chloride channels, calcium channels, proton channels, nonselective channels), gating mechanism (voltage-gated, ligand-gated, cyclic nucleotide-gated, light-gated, mechanosensitive) or localization (plasma membrane or intracellular).Citation30
Recently, ion channels have emerged as important regulators of both basal and induced autophagy in different cell types. However, despite the well-established role of different ion channels in a plethora of cellular processes, including apoptosis, proliferation, differentiation, and migration, information on the molecular nature of channels involved in the regulation of autophagy and the mechanisms of such regulation is still limited. Here, we discuss the existing data on the involvement of ion channels in autophagy regulatory pathways.
Calcium-permeable channels in the regulation of autophagy
To date, most reports considering ion channels as autophagy regulators are focused on calcium-permeable ion channels.Citation31 This is not a surprising fact when taking into account the particular significance of calcium ions for the cell. Calcium is a ubiquitous intracellular messenger that influences almost every aspect of cellular life.Citation32 Calcium is maintained at very low concentration in the cytosol (∼100 nM) and a steep (∼20,000 fold) Ca2+ gradient exists across the plasma membrane.Citation33 No other ion can boast such characteristics. Increases of cytosolic Ca2+ concentration organized in space, time and amplitude affect a variety of cellular processes including gene transcription, cell proliferation, differentiation, exocytosis, excitability and cell death. To ensure proper signaling, calcium signals are managed by a complex set of regulatory elements known as the “calcium signaling toolkit”.Citation32 Calcium-permeable ion channels, being the components of the “calcium signaling toolkit,” contribute to changes in cytosolic calcium concentration by providing Ca2+ entry pathways, by modulating the driving force for the Ca2+ entry, and also by providing intracellular pathways for Ca2+ uptake/release into/from cellular organelles.Citation34 In this way calcium-permeable channels indirectly control a variety of calcium-dependent cellular processes such as cell proliferation, apoptosis and autophagy.
The role of calcium in the regulation of autophagy is rather complicated and controversial with many reports suggesting an inhibitory effect of calcium on autophagy, contrasting with the stimulatory action of calcium reported by others.Citation31,35–37 However, careful analysis of available data shows that these controversies arise from the great complexity of intracellular calcium signaling. In reality, the final effect of calcium on autophagy greatly depends on spatiotemporal characteristics and the amplitude of calcium signals as well as on cell growth conditions (e.g., nutrient and growth factor availability) and type of autophagy.
Early approaches to study the role of calcium in autophagy were based on the use of calcium chelators and calcium mobilizing agents to alter the cytosolic calcium concentration. And from the very beginning these approaches proved that the role of calcium in the regulation of autophagy is very complex. In their first report linking autophagy and calcium, Gordon et al. demonstrated that both a decrease and an increase in cytosolic calcium inhibit hepatocytic autophagy.Citation38 Inexplicably, despite the authors' suggestion that autophagy is dependent on calcium compartmentalization, this area of research was not developing until more than a decade later when Høyer-Hansen et al. demonstrated that a rise in free cytosolic calcium is a potent inducer of macroautophagy.Citation39 The authors provided evidence that Ca2+-mobilizing agents (vitamin D3, thapsigargin, ATP and ionomycin) induce autophagosome accumulation by inhibiting the activity of MTOR via a signaling pathway involving CAMKK2/CaMKKβ (calcium/calmodulin dependent protein kinase kinase 2) and AMPK.Citation39 Later on, the Ca2+-CAMKK2-AMPK pathway (both MTOR dependent and MTOR independent) became a “classical” pathway for cytosolic calcium-induced autophagy. Indeed, a number of studies suggest that this pathway is involved in autophagy induction following an increase in cytosolic Ca2+ arising from both the extracellular medium and intracellular stores. For example, the Ca2+-CAMKK2-AMPK pathway has been reported to be involved in amyloid-beta peptide-induced autophagosome formation as well as in autophagy induced by amino acid starvation, alisol B or LRRK2 (leucine rich repeat kinase 2).Citation40–Citation43
Naturally, the Ca2+-CAMKK2-AMPK pathway is not a unique pathway discovered for calcium-induced autophagy. It has been reported that Ca2+-dependent activation of PRKCQ (protein kinase C theta) is specifically required for autophagy in response to ER-stress but not in response to amino acid starvation, confirming the complexity of autophagy regulatory mechanisms.Citation44 Further, it has been shown that Ca2+ signaling can induce autophagy independently of the Ca2+-mediated activation of AMPK.Citation45 Moreover, direct exogenous introduction of Ca2+ in the form of calcium phosphate precipitates to mammalian cells induces BECN1-, ATG5- and phosphatidylinositol 3-kinase (PtdIns3K)-dependent autophagy.Citation46 It should be noted, that in all the cases discussed above, cytosolic Ca2+ chelation with BAPTA-AM strongly inhibits accumulation of autophagic vesicles, thus confirming the role of Ca2+ in autophagy activation.
Conversely, a number of studies report that Ca2+ acts as an inhibitor of autophagy. For example, amino acids induce a rise in intracellular Ca2+ resulting in a direct binding of Ca2+-CALM/calmodulin to PIK3C3/VPS34 and subsequent MTORC1 activation.Citation47 In line with this, Khan and Joseph reported that the ITPR (inositol 1,4,5-trisphosphate receptor)-dependent Ca2+ signals act to maintain an elevated MTORC1 activity, and in this way negatively regulate basal autophagy.Citation48
Overall, it is clear that calcium is an important regulator of autophagy, although, it seems that multiple parameters could determine whether calcium will activate or inhibit autophagy in each particular case.
Considering that cytosolic and organellar calcium levels are regulated by calcium-permeable ion channels, the increasing interest in calcium-dependent mechanisms of autophagy regulation inevitably led to the discovery of connections between calcium channels and autophagy.
Inositol 1,4,5-trisphosphate receptor
ITPR, the main intracellular Ca2+ release channel, represents one of the most studied calcium-permeable channels with regard to the regulation of autophagy. ITPR is located on the membrane of the endoplasmic reticulum (ER) and mediate calcium release from the ER in response to inositol 1,4,5-trisphosphate (InsP3). ITPR-mediated calcium signaling has been reported to regulate a variety of cellular processes, such as cell growth, survival and death.Citation49 The complex role of ITPR in autophagy is thoroughly discussed in recent reviews to which the reader is referred.Citation50,51 In fact, available data suggest that ITPR can have a dual impact on autophagy that could result in both activation and inhibition of this process.
Originally, ITPR was proposed as an autophagy suppressor. Specifically, a decrease in InsP3 levels,Citation52 pharmacological inhibition of ITPR and knockdown/knockout of ITPR have been reported to induce autophagy via different mechanisms.Citation48,53–55 In DT40 cells basal autophagy is negatively regulated by ITPR-dependent Ca2+ signals, which act to maintain an elevated MTORC1 activity through an AMPK-independent pathway. Consistent with this, triple knockout DT40 cells (containing targeted deletions of all 3 ITPR isoforms) exhibit enhanced basal autophagic flux even under nutrient-replete conditions.Citation48 In line with these results, Cardenas et al. also reported that ITPR-mediated Ca2+ signaling is required for autophagy suppression in DT40 cells in nutrient-rich media. The authors, however, proposed another MTOR-independent mechanism for this regulation. More specifically, the authors demonstrated that constitutive ITPR-mediated Ca2+ release from the ER and subsequent uptake of this Ca2+ by mitochondria are fundamentally required to maintain mitochondrial bioenergetics and ATP production in resting cells. Absence of this Ca2+ transfer results in reduction of ATP production and activation of AMPK, which in turn activates prosurvival autophagy in an MTOR-independent manner.Citation55 Indeed, previous studies suggested that ITPR-mediated Ca2+ signals can be transmitted to mitochondria at ER-mitochondria contact sites.Citation56–Citation58 It has now become evident that by allowing bidirectional trafficking of factors between the 2 organelles these contact sites play an important role in the regulation of such cellular processes as lipid biosynthesis, mitochondrial fission and fusion, mitochondrial dynamics and function.Citation56
The Ca2+ transfer from the ER to the mitochondria is proposed to occur through the ITPR on the ER to VDAC1 (voltage dependent anion channel 1) on the outer mitochondrial membrane.Citation57,59 The passage of calcium through the inner mitochondrial membrane is mediated by a highly selective, low-affinity Ca2+ channel, known as MCU (mitochondrial calcium uniporter).Citation60 It has been proposed that Ca2+ release from the ER to the mitochondria serves to regulate mitochondrial ATP production, mitochondrial division and mitochondrial apoptosis.Citation56 The work of Cardenas et al. introduced basal autophagy as yet another process which is indirectly regulated by ER-mitochondria contact sites and related ITPR Ca2+ signaling.Citation55 Interestingly, recent studies revealed that ER-mitochondria contact sites are directly involved in autophagosome formation under starvation conditions.Citation12,61 Specifically, it has been demonstrated that during starvation mitochondria-associated ER membrane (MAM) recruits major protein complexes essential for the initiation of autophagosome formation and serves as a platform for autophagosome assembly.Citation61 Hence, taking into account that MAMs are enriched with ITPR, it is natural to assume that ITPR could potentially be involved in autophagosome formation under starvation conditions. Several studies suggesting the necessity of ITPR for autophagy, support this hypothesis. One study reported that the ITPR inhibitor xestospongin B as well as the Ca2+ chelator BAPTA-AM abolish starvation-induced autophagic flux, suggesting that ITPR-mediated Ca2+ signaling is critical for starvation-induced autophagy both in cancer and noncancer cells. Furthermore, starvation leads to ITPR sensitization through the enhanced association of BECN1 with the ITPR.Citation62 Another study suggests that ITPR is required for differentiation factor DIF-induced autophagic cell death in Dictyostelium discoideum. Through random insertional mutagenesis, the authors showed that inactivation of the iplA gene, the only gene encoding an ITPR in this organism, prevents autophagic cell death.Citation63 In line with this, ITPR1 protects renal cancer cells against natural killer (NK) cells by supporting NK-induced autophagy (ITPR1 knockdown inhibits NK-mediated autophagy induction in renal cancer cells).Citation64
Importantly, ITPR has been also reported to regulate autophagy independently of its Ca2+ channel function. More specifically, the ITPR antagonist xestospongin B induces autophagy in a Ca2+-independent manner by disrupting a molecular complex formed by the ITPR, BECN1, and BCL2 (BCL2, apoptosis regulator) proteins.Citation53 Therefore, ITPR has been proposed to act as a regulator of the BECN1 complex that bridges signals converging on the ER and initial phagophore formation.Citation53
Apart from mitochondria, the ER forms numerous membrane contact sites with other organelles (endosomes, lysosomes, Golgi) as well as the plasma membrane.Citation65 These contact sites might allow the formation of Ca2+ microdomains and the generation of complex Ca2+ signals influencing organelle function. Considering that lysosomal Ca2+ is an important regulator of autophagy the bidirectional Ca2+ signaling between the ER and lysosomes at the ER-lysosome contact sites could have a great impact on autophagy.Citation66,67 Interestingly, a recent study revealed an important role of ITPRs in lysosome calcium refilling. More specifically, antagonists of ER ITPRs rapidly and completely blocked Ca2+ refilling of lysosomes and caused lysosomal dysfunction and a lysosomal storage disease-like phenotype.Citation68
Taken together, these diverse results indicate a complex action of the ITPR in autophagy regulation. Apparently, ITPRs can regulate autophagy at different stages (initiation as well as fusion of autophagosomes with lysosomes), and the final effect of ITPRs on autophagy depends on multiple factors, including cellular state (e.g., starved or fed), spatiotemporal characteristics of ITPR-related calcium signals (e.g., mitochondrial, lysosomal or cytoplasmic localization), regulation by associated proteins, and others.
RYR (ryanodine receptor)
The role of another major mediator of ER Ca2+-release, RYR (ryanodine receptor), in the regulation of autophagy is much less studied compared to ITPR. Similar to ITPR, RYR is also involved in Ca2+-transfer from the sarcoplasmic reticulum (SR) to mitochondria in the heart.Citation69 Moreover, apart from SR, RYR also localizes on mitochondria, where it plays an essential role in mitochondrial Ca2+ uptake.Citation70 Overexpression of RYR1 (ryanodine receptor 1) enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts.Citation71 The results of these studies indirectly suggest that RYR could be potentially involved in the regulation of basal/induced autophagy via mechanisms similar to ITPR. In line with this, depletion of cardiac RYR2 (ryanodine receptor 2) leads to a reduction in mitochondrial Ca2+ and oxidative metabolism, and increased autophagy in cardiomyocytes.Citation72 The inhibitory action of RYR2 on autophagy was also indirectly demonstrated by Yuan et al. in their study of the mechanistic role of FKBP1B/Calstabin2 (an inhibitory component of the cardiac RYR macromolecular complex, that binds to RYR and stabilizes its closed state) in the process of cardiac aging.Citation73 The authors demonstrated that Fkbp1b deletion (which leads to RYR channel leakage) results in activation of MTOR and inhibition of autophagy in the cardiomyocytes of mice.Citation73 Further, the RYR antagonist dantrolene reduces LC3-II levels in the brain of neuronopathic Gaucher disease mice exhibiting abnormal autophagy.Citation74 In accordance with this, a very recent study showed that basal RYR3 activity suppresses autophagic flux at the level of lysosomes. Specifically, pharmacological inhibition of endogenous (in C2C12 myoblasts and primary hippocampal neurons) or ectopically expressed (in HEK293 cells) RYRs augments autophagic flux by increasing lysosomal turnover.Citation75
Interestingly, RYR3 (ryanodine receptor 3) colocalizes with lysosomal markers in rat pulmonary arterial smooth muscle, suggesting that lysosomes form junctions with SR to support Ca2+ signaling between 2 organelles.Citation76 However, although Ca2+ is essential for lysosomal fusion events, high levels of Ca2+ suppress fusion, suggesting that tight regulation of Ca2+ concentrations is of crucial importance for effective fusion of autophagosomes with lysosomes and subsequent autophagosomal cargo degradation. Citation77,78 In contrast to the inhibitory action of RYRs on autophagy discussed above, proautophagic function of RYR3 in hippocampal neural stem cells has recently been reported.Citation79 It was demonstrated that pharmacological inhibition or genetic ablation of RYR reduces, whereas the RYR agonist caffeine enhances, autophagy induced by insulin withdrawal.Citation79 In accordance with this, the myocardium of RYR2-deficient mice displays less expression of autophagic markers following pressure overload than their wild-type counterparts.Citation80 Interestingly, in contrast to ITPR, overexpression of RYR2 in ITPR triple knockout chicken B lymphocytes is without effect on constitutive autophagy.Citation55 Further, ITPRs but not RYRs have recently been shown to be essential for calcium refilling of lysosomes.Citation68 These data suggest that RYR and ITPR are not interchangeable when it comes to their effect on autophagy and lysosomes, even though both receptors mediate calcium release from the ER.
Overall, the existing data indicates that similar to ITPRs, RYRs can regulate autophagy at different stages (early as well as late stages). Nevertheless, despite apparent similarities between RYRs and ITPRs both receptors differentially influence autophagy. The most likely reasons of this are differential localization in distinct microdomains as well as different mechanisms of activation and regulation, which taken together determine the spatiotemporal and amplitude characteristics of the corresponding calcium signals.
MCU (mitochondrial calcium uniporter)
The MCU complex is comprised of a Ca2+ channel-forming subunit (MCU itself) as well as regulatory subunits.Citation81 MCU is located on the inner mitochondrial membrane and is responsible for Ca2+ uptake into the matrix.Citation60,82 By controlling the mitochondrial Ca2+ concentration, MCU plays a critical role in the regulation of aerobic metabolism and cell survival.Citation83,84 Given that MCU controls ATP production, the discovery of its role in autophagy was just a matter of time. In 2010, Cardenas et al. showed that the uniporter inhibitor Ru360 inhibited cell O2 consumption rate, activated AMPK, and induced MTOR-independent autophagy.Citation55 Later on, the role of MCUR1 (mitochondrial calcium uniporter regulator 1) in autophagy was revealed. MCUR1 is an integral membrane protein that binds to MCU and is required for MCU-dependent mitochondrial Ca2+ uptake.Citation85 MCUR1 knockdown (similar to MCU knockdown) abrogates mitochondrial Ca2+ uptake, disrupts oxidative phosphorylation, lowers cellular ATP and activates AMPK-dependent prosurvival autophagy in HEK293T and HeLa cells.Citation85
The important role of MCU and MCUR1 in mitochondrial bioenergetics and autophagy has been recently confirmed by Tomar et al.Citation86 The authors reported that mouse cardiomyocytes and endothelial cells lacking MCUR1 have severely impaired mitochondrial Ca2+ uptake and current through MCU. Loss of MCU or MCUR1 perturbs the MCU heterooligomeric complex, impairs mitochondrial bioenergetics, cell proliferation and migration, and elicits AMPK-dependent autophagy.Citation86 Besides mammalian cells, the involvement of MCU in the regulation of cell bioenergetics and autophagy in Trypanosoma brucei has been recently reported.Citation87 The authors demonstrated that ablation of Trypanosoma brucei MCU in RNAi or conditional knockout trypanosomes reduces mitochondrial Ca2+ uptake, increases the AMP:ATP ratio, stimulates autophagosome formation and produces marked defects in growth in vitro and infectivity in mice.Citation87
Taken together, available data suggest that MCU negatively regulates basal autophagy through the regulation of ATP production and, consequently, AMPK activation status.
Voltage-gated calcium channels
Voltage-gated calcium channels (VGCCs) mediate Ca2+ entry into cells in response to membrane depolarization.Citation88 Based on their activation threshold, they can be divided into 2 subgroups: high-voltage-activated (including L-, P/Q-, N- and R-types) and low-voltage-activated (T-type) channels.Citation89 VGCC regulate a variety of physiological processes, such as contraction, secretion, gene expression, synaptic transmission and many others.Citation88 Several reports have connected VGCC to autophagy regulation. Williams et al. reported that L-type Ca2+ channel antagonists induce autophagy via an MTOR-independent pathway.Citation90 Conversely, the L-type Ca2+ channel agonist (±)-BAYK8644 inhibits autophagy. The authors provided evidence that elevated cytosolic Ca2+ can activate CAPNs/calpains, a family of Ca2+-dependent cysteine proteases, which cleave and activate the α-subunit of heterotrimeric G proteins, GNAS/Gsα, leading to ADCY/adenylyl cyclase activation and increase in 3′-5′-cyclic adenosine monophosphate (cAMP) levels. The rise in cAMP levels positively regulate InsP3 production via a cAMP-activated pathway involving RAPGEF3/EPAC (Rap guanine nucleotide exchange factor 3), the small G protein RAP2B, and PLCE1 (phospholipase C epsilon 1). InsP3, in turn, activates ITPR resulting in elevated cytosolic Ca2+ levels, which again activate CAPNs/calpains, thus creating a potential positive feedback loop for autophagy inhibition.Citation90
In line with this, fluspirilene (an antagonist of different types of VGCC) induces autophagy via inhibition of CAPN1-mediated cleavage of ATG5.Citation91 Several other studies confirmed the effects of VGCC blockers on autophagy in different cell types. More specifically, isradipine (an L-type calcium channel blocker) improves autophagy function in the brain;Citation92 the T-type calcium channel blocker, KYS05090 induces autophagy and apoptosis in human lung adenocarcinoma A549 cells;Citation93 verapamil (an L-type calcium channel blocker) suppresses obesity-induced accumulation of protein aggregates and restores lysosomal degradation of autophagosomes in the liver of obese mice.Citation77 Interestingly, in contrast to all these data, verapamil treatment has been reported to be without effect on autophagy in myotubes,Citation94 whereas T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells.Citation95
Considering these conflicting results, it should be noted, that the effects of calcium channel blockers on autophagy could not be related only to inhibition of calcium channel function, and nonspecific effects cannot be excluded. For example, different L-type channel antagonists, including nimodipine, nifedipine, verapamil, and diltiazem have been previously reported to inhibit glucose transport through mechanisms other than the antagonism of calcium channels.Citation96 Moreover, a number of these blockers possess weak base properties and thus could have a direct impact on lysosomal function. For example, verapamil passively diffuses into the lysosome, where it becomes protonated and could cause an increase in lysosomal pH.Citation97 Therefore, the effects of channel blockers on autophagy should be compared to the effects induced by channel knockdown/knockout.
Recently, it was demonstrated that L-type and T-type calcium channel blockers (nifedipine and mibefradil, respectively) differentially regulate autophagy in cardiomyocytes.Citation98 Specifically, nifedipine triggers the activation of autophagy, whereas mibefradil inhibits constitutive autophagy. The effect of mibefradil is mimicked by siRNA-mediated knockdown of T-type CACNA1H/Cav3.2 (calcium voltage-gated channel subunit alpha1 H) but not CACNA1G/Cav3.1 (calcium voltage-gated channel subunit alpha1 G) channels. In contrast, silencing of L-type CACNA1C/Cav1.2 (calcium voltage-gated channel subunit alpha1 C) and CACNA1D/Cav1.3 (calcium voltage-gated channel subunit alpha1 D) channels results in accumulation of both LC3-II and SQSTM1/p62 (sequestosome 1), contrasting with the effect of nifedipine.Citation98 One of the possible explanation for this discrepancy is related to the fact that nifedipine (which is known for its L-type-channel specific action) is capable of blocking T-type channels as well.Citation99 Recently, the role of intracellular VGCC in autophagy has been revealed. Tian et al. reported that the P/Q-type calcium channel CACNA1A/Cav2.1 (calcium voltage-gated channel subunit alpha1 A) is localized to the lysosomes and loss of lysosomal CACNA1A in cerebellar cultured neurons leads to a failure of lysosomes to fuse with endosomes and autophagosomes.Citation100 The authors showed that it is lysosomal CACNA1A that is required for lysosomal fusion and not plasma membrane resident CACNA1A.Citation100
Overall, the specific roles of VGCCs in autophagy continue to be unclear with potential involvement in both early and late stages of autophagy. Most of the existing reports converge in that L-type calcium channel inhibitors induce accumulation of autophagic markers. Interestingly, in contrast to L-type channels, several blockers of T-type channels inhibit autophagy. However, nonspecific effects reported as well as chemical properties suggest that all these inhibitors could potentially regulate autophagy in a calcium-independent manner. This hypothesis is further supported by the fact that the action of VGCC inhibitors on autophagy is not limited to excitable cells, and along with neurons and muscle cells could also be observed in fibroblasts, hepatocytes as well as various cancer cell lines. Therefore, further studies should primarily implement a channel knockdown/knockout approach rather than a pharmacological approach to assess the involvement of different VGCCs in the regulation of autophagy.
TPCNs (2 pore segment channels)
TPCNs/TPCs are voltage-gated ion channels localized intracellularly to the endolysosomal system. Human TPCNs are represented by TPCN1 (2 pore segment channel 1) and TPCN2 (2 pore segment channel 2), with TPCN2 being specifically localized to lysosomes.Citation101 Although originally TPCNs were proposed as Ca2+ channels, some groups reported that under some experimental conditions TPCNs behave as Na+- or H+-selective channels.Citation102 Ample evidence indicates that TPCNs mediate endolysosomal calcium release in response to the elevation of the second messenger nicotinic acid adenine dinucleotide phosphate (NAADP).Citation103,104 The involvement of TPCNs in autophagy regulation has been revealed by studying the role of NAADP-mediated Ca2+ signals in autophagy.Citation105 NAADP increases acidic vesicular organelle formation and levels of the autophagic markers LC3-II and BECN1 in rat astrocytes, whereas expression of a dominant-negative TPCN2 construct substantially reduces NAADP-mediated increases in LC3-II levels.Citation105 The expression of other autophagy markers as well as autophagic flux were not assessed in this study, thus it is not clear whether LC3-II accumulation in this case is due to increased autophagosome production or inhibited degradation.
Further, LRRK2, mutation of which causes an autosomal dominant form of Parkinson disease, has been reported to activate autophagy through a calcium-dependent pathway involving NAADP, TPCN2, CAMKK2 and AMPK.Citation41 Interestingly, the authors also reported LRRK2-induced SQSTM1 accumulation and lysosome alkalization. The authors, however, suggested that the LRRK2-mediated effects on SQSTM1 levels may be due to Ca2+-dependent changes in protein synthesis and not due to late-stage autophagy arrest. Of note, LRRK2 overexpression has been previously shown to induce accumulation of multivesicular bodies and autophagosomes containing incompletely degraded material and SQSTM1.Citation106 Thus, it seems that LRRK2 not only stimulates autophagy via the AMPK pathway, but also can influence autophagic cargo degradation at the lysosomal level.
The NAADP-TPCN-AMPK (MTOR independent) axis has been also reported to mediate glutamate-induced accumulation of LC3-II.Citation107 Although the authors clearly show the activation of the AMPK pathway by glutamate, it is not clear that lysosomal function is not affected here. Lu et al. demonstrate that overexpression of TPCN2 in HeLa or mouse embryonic stem cells alkalinizes lysosomal pH and inhibits autophagosomal-lysosomal fusion, thereby resulting in the accumulation of autophagosomes.Citation108 In turn, inhibition of TPCN2 signaling either by TPCN2 knockdown or by treatment with the TPCN2 pharmacological inhibitor Ned-19, facilitates autophagosomal-lysosomal fusion and markedly decreases accumulation of autophagosomes.Citation108 Interestingly, animals lacking TPCN2 demonstrate an enhanced autophagic flux accompanied by reduced AKT (AKT serine/threonine kinase) and MTOR activity. Moreover, MTOR reactivation during prolonged starvation is delayed in the absence of TPCN2 suggesting a role for TPCN2 in MTOR reactivation and autophagy termination.Citation109
Further, both TPCN1 and TPCN2 are essential for appropriate basal and induced autophagic flux in cardiomyocytes. However, in contrast to the studies discussed above, silencing of either TPCN channel in cardiomyocytes provokes lysosomal and autophagic alterations manifested in the accumulation of both LC3-II and SQSTM1 proteins under severe energy-depletion conditions.Citation110
Taken together, accumulated data suggest that TPCNs are important regulators of autophagy in different cell types. Their endolysosomal localization implies that these channels could primarily regulate late stages (fusion of autophagosomes with endosomes/lysosomes and subsequent degradation) of autophagy. Nevertheless, available data indicate that TPCN channels can control the activation status of AMPK and MTOR, and as such influence autophagy initiation and termination during prolonged starvation.
Transient receptor potential channels
The mammalian TRP (transient receptor potential) channel superfamily encompasses 28 members with diverse physiological functions and cellular distributions.Citation111,112 Based on their structural homology TRP channels are grouped into 6 subfamilies (TRPC/canonical, TRPM/melastatin, TRPV/vanilloid, TRPA/ankyrin-like, PKD2/TRPP/polycystin, MCOLN/TRPML/mucolipin). TRP channels are nonselective Ca2+-permeable cation channels, with the exception of TRPM4 and TRPM5, which are calcium-impermeable. TRP channels are activated by a wide range of stimuli (including mechanical force, osmotic stress, temperature changes, extra- and intracellular messengers, and many others), can be localized on the plasma membrane or in intracellular membranes, and are involved in numerous fundamental cell functions.Citation111,112 Current evidence supports the idea that TRP channels are also implicated in the regulation of autophagy.
MCOLN/TRPML channels. MCOLN1/TRPML1 (mucolipin 1) represents the most studied TRP channel with regard to the regulation of autophagy. MCOLN1 is ubiquitously expressed and primarily localizes on the membranes of late endosomes and lysosomes.Citation113 The main physiological function of the MCOLN1 channel is considered to be as a late endosomal/lysosomal Ca2+-release channel, although permeability to Na+, K+, Fe2+, Mn2+ and Zn2+ has also been reported.Citation114,115
Loss-of-function mutations in the human MCOLN1 gene are responsible for the severe neurodegenerative lysosomal storage disorder mucolipidosis type IV.Citation116,117 MCOLN1−./− cells from mucolipidosis type IV patients are characterized by an impairment in lysosomal pH; abnormal mitochondria; accumulation of enlarged endosomal/lysosomal compartments and autophagosomes; accumulation of undigested material, SQSTM1 and ubiquitin proteins, all of which suggest defective autophagic-lysosomal function.Citation113,118–121
A number of studies report the involvement of MCOLN1 in autophagy regulation. Accumulation of autophagosomes in MCOLN1-deficient fibroblasts from mucolipidosis type IV patients has been demonstrated by Vergarajauregui et al.Citation121 The authors showed that this accumulation is due to increased de novo autophagosome formation and to the delayed fusion of autophagosomes with late endosomes/lysosomes, suggesting that MCOLN1 is required for the efficient fusion of autophagosomes with lysosomes.Citation121 Further, it was reported that loss of MCOLN1 leads to the impairment in both chaperone-mediated autophagy and macroautophagy.Citation118,122 The authors suggest that the interaction between MCOLN1 and members of the lysosomal chaperone complex (namely, HSPA8/Hsc70 and DNAJ/Hsp40) may be required for proper CMA function. More specifically, MCOLN1 may act as a docking site for intralysosomal HSPA8 allowing it to more efficiently pull in substrates for CMA.Citation122 Later, the same group reported that macroautophagy is defective in MCOLN1-deficient mouse neurons.Citation118 These cells display bigger lysosomes as well as higher levels of autophagy markers LC3-II and SQSTM1 compared to wild-type cells, suggesting impairments in autophagosome-lysosome function.Citation118
Similar effects are observed in C. elegans harboring mutations in cup-5, encoding the C. elegans ortholog of mammalian MCOLN1. The authors report the accumulation of autophagy substrates in enlarged vacuoles in cup-5 mutants and suggested that CUP-5 is required for proteolytic degradation in autolysosomes.Citation123 Interestingly, the Drosophila Trpml/MCOLN1 channel is required for TORC1 activation.Citation124 Trpml-deficient flies display decreased torc1 signaling, upregulation of autophagy induction, and accumulation of large autophagic vacuoles. The authors demonstrated that Trpml, which is localized to the late-endosomal/lysosomal membranes, is required for fusion between amphisomes and lysosomes. Interestingly, torc1 exerts reciprocal control on Trpml function by regulating its subcellular localization. Specifically, activated torc1 diminishes endocytosis of Trpml, thereby promoting its plasma membrane localization.Citation124 Recently, Onyenwoke et al. confirmed that MCOLN1 is regulated by the TOR kinase. The authors demonstrated, however, that TOR kinase directly targets and inactivates the MCOLN1 channel and thereby functional autophagy, through phosphorylation.Citation125
The MCOLN1 channel has also been proposed to be essential for lysosomal adaptation to nutrient starvation.Citation126 Starvation, MTOR inhibition or activation of TFEB (transcription factor EB) lead to a significant upregulation of MCOLN1-mediated lysosomal calcium release. Genetic deletion or pharmacological inhibition of MCOLN1 completely abolishes the effects of starvation on boosting the degradation capability of lysosomes. Inversely, overexpression of MCOLN1 mimics the starvation effect.Citation126 In line with this, dominant-negative MCOLN1 channels were reported to inhibit starvation-induced autophagy by interacting with and affecting native MCOLN channel function.Citation127
Recently, Ballabio's group demonstrated that MCOLN1-mediated lysosomal calcium release controls the activity of TFEB (a master transcriptional regulator of lysosomal biogenesis and autophagy) and in this way regulates autophagy.Citation128 Starvation-induced lysosomal Ca2+ release through MCOLN1 activates PPP3/calcineurin, which in turn binds and dephosphorylates TFEB, thus promoting its nuclear translocation and autophagy upregulation. Silencing of MCOLN1 inhibits starvation-mediated autophagy. Interestingly, MCOLN1 is a direct transcriptional target of TFEB, suggesting the presence of a positive feedback loop by which TFEB regulates the expression of MCOLN1, which in turn promotes TFEB activity.Citation128 In a recent study, Zhang et al. proposed an MCOLN1-dependent mechanism by which reactive oxygen species (ROS) activate autophagy.Citation129 The authors demonstrated that ROS directly and specifically activate lysosomal MCOLN1 channels, inducing lysosomal Ca2+ release and subsequent TFEB activation. Genetic inactivation or pharmacological inhibition of MCOLN1 prevent the clearance of damaged mitochondria as well as removal of excess ROS, suggesting that MCOLN1 is an important player in oxidative stress-induced autophagy.Citation129
Along with MCOLN1, another member of the mucolipin family, MCOLN3/TRPML3 (mucolipin 3) is involved in autophagy regulation. As with MCOLN1, MCOLN3 localizes to late endosomes/lysosomes, although some reports suggest that MCOLN3 could also be found in early endosomes and the plasma membrane.Citation113 Gain-of-function mutations in the mouse MCOLN3 have been reported to result in the varitint-waddler phenotype with hearing and pigmentation defects.Citation130 Consistent with its intracellular localization, MCOLN3 has been reported to be an important regulator of endocytosis, membrane trafficking and autophagy.Citation131 Specifically, overexpression of MCOLN3 leads to increased autophagy in HeLa cells, whereas channel silencing inhibits autophagy.Citation131 Interestingly, MCOLN3 has been proposed to be recruited to phagophores or autophagosomes upon induction of autophagy. The authors speculate that MCOLN3-mediated calcium efflux is required for fusion and fission events in autophagy.Citation131 Recently, it has been reported that MCOLN3 specifically interacts with the mammalian Atg8 homolog GABARAPL2 to regulate autophagosome maturation. However, despite the direct binding of MCOLN3 to GABARAPL2, GABARAPL2 is not required for the presence of MCOLN3 in autophagosome membranes.Citation132 Finally, heteromultimerization of MCOLN channels (MCOLN1, 2, 3) influences lysosomal integrity and autophagy, suggesting that interaction between these channels is a determining factor that should be taken into account when assessing the role of a particular MCOLN channel in autophagy.Citation127
The role of another member of the MCOLN family, MCOLN2/TRPML2 (mucolipin 2) in autophagy is not clear. Despite its endolysosomal localization and the fact that MCOLN2 knockdown cells exhibit lysosomal inclusions,Citation133 MCOLN2 (unlike MCOLN3) does not appear to play an active role in starvation-induced autophagy.Citation127 Further studies are needed to clarify the specific role of MCOLN2 in the regulation of autophagy.
Overall, ample evidence suggests that MCOLN1 channel is a positive regulator of autophagy. Due to its endolysosomal localization MCOLN1 regulates fusion of autophagosomes with late endosomes/lysosomes, and lysosomal function as well as lysosomal adaptation to nutrient starvation. However, MCOLN1 also influences autophagosome biogenesis by controlling the activation status of TFEB.
MCOLN3 has been also proposed to regulate both autophagosome production and fusion with lysosomes. To our knowledge, at present the MCOLN3 channel represents the only ion channel that has been reported to colocalize with autophagosomal markers.
TRPM channels. In addition to its role as a plasma membrane channel, TRPM2 (transient receptor potential cation channel subfamily M member 2) also functions as an endolysosomal Ca2+-release channel activated by numerous stimuli including adenosine diphosphoribose, hydrogen peroxide (H2O2), NAADP, pH and others.Citation134 Due to the presence of enzymatically active adenosine diphosphoribose hydrolase domain in its C terminus, TRPM2 can function as both ion channel and enzyme (so called chanzyme), and contributes to various cellular functions that include cytokine production, insulin release, cell motility and cell death.Citation135 Several studies suggest that the TRPM2 channel is involved in the regulation of oxidative stress-induced autophagy. For example, the nuclear enzyme PARP1 (poly[ADP-ribose] polymerase 1) influences autophagy via TRPM2 channels located in the plasma membrane.Citation136 The authors showed that activated PARP1 (which activates TPRM2-mediated calcium influx) operates as an autophagy suppressor after oxidative stress, thus suggesting the inhibitory action of plasmalemmal TRPM2 toward autophagy.Citation136 In line with this, Wang et al. reported that plasmalemmal TRPM2 inhibits oxidative stress-induced autophagy via activation of CAMK2 (calcium/calmodulin dependent protein kinase II) and subsequent phosphorylation of BECN1 on Ser295. Phosphorylated BECN1 subsequently dissociates from PIK3C3 to bind with BCL2, and thereby inhibits autophagy.Citation137 Overall, the available data demonstrate that plasmalemmal TRPM2 negatively regulates oxidative stress-induced autophagy at an early stage (initiation).
In contrast to TRPM2, another member of TRPM family, TRPM3 (transient receptor potential cation channel subfamily M member 3), has been demonstrated to be an important autophagy stimulator in clear cell renal cell carcinoma (ccRCC). Increased expression of TRPM3 in ccRCC leads to Ca2+ influx, activation of CAMKK2, AMPK, and ULK1, and phagophore formation. Moreover, TRPM3-mediated Ca2+ and Zn2+ fluxes inhibit MIR214, which directly targets LC3A and LC3B.Citation138 Thus, TRPM3 positively regulates autophagy on the level of phagophore formation and indirectly controls phagophore elongation via regulation of expression levels of LC3 proteins.
TRPM7 (transient receptor potential cation channel subfamily M member 7) is a ubiquitous ion channel with enzyme activity, harboring a Ser/Thr kinase domain at its carboxyl terminus.Citation139 TRPM7 plays a prominent role in cellular and systemic magnesium homeostasis and contribute to several fundamental physiological processes.Citation140–Citation142 The role of TRPM7 in the regulation of basal autophagy has been recently revealed by Oh et al. The authors demonstrated that TRPM7 stimulates basal autophagy via the CAMKK2-AMPK pathway, whereas channel silencing by shRNA attenuates basal autophagy in SH-SY5Y cells.Citation143 This study suggests that TRPM7 represents a positive regulator of autophagy at an early stage.
TRPV channels. Several members of the vanilloid subfamily of TRP channels have also been implicated in the regulation of autophagy. For example, TRPV1 (transient receptor potential cation channel subfamily V member 1), which was originally identified in sensory neurons as a heat-activated ion channel,Citation144 has been proposed to regulate autophagy in thymocytes.Citation145 The authors demonstrated that capsaicin, an activator of TRPV1, induces BECN1-dependent autophagy in murine thymocytes via ROS-regulated AMPK and ATG4C (autophagy related 4C cysteine peptidase) pathways. The effect of capsaicin is antagonized by the TRPV1 inhibitor capsazepine as well as by compound C, an AMPK inhibitor.Citation145 Of note, the ATG4 family of cysteine proteases serve to cleave LC3-I near the C terminus, thus allowing its subsequent conjugation to phosphatidylethanolamine (PE) and incorporation into the phagophore membrane.Citation146 Considering that LC3 plays an essential role in phagophore membrane expansion, and closure as well as fusion of the autophagosome with a lysosome, we can conclude that TRPV1 could be implicated not only in initiation of autophagy, but also in phagophore membrane expansion, and closure and probably fusion with lysosomes.
The TRPV1-AMPK signaling cascade has also been implicated in capsaicin-induced autophagy in vascular smooth muscle cells as well as in pressure overload- and cold exposure-induced cardiac autophagy.Citation147,148 In line with this, dietary capsaicin has been reported to increase the expression of autophagy-related proteins LC3-II, BECN1, ATG5, and ATG7 in HepG2 liver carcinoma cells via TRPV1-mediated activation of PPARD/PPARδ (peroxisome proliferator activated receptor delta). In vivo studies confirmed that chronic dietary capsaicin increases hepatic autophagy in wild-type mice, whereas in trpv1-null mice capsaicin is without effect on autophagy, thus suggesting the critical involvement of TRPV1 in capsaicin induced-autophagy.Citation149 Further, it has been suggested that capsaicin induces autophagy in osteosarcoma cells via activation of ER-localized TRPV1.Citation150 In astrocytes, however, evodiamine has been reported to induce autophagy mediated by TRPV1 localized on the plasma membrane. The authors speculated that evodiamine-induced autophagy may occur through a TRPV1-Ca2+-JUN/c-Jun pathway.Citation151 Interestingly, Ahn et al. reported that TRPV1 is degraded by autophagy,Citation152 suggesting the existence of a reciprocal control of TRPV1 and autophagy.
The involvement of another heat-activated channel, TRPV2 (transient receptor potential cation channel subfamily V member 2), in autophagy regulation has been recently demonstrated. Cannabidiol induces autophagy in glioma stem-like cells by activating TRPV2. Considering that cannabidiol treatment results in AKT inhibition, the authors speculated that cannabidiol regulates autophagy trough inhibition of MTOR.Citation153 Further, the TRPV4 channel has been proposed to inhibit rat hepatic stellate cell apoptosis through autophagy induction, presumably through the AKT pathway.Citation154
Taken together, the available evidence suggests that the TRPV1, TRPV2 and TRPV4 channels positively regulate autophagy.
TRPC channels. Recently, prosurvival and proautophagic functions of TRPC1 (transient receptor potential cation channel subfamily C member 1) in salivary gland cells and neuroblastic cells have been revealed. Specifically, Sukumaran and coauthors reported that TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia- and nutrient depletion-dependent autophagy.Citation155 The mechanism of such regulation, however, was not addressed in this study. In line with this work, TRPC1 silencing has been reported to significantly reduce hypoxia-induced LC3-II accumulation in breast cancer cell lines, however, the mechanism of this regulation remains unexplored.Citation156
Upregulation of TRPC4 (transient receptor potential cation channel subfamily C member 4) has been demonstrated to induce autophagy in vascular endothelial cells. Mechanistic studies revealed that the upregulation of TRPC4 increases the intracellular Ca2+ concentration, which, in turn, activates the Ca2+-CAMKK2-AMPK pathway, leading to MTOR inhibition and autophagy.Citation157 Thus, TRPC1 and TRPC4 channels function as positive regulators of autophagy.
TRPP channels. The stimulatory action of PKD2/TRPP2 (polycystin 2, transient receptor potential cation channel) toward autophagy has been recently reported by the group of P. Codogno.Citation158 PKD2, the ion channel frequently mutated in autosomal dominant polycystic kidney disease, has been previously shown to regulate calcium influx at the primary cilium.Citation159 Downregulation of PKD2 impairs the activation of autophagy in response to fluid flow. The authors hypothesized that PKD2 mediates the MTOR-independent early autophagic response most probably in a complex with PKD1/TRPP1 (polycystin 1, transient receptor potential channel interacting), an integral membrane protein that regulates the calcium channel function of PKD2.Citation158 Interestingly, PKD1 has been previously demonstrated to negatively regulate PKD2 expression via the aggresome-autophagosome pathway.Citation160 Specifically, PKD1 upregulation leads to increased degradation of PKD2 via autophagy. Of note, overexpression of PKD1 is without effect on autophagy in general.Citation160
Store-operated calcium channels
The store-operated calcium (SOC) channels represent one of the major calcium-entry pathways in nonexcitable cells and are widely distributed in various cell types. As predicted from their name, activation of SOC channels is controlled by the filling state of intracellular calcium stores, namely the ER. SOC channels reside on the plasma membrane and are activated by the depletion of ER Ca2+ stores to provide Ca2+ for ER store refilling as well as for signaling purposes.Citation161 The major players in store-operated calcium entry (SOCE) are the ER calcium sensor STIM1 (stromal interaction molecule 1) and the plasma membrane ORAI1 protein, which forms the Ca2+ selective pore of the SOC channel.Citation162,163 Depletion of Ca2+ from the ER causes STIM1 to translocate to the plasma membrane, where it binds to and activates ORAI1 leading to calcium entry.Citation161 SOC channels are implicated in a number of physiological and pathological processes including gene expression, proliferation, apoptosis, cell migration, invasion and others.Citation161
The role of SOC channels in the regulation of autophagy remains poorly understood. Abdelmohsen et al. reported that ORAI1 regulates MIR519-induced autophagy. The authors showed that MIR519 reduces the expression of several Ca2+ transport proteins (including ORAI1) resulting in aberrant elevation of cytosolic Ca2+ levels and autophagy induction. Overexpression of ORAI1 blunts the MIR519-mediated increase in LC3-II levels, suggesting an inhibitory role for ORAI1 in MIR519-induced autophagy.Citation164 This role of ORAI1 has been confirmed in a very recent study showing that ORAI1 negatively regulates 5-fluorouracil (5-FU)-induced autophagy in hepatocarcinoma cells.Citation165 The authors demonstrated that knockdown of ORAI1 or pharmacological inhibition of SOCE enhances 5-FU-induced inhibition of phosphoinositide 3-kinase-AKT-MTOR pathway and potentiates 5-FU-activated autophagy and autophagic cell death. In contrast, ectopic overexpression of ORAI1 antagonizes 5-FU-induced autophagy.Citation165 In line with these studies, inhibition of SOCE by SKF-96365 has been recently proposed to stimulate autophagy in cervical cancer cell lines.Citation166 Further, it has been demonstrated that resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 (and a decrease in SOCE) and the MTOR pathway. Treatment of prostate cancer cells with SKF-96365 results in increases in LC3-II levels, inhibition of the AKT-MTOR pathway and decreases cell proliferation/survival.Citation167 Interestingly, the same group previously reported that SKF-96365 efficiently inhibits autophagy induced by the hypoxia-mimetic agents DMOG and DFO in human salivary gland and neuroblastic cells. In this study the authors attributed this effect to the inhibition of the TRPC1 channel by SKF-96365.Citation155 It should be noted, that besides SOCE, SKF-96365 has been reported to inhibit TRPC channels, voltage-gated Ca2+ channels and others.Citation168 Thus, additional experiments using more specific antagonists as well as siRNA-mediated knockdown are needed.
Recently, we reported that a SOCE inhibitor, ML-9, stimulates autophagosome formation (by downregulating the AKT-MTOR pathway) and inhibits autophagosome degradation (by increasing lysosomal pH). We demonstrated that these effects were SOCE and STIM1 independent, as neither the SOCE inhibitor BTP2 nor STIM1 silencing by siRNA induce accumulation of autophagic markers in prostate cancer cells.Citation169
Several studies suggested that SOC channels can regulate AMPK activity under hypoxic conditions. It has been proposed that hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of SOC channels, leading to increases in cytosolic calcium and subsequent activation of the CAMKK2-AMPK pathway.Citation170,171 The impact of SOC channel activation on autophagy was not assessed in these studies, however. Finally, a very recent study demonstrated that SOCE positively regulates autophagy in endothelial progenitor cells via the CAMKK2-MTOR pathway. The authors showed that either Ca2+ chelators or deficiency in STIM1 attenuate oxidized low-density lipoprotein (ox-LDL)-induced autophagy activation, confirming the involvement of SOCE in the process.Citation172
Taken together, the available data suggest that apparently SOC channels can differentially influence autophagy depending on cell type and autophagy inducers. Considering the conflicting results from different groups, further studies should investigate the role of SOC channels in autophagy by using a knockdown/knockout approach and not by using pharmacological SOCE inhibitors, which are often highly nonspecific.
Sodium channels in the regulation of autophagy
Sodium channels represent integral membrane proteins that mediate sodium influx into the cell or intracellular organelles. According to the gating mechanism, sodium channels can be classified into voltage-gated sodium channels (VGSC) and voltage-independent sodium channels comprising SCNN1/ENaC (sodium channel epithelial 1), ASICs (acid sensing ion channels) and NALCN (sodium leak channel, nonselective).Citation173–Citation176 Despite understanding the important role Na+ ions play in cell physiology, data on the involvement of sodium channels in autophagy regulation are virtually absent. Inhibition of ASICs by paeoniflorin or amiloride has been reported to enhance the autophagic degradation of SNCA/α-synuclein in rat pheochromocytoma PC12 cells.Citation177 However, additional experiments using more specific ASIC channel inhibitors as well as channel knockdown/knockout are needed to confirm the role of ASIC channels in autophagy regulation.
A recent study identified the lysosomal ATP-sensitive 2-pore Na+ channel (lysoNaATP) as an important regulator of endolysosomal function.Citation178 The authors reported that the novel channel represents a complex formed by the 2-pore channels TPCN1 and TPCN2 (discussed above as calcium-permeable channels). LysoNaATP controls the lysosome membrane potential, pH stability, and amino acid homeostasis. Interestingly, although there is a significant shift of lysosomal pH toward alkalinization in mutant mice (lacking lysoNaATP) during starvation stress, no gross defects in autophagy in mutant mice have been reported.Citation178
It should be noted that nonselective cation channels, e.g. the members of TRP channels family, are also permeable for Na+ ions, suggesting that these channels could potentially regulate autophagy by Na+-dependent mechanisms.
Potassium channels in the regulation of autophagy
Potassium channels mediate the flow of potassium ions down their electrochemical gradient. K+ channels are widely distributed in a vast variety of cell types and are involved in a plethora of physiological processes including maintenance of membrane potential, cell proliferation and apoptosis. Based on structural criteria, conductance properties as well as activation mechanisms, potassium channels are divided into 4 classes: voltage-gated, calcium-activated, inward-rectifier and 2-pore-domain potassium channels.Citation179
Several studies suggested that potassium ions as well as potassium channels could be implicated in autophagy regulation. Perturbation of intracellular K+ homeostasis with the K+-selective ionophore valinomycin promotes autophagy in several cell types.Citation180 The authors, however, showed that valinomycin induces mitochondrial swelling and hypothesized that autophagy is activated in response to mitochondrial damage, and no direct role of K+ in the regulation of autophagy has been proposed.Citation180 Consistent with these data, another potassium ionophore, salinomycin, has been recently demonstrated to induce autophagy through generation of ROS in osteosarcoma U2OS cells. The role of K+ ions in this process was not assessed in this study, however.Citation181
Several potassium channels have been proposed to regulate autophagy. Opening of ATP-sensitive K+ (K+ATP) channels has been reported to induce autophagy in different cell types.Citation90,182 Williams et al. reported that minoxidil, a K+ATP channel agonist, enhances autophagy in PC12 cells, whereas K+ATP channel blockers, such as quinine sulphate and tolazamide slow down clearance of autophagy substrates.Citation90 In line with this, the mitochondrial K+ATP channel inhibitor 5-hydroxydecanoate inhibits angiotensin II-induced autophagy in vascular smooth muscle cells.Citation182 Recently, an involvement of the KCNH7/Kv11.3/HERG3 (potassium voltage-gated channel subfamily H member 7) plasma membrane potassium channel in the regulation of autophagy and senescence in melanoma has been suggested.Citation183 More specifically, pharmacological stimulation of the KCNH7 channel with a small molecule activator, NS1643, induces autophagy via activation of an AMPK-dependent signaling pathway in a melanoma cell line.Citation183 Another potassium channel that has been reported to contribute to autophagy regulation is the recently-identified endolysosomal TMEM175 (transmembrane protein 175).Citation184 Cang et al. reported that TMEM175 regulates endolysosomal function, membrane potential, and pH stability, as well as fusion with autophagosomes during autophagy. Lysosomes lacking TMEM175 exhibit no K+ conductance, have a markedly depolarized membrane potential, have compromised lumenal pH stability and abnormal fusion with autophagosomes during autophagy.Citation184
Overall, the data on the role of potassium channels in autophagy are scarce. Nevertheless, available evidence suggests that both plasmalemmal and intracellular potassium channels act as positive regulators of autophagy in different cell types.
Chloride channels in the regulation of autophagy
Chloride channels mediate the flow of chloride ions across cell membranes and reside both in the plasma membrane and in intracellular organelles. Chloride channels are ubiquitously expressed and contribute to a variety of fundamental cellular processes including apoptosis, volume regulation, cell cycle, acidification of intracellular organelles, and others.Citation185 According to the gating mechanisms, chloride channels can be classified into CLCN/CLC (chloride voltage-gated channel), CFTR (cystic fibrosis transmembrane conductance regulator), calcium-activated chloride channels, volume regulated anion channel LRRC8/VRAC/VSOR (leucine rich repeat containing 8 VRAC), and ligand-gated anion channels.Citation185
In fact, Cl− channels exhibit very low selectivity among anions and allow various anions to permeate. However, considering the fact that Cl− is the predominant anion in all organisms it is widely accepted that Cl− constitutes the prevalent ion passing through chloride channels.Citation186
The data on the role of chloride channels in the regulation of autophagy are scarce.
siRNA-mediated knockdown of CLIC4/mtCLIC (chloride intracellular channel 4) has been reported to enhance starvation-induced BECN1-dependent autophagy and apoptosis in human glioma U251 cells.Citation187
CFTR channel is yet another chloride channel that has been proposed to regulate autophagy. Loss-of-function mutations in the human CFTR gene cause the autosomal recessive lethal disease cystic fibrosis. Interestingly, accumulation of misfolded protein aggregates is frequently observed in airways of patients with cystic fibrosis, suggesting defective autophagic function. In accordance with this, Luciani et al. demonstrated that defective CFTR results in defective autophagy leading to the accumulation of misfolded proteins and protein aggregates.Citation188 More specifically, defects in CFTR function (or CFTR knockdown) result in upregulation of ROS and TGM2/TG2 (transglutaminase 2) leading to crosslinking of BECN1, sequestration of PtdIns3K complex III and accumulation of SQSTM1.Citation188 These results suggest that CFTR positively regulates autophagy. In line with this, CFTR has recently been shown to promote BECN1-dependent autophagosome formation both in normal and stress states in tumor cells.Citation189
Recently, volume-sensitive outwardly rectifying (LRRC8/VSOR) chloride channel has been shown to promote high glucose-induced apoptosis of cardiomyocytes through inhibition of autophagy. The authors demonstrated that LRRC8/VSOR channel blockers induce autophagy via MTOR inhibition.Citation190
Finally, 2 members of the CLCN family of chloride channels, namely CLCN3/CLC-3 and CLCN7/CLC-7, have been reported to contribute to autophagy regulation. It was demonstrated that loss of CLCN7 leads to lysosomal storage disease and an increase in LC3-II levels.Citation191 These results are in line with the previously proposed role of CLCN7 in control of lysosomal acidification.Citation192 Similarly, the CLCN3 channel also resides mainly on endosomal compartmentsCitation193 and positively regulates autophagy.Citation194 It should be noted, however, that both CLCN3 and CLCN7 function as Cl−/H+ exchangers rather than voltage-dependent anion channels.Citation186
Taken together, the available data suggest that chloride channels can regulate autophagy at both early and late stages.
Conclusions
Since the identification of autophagy as a crucial player in cell physiology, the autophagic machinery has been studied in great detail and a variety of molecular components of this machinery has been revealed. Furthermore, autophagy appears to be tightly regulated by a number of intracellular signaling pathways and molecules.
Recently, cytoplasmic and extracellular ion concentrations as well as ion channels, mediating ion fluxes across cellular membranes, have emerged as important regulators of both basal and induced autophagy. Interestingly, accumulated data prove that both ion channel dysfunctions and deregulation of autophagy are implicated in various diseases, making it possible to consider ion channels and autophagy as potential therapeutic targets. Indeed, a number of clinical trials are currently evaluating the efficacy of autophagy modulators in the treatment of different diseases, including neurodegenerative diseases and cancer. In addition, several ion channel modulators are already in clinical use or are being tested in clinical trials.
The identification of ion channels as autophagy regulators opens new ways for targeting autophagy in disease treatment. In cancer, for example, an altered expression of ion channels has been linked to a number of cancer-related processes, including autophagy. For instance, targeting of the oncogenic TRPM3 channel (which is overexpressed in clear cell renal cell carcinoma and positively regulates autophagy) could potentially be used to modulate autophagy and tumor growth.Citation138
Conversely, stimulation or inhibition of “selective autophagy of ion channels” (which could be called “channelophagy”) could provide a new tool for regulation of ion channel cellular expression that could potentially have therapeutic implications.
Accumulated data suggest that ion channels are implicated in different stages of autophagy, including initiation of phagophore formation, and phagophore elongation as well as autophagosome-lysosome fusion. Here, apparently, channel localization matters. Autophagosome-lysosome fusion is mainly regulated by endolysosomal ion channels (e.g., chloride channels CLCN3 and CLCN7; the potassium channel TMEM175; the calcium channels CACNA1A, TPCNs, MCOLN1, MCOLN3, and TRPM2), and autophagosomal ion channels (presumably, MCOLN3), but also by nonlysosomal ion channels localized to contact sites between lysosomes and other organelles, such as the ER (e.g., ER-localized ITPR, RYR). By controlling lysosomal-autophagosomal ionic homeostasis, all these channels influence lysosomal-autophagosomal function and as such can affect fusion between 2 organelles. Moreover, lysosomal-autophagosomal calcium-permeable channels function to provide Ca2+ required for organelle fusion. Along with the regulation of late-stages of autophagy, some endolysosomal ion channels (e.g., TPCNs, MCOLN1, TRPM2) have also been reported to control autophagy initiation and autophagosome biogenesis by influencing the activation status of AMPK, MTOR, and TFEB as well as the phosphorylation status of BECN1. A graphic overview of the ion channel-related mechanisms of autophagy regulation is presented in . Moreover, for clarity the data discussed in this review are summarized in Table S1.
Figure 1. Ion channels in the regulation of autophagy. Inhibitory and stimulatory actions of various ion channels on autophagy are depicted. Ion channels that stimulate autophagy are shown in green, whereas ion channels that inhibit autophagy are shown in red. Ion channels for which both stimulatory and inhibitory roles have been reported are shown in green/red. For a detailed description, please refer to the specific sections in the text or Table S1.
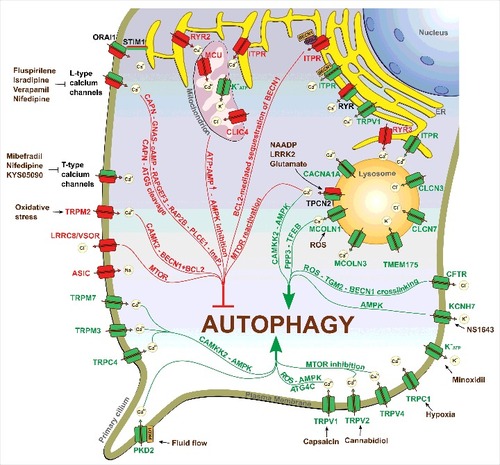
Plasma membrane channels have mostly been implicated in control of autophagy initiation by influencing the activity of master regulators of autophagy, AMPK or MTOR, whereas ER-resident channels exhibit the potential to influence autophagy at different stages (initiation as well as fusion of autophagosomes with lysosomes). Such properties of ER-localized ion channels could be explained by a special role the ER plays in autophagy. In fact, the ER may serve as a platform for autophagosome nucleation. More specifically, ER-mitochondria contact sites recruit major protein complexes essential for the initiation of autophagosome formation.Citation12,61 At the same time, the ER forms numerous membrane contact sites with other organelles, including lysosomes.Citation65 As such, the bidirectional Ca2+ signaling between the ER and lysosomes at the ER-lysosome contact sites could have a great impact on the late stage of autophagy (fusion of autophagosomes with lysosomes or cargo degradation).Citation66,67 Therefore, channels localized to ER-mitochondria contact sites would primarily influence autophagy initiation, whereas channels localized to ER-lysosome contact sites would preferentially affect lysosomal function, fusion with autophagosomes and autophagic cargo degradation. Thus, channels which are localized to both ER-mitochondria and ER-lysosome contact sites could potentially have a double role in autophagy. Indeed, several ER-localized ion channels (e.g., ITPRs, RYRs) have been reported to have a dual paradoxical impact on autophagy that could result in both activation and inhibition of this process.
As for plasma membrane channels, the mechanisms by which these channels influence autophagy are not necessarily cytosolic. Ample evidence suggests the existence of plasma membrane-ER and plasma membrane-mitochondria contact sites, through which plasmalemmal ion channels could regulate ionic homeostasis and the function of the ER and mitochondria, respectively.Citation195,196 Interestingly, a very recent study revealed that ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PtdIns3P synthesis.Citation197 Thus, it seems that membrane contact sites play an important role in the regulation of autophagy, and ion channels localized to these contact sites could be essential components of such regulation.
Another important point to be mentioned is regulation of membrane potential by ion channels. Indeed, by mediating influx/efflux of ions into/from the cell or organelles, ion channels control potential difference across cellular membranes. Maintenance of membrane potential is crucially important for proper functioning of the cell and its organelles. For example, H+ pumping for lysosomal acidification is dependent on the lysosomal membrane potential as well as on the efflux of countercations and influx of counteranions.Citation198 Further, mitochondrial transmembrane potential is required for ATP synthesis.Citation199 Thus, it seems that modulation of membrane potential represents another possible mechanism by which ion channels could influence autophagy.
Another potential mechanism by which ion channels (in particular calcium-permeable channels) could control autophagosome biogenesis is regulation of lipid transfer from intracellular organelles and the plasma membrane to phagophore assembly sites. In fact, many intracellular organelles (including the ER, mitochondria, Golgi) as well as the plasma membrane have been reported to contribute membranes for phagophore expansion.Citation12 It was also demonstrated that maturation and elongation of phagophores requires a series of homotypic (ATG16L1) and heterotypic (ATG9-ATG16L1) fusions of vesicles containing lipids from different cellular compartments.Citation200,201 These fusions depend on SNARE proteins and could potentially be regulated by calcium. Thus, calcium-permeable ion channels could apparently regulate vesicle fusion and as such influence autophagosome precursor maturation and expansion.
At present, no report directly and specifically links ion channels to phagophore elongation. However, existing evidence suggests that some ion channels could potentially have a role in the regulation of phagophore membrane expansion. For example, the TRPM3 channel regulates LC3A/B expression at the translation level in clear cell renal cell carcinoma.Citation138 Considering that LC3 is a crucial regulator of phagophore membrane expansion, these results suggest that TRPM3 not only controls autophagy initiation but also could potentially influence the phagophore elongation process. Further, by regulating the activity of ATG4, the TRPV1 channel could regulate incorporation of LC3 into the phagophore membrane, thereby influencing membrane expansion.Citation145
The field of “ion channels and autophagy” is very young and needs further development. To date, most reports considering ion channels as autophagy regulators are focused on calcium-permeable ion channels, whereas the information on autophagy regulation by other types of channels (potassium, sodium, chloride) is still very limited. Despite the essential progress achieved in this field, the mechanisms by which ion channels regulate autophagy are poorly understood and on the whole the field is developing rather slowly. This is due to the fact that studying the implication of ion channels in autophagy regulation appears to be a much more complex task than it may seem at first glance. One of the reasons is that most cells co-express multiple types of channels with the same ion selectivity. For example, most cells possess several types of calcium-permeable channels (including SOCs, TRPs, ITPRs and others) that greatly complicates studying the impact of one given channel type on autophagy. Also, it seems that the effect of one specific channel on autophagy in different cell types could be different. This could be explained by differences in channel basal expression levels, localization, and regulation by partner proteins. Moreover, variations in the abundance of specific cellular organelles will also make a difference. The existence of alternatively spliced variants of many channels adds further complexity to the situation.
Overall, the data presented here clearly demonstrate the diversity and particular importance of ion channels in autophagy regulation. These channels belong to different families, control the flow of different classes of ions across cell membranes, and have different gating mechanisms as well as biophysical properties, and show different localization patterns. Ion channels have been implicated in virtually all stages of autophagy, from initiation of autophagosome formation to final fusion with lysosomes. Importantly, the contribution of specific ion channels to autophagy is rather complex and depends on multiple factors including cell type and state (e.g., starved or fed), ion channel type, localization, expression levels, and isoform existence as well as regulation by associated proteins, which, taken together, determine the spatiotemporal and amplitude characteristics of the corresponding ionic signals.
Abbreviations
| 5-FU | = | 5-fluorouracil |
| AKT | = | AKT serine/threonine kinase |
| AMPK | = | 5′ adenosine monophosphate-activated protein kinase |
| ASIC | = | acid sensing ion channel |
| ATG | = | autophagy related |
| BECN1/Beclin 1 | = | Beclin 1 |
| Ca2+ | = | calcium |
| CAMKK2/CaMKKβ | = | calcium/calmodulin dependent kinase kinase 2 |
| cAMP | = | 3′-5′-cyclic adenosine monophosphate |
| ccRCC | = | clear cell renal cell carcinoma |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| CMA | = | chaperone-mediated autophagy |
| ER | = | endoplasmic reticulum |
| InsP3 | = | inositol 1,4,5-trisphosphate |
| ITPR | = | inositol 1,4,5-trisphosphate receptor |
| KCNH7/HERG3 | = | potassium voltage-gated channel subfamily H member 7 |
| LRRK2 | = | leucine rich repeat kinase 2 |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| MCOLN1/TRPML1 | = | mucolipin 1 |
| MCOLN2/TRPML2 | = | mucolipin 2 |
| MCOLN3/TRPML3 | = | mucolipin 3 |
| MCU | = | mitochondrial calcium uniporter |
| MCUR1 | = | mitochondrial calcium uniporter regulator 1 |
| MTOR | = | mechanistic target of rapamycin |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| NAADP | = | nicotinic acid adenine dinucleotide phosphate |
| PIK3C3/VPS34 | = | phosphatidylinositol 3-kinase catalytic subunit type 3 |
| PKD1/TRPP1 | = | polycystin 1, transient receptor potential channel interacting |
| PKD2/TRPP2 | = | polycystin 2, transient receptor potential cation channel |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| ROS | = | reactive oxygen species |
| RYR | = | ryanodine receptor |
| SOC | = | store-operated calcium |
| SOCE | = | store-operated calcium entry |
| SQSTM1/p62 | = | sequestosome 1 |
| STIM1 | = | stromal interaction molecule 1 |
| TFEB | = | transcription factor EB |
| TMEM175 | = | transmembrane protein 175 |
| TPCN/TPC | = | 2-pore segment channel |
| TRP | = | transient receptor potential |
| TRPC1 | = | transient receptor potential cation channel subfamily C member 1 |
| TRPC4 | = | transient receptor potential cation channel subfamily C member 4 |
| TRPM2 | = | transient receptor potential cation channel subfamily M member 2 |
| TRPM3 | = | transient receptor potential cation channel subfamily M member 3 |
| TRPM7 | = | transient receptor potential cation channel subfamily M member 7 |
| TRPV1 | = | transient receptor potential cation channel subfamily V member 1 |
| TRPV2 | = | transient receptor potential cation channel subfamily V member 2 |
| ULK | = | unc-51 like autophagy activating kinase |
| VGCC | = | voltage-gated calcium channels |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2017AUTO0125R3-s02_1384887.docx
Download MS Word (47.4 KB)Funding
We acknowledge financial and technical support from the INSERM, INCA, FRABio, SIRIC ONCOLille, la Ligue Nationale Contre le Cancer, le Ministère de l‘Education Nationale, the Region Nord/Pas-de-Calais and the National Institutes of Health (GM053396).
Related Research Data
References
- Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013;5:427–33. doi:10.1159/000351979. PMID:23774579.
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi:10.1152/physrev.00030.2009. PMID:20959619.
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi:10.1016/j.cmet.2011.04.004. PMID:21531332.
- Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–6. doi:10.1056/NEJMra1205406. PMID:23656658.
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi:10.1016/j.molcel.2010.09.023. PMID:20965422.
- Li W, Li J, Bao J. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–36. doi:10.1007/s00018-011-0865-5. PMID:22080117.
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. doi:10.1038/cr.2013.153. PMID:24281265.
- Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb cell (Graz, Austria). 2016;3:588–96. doi:10.15698/mic2016.12.546.
- Gallagher LE, Chan EYW. Early signalling events of autophagy. Essays Biochem. 2013;55:1–15. doi:10.1042/bse0550001. PMID:24070467.
- Tooze SA. Current views on the source of the autophagosome membrane. Essays Biochem. 2013;55:29–38. doi:10.1042/bse0550029. PMID:24070469.
- Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014;15:839–52. doi:10.15252/embr.201439076. PMID:25027988.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–74. doi:10.1038/nrm3696. PMID:24201109.
- Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi:10.1038/ncb2152. PMID:21258367.
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S -i., Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 Association with the ULK1-Atg13-FIP200 Complex Required for Autophagy. Mol Biol Cell. 2009;20:1981–91. doi:10.1091/mbc.E08-12-1248. PMID:19211835.
- Inoki K, Zhu T, Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi:10.1016/S0092-8674(03)00929-2. PMID:14651849.
- Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, Kim H, Neufeld TP, Dillin A, Guan K-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–50. doi:10.1038/ncb2757. PMID:23685627.
- Mizushima N, Yoshimori T, Ohsumi Y. The Role of Atg Proteins in Autophagosome Formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi:10.1146/annurev-cellbio-092910-154005. PMID:21801009.
- Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128:207–17. doi:10.1242/jcs.146258. PMID:25568150.
- Polson HEJ, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, Tooze SA. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–22. doi:10.4161/auto.6.4.11863. PMID:20505359.
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi:10.1074/jbc.C700195200. PMID:17986448.
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–100. doi:10.1091/mbc.E07-12-1257. PMID:18321988.
- Lee Y-K, Lee J-A. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016;49:424–30. doi:10.5483/BMBRep.2016.49.8.081. PMID:27418283.
- Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol Cell. 2014;53:167–78. doi:10.1016/j.molcel.2013.12.014. PMID:24462201.
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652.
- Arcangeli A, Becchetti A. Ion Channels and the Cell Cycle. In: Janigro D, editor. The Cell Cycle in the Central Nervous System. Totowa (NJ): Humana Press; 2006. p. 81–94.
- Lang F, Föller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, Huber SM. Ion channels in cell proliferation and apoptotic cell death. J Membr Biol. 2005;205:147–57. doi:10.1007/s00232-005-0780-5. PMID:16362503.
- Kondratskyi A, Kondratska K, Skryma R, Prevarskaya N. Ion channels in the regulation of apoptosis. Biochim Biophys Acta. 2015;1848:2532–46. doi:10.1016/j.bbamem.2014.10.030. PMID:25450339.
- Jentsch TJ, Hübner CA, Fuhrmann JC. Ion channels: Function unravelled by dysfunction. Nat Cell Biol. 2004;6:1039–47. doi:10.1038/ncb1104-1039. PMID:15516997.
- Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, et al. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J Med Chem. 2013;56:593–624. doi:10.1021/jm3011433. PMID:23121096.
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C, Buneman OP, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol. 2015;172:5729–43. doi:10.1111/bph.13347. PMID:26650438.
- Kondratskyi A, Yassine M, Kondratska K, Skryma R, Slomianny C, Prevarskaya N. Calcium-permeable ion channels in control of autophagy and cancer. Front Physiol. 2013;4:272. doi:10.3389/fphys.2013.00272. PMID:24106480.
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi:10.1038/35036035. PMID:11413485.
- Clapham DE, Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR, Tabin CJ, Alvarez VA, Sabatini BL, Bamji SX, et al. Calcium Signaling. Cell. 2007;131:1047–58. doi:10.1016/j.cell.2007.11.028. PMID:18083096.
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi:10.1038/nrm1155. PMID:12838335.
- La Rovere RML, Roest G, Bultynck G, Parys JB. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 2016;60:74–87. doi:10.1016/j.ceca.2016.04.005. PMID:27157108.
- East DA, Campanella M. Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy. 2013;9:1710–9. doi:10.4161/auto.25367. PMID:24121708.
- Decuypere J-P, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium. 2011;50:242–50. doi:10.1016/j.ceca.2011.04.001. PMID:21571367.
- Gordon PB, Holen I, Fosse M, Røtnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem. 1993;268:26107–12. PMID:8253727.
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi:10.1016/j.molcel.2006.12.009. PMID:17244528.
- Law BYK, Wang M, Ma D-L, Al-Mousa F, Michelangeli F, Cheng S-H, Ng MHL, To K-F, Mok AYF, Ko RYY, et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol Cancer Ther. 2010;9:718–30. doi:10.1158/1535-7163.MCT-09-0700. PMID:20197400.
- Gómez-Suaga P, Luzón-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet. 2012;21:511–25. doi:10.1093/hmg/ddr481. PMID:22012985.
- Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J Biol Chem. 2012;287:38625–36. doi:10.1074/jbc.M112.365767. PMID:23027865.
- Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol Aging. 2012;33:1006.e11–23. doi:10.1016/j.neurobiolaging.2011.09.039.
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem. 2008;283:15370–80. doi:10.1074/jbc.M710209200. PMID:18356160.
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010;22:914–25. doi:10.1016/j.cellsig.2010.01.015. PMID:20114074.
- Gao W, Ding W-X, Stolz DB, Yin X-M. Induction of macroautophagy by exogenously introduced calcium. Autophagy. 2008;4:754–61. doi:10.4161/auto.6360. PMID:18560273.
- Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, Kozma SC, Thomas AP, Thomas G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–65. doi:10.1016/j.cmet.2008.03.002. PMID:18460336.
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem. 2010;285:16912–20. doi:10.1074/jbc.M110.114207. PMID:20308071.
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014;1843:2164–83. doi:10.1016/j.bbamcr.2014.03.007. PMID:24642269.
- Parys JB, Decuypere J-P, Bultynck G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun Signal. 2012;10:17. doi:10.1186/1478-811X-10-17. PMID:22770472.
- Decuypere J-P, Parys JB, Bultynck G. ITPRs/inositol 1,4,5-trisphosphate receptors in autophagy: From enemy to ally. Autophagy. 2015;11:1944–8. doi:10.1080/15548627.2015.1083666. PMID:26291777.
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi:10.1083/jcb.200504035. PMID:16186256.
- Vicencio JM, Ortiz C, Criollo A, Jones AWE, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16:1006–17. doi:10.1038/cdd.2009.34. PMID:19325567.
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgó J, Díaz J, Lavandero S, Harper F, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–39. PMID:17256008.
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K-H, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–83. doi:10.1016/j.cell.2010.06.007. PMID:20655468.
- Rowland AA, Voeltz GK. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–25. doi:10.1038/nrm3440. PMID:22992592.
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. doi:10.1126/science.280.5370.1763. PMID:9624056.
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–7. doi:10.1126/science.8235595. PMID:8235595.
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–11. doi:10.1083/jcb.200608073. PMID:17178908.
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–40. doi:10.1038/nature10230. PMID:21685888.
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER–mitochondria contact sites. Nature. 2013;495:389–93. doi:10.1038/nature11910. PMID:23455425.
- Decuypere J-P, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgó J, Agostinis P, Missiaen L, De Smedt H, Parys JB, et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy. 2011;7:1472–89. doi:10.4161/auto.7.12.17909. PMID:22082873.
- Lam D, Kosta A, Luciani M-F, Golstein P. The Inositol 1,4,5-Trisphosphate Receptor Is Required to Signal Autophagic Cell Death. Mol Biol Cell. 2008;19:691–700. doi:10.1091/mbc.E07-08-0823. PMID:18077554.
- Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M, Baud V, Viry E, Billot K, Nanbakhsh A, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014;74:6820–32. doi:10.1158/0008-5472.CAN-14-0303. PMID:25297632.
- Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2015;17:69–82. doi:10.1038/nrm.2015.8. PMID:26627931.
- Medina DL, Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. 2015;11:970–1. doi:10.1080/15548627.2015.1047130. PMID:26000950.
- Morgan AJ, Davis LC, Wagner SKTY, Lewis AM, Parrington J, Churchill GC, Galione A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol. 2013;200:789–805. doi:10.1083/jcb.201204078. PMID:23479744.
- Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife. 2016;55. pii: e15887. doi:10.7554/eLife.15887.
- Min CK, Yeom DR, Lee K-E, Kwon H-K, Kang M, Kim Y-S, Park ZY, Jeon H, Kim DH. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem J. 2012;447:371–9. doi:10.1042/BJ20120705. PMID:22867515.
- Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276:21482–8. doi:10.1074/jbc.M101486200. PMID:11297554.
- O-Uchi J, Jhun BS, Hurst S, Bisetto S, Gross P, Chen M, Kettlewell S, Park J, Oyamada H, Smith GL, et al. Overexpression of ryanodine receptor type 1 enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts. Am J Physiol Heart Circ Physiol. 2013;305:H1736–51. doi:10.1152/ajpheart.00094.2013. PMID:24124188.
- Bround MJ, Wambolt R, Luciani DS, Kulpa JE, Rodrigues B, Brownsey RW, Allard MF, Johnson JD. Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J Biol Chem. 2013;288:18975–86. doi:10.1074/jbc.M112.427062. PMID:23678000.
- Yuan Q, Chen Z, Santulli G, Gu L, Yang Z-G, Yuan Z-Q, Zhao Y-T, Xin H-B, Deng K-Y, Wang S-Q, et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci Rep. 2014;4:7425. doi:10.1038/srep07425. PMID:25502776.
- Liou B, Peng Y, Li R, Inskeep V, Zhang W, Quinn B, Dasgupta N, Blackwood R, Setchell KDR, Fleming S, et al. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum Mol Genet. 2016;25:5126–41. PMID:27655403.
- Vervliet T, Pintelon I, Welkenhuyzen K, Bootman MD, Bannai H, Mikoshiba K, Martinet W, Nadif Kasri N, Parys JB, Bultynck G. Basal ryanodine receptor activity suppresses autophagic flux. Biochem Pharmacol. 2017;132:133–42. doi:10.1016/j.bcp.2017.03.011. PMID:28322744.
- Kinnear NP, Wyatt CN, Clark JH, Calcraft PJ, Fleischer S, Jeyakumar LH, Nixon GF, Evans AM. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium. 2008;44:190–201. doi:10.1016/j.ceca.2007.11.003. PMID:18191199.
- Park H-W, Park H, Semple IA, Jang I, Ro S-H, Kim M, Cazares VA, Stuenkel EL, Kim J-J, Kim JS, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun. 2014;5:4834. doi:10.1038/ncomms5834. PMID:25189398.
- Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi:10.1038/ncomms8007. PMID:25959678.
- Chung KM, Jeong E-J, Park H, An H-K, Yu S-W. Mediation of Autophagic Cell Death by Type 3 Ryanodine Receptor (RyR3) in Adult Hippocampal Neural Stem Cells. Front Cell Neurosci. 2016;10:116. doi:10.3389/fncel.2016.00116. PMID:27199668.
- Zou Y, Liang Y, Gong H, Zhou N, Ma H, Guan A, Sun A, Wang P, Niu Y, Jiang H, et al. Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertens (Dallas, Tex 1979). 2011;58:1099–110. doi:10.1161/HYPERTENSIONAHA.111.173500.
- Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol. 2015;16:545–53. doi:10.1038/nrm4039. PMID:26285678.
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5. doi:10.1038/nature10234. PMID:21685886.
- Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium. 2012;52:36–43. doi:10.1016/j.ceca.2012.02.008. PMID:22480931.
- Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, Giorgi C, Marchi S, Missiroli S, Poletti F, et al. ATP synthesis and storage. Purinergic Signal. 2012;8:343–57. doi:10.1007/s11302-012-9305-8. PMID:22528680.
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–43. doi:10.1038/ncb2622. PMID:23178883.
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016;15:1673–85. doi:10.1016/j.celrep.2016.04.050. PMID:27184846.
- Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun. 2013;4:2865. doi:10.1038/ncomms3865. PMID:24305511.
- Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi:10.1101/cshperspect.a003947. PMID:21746798.
- Lacinová L. Voltage-dependent calcium channels. Gen Physiol Biophys. 2005;24 Suppl 1:1–78. PMID:16096350.
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi:10.1038/nchembio.79. PMID:18391949.
- Xia H-G, Zhang L, Chen G, Zhang T, Liu J, Jin M, Ma X, Ma D, Yuan J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6:61–6. doi:10.4161/auto.6.1.10326. PMID:19901552.
- Anekonda TS, Quinn JF. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: the case for isradipine. Biochim Biophys Acta. 2011;1812:1584–90. doi:10.1016/j.bbadis.2011.08.013. PMID:21925266.
- Rim H-K, Cho S, Shin D-H, Chung K-S, Cho Y-W, Choi J-H, Lee JY, Lee K-T. T-type Ca2+ channel blocker, KYS05090 induces autophagy and apoptosis in A549 cells through inhibiting glucose uptake. Molecules. 2014;19:9864–75. doi:10.3390/molecules19079864. PMID:25006791.
- Lim J-A, Li L, Kakhlon O, Myerowitz R, Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy. 2015;11:385–402. doi:10.1080/15548627.2015.1009779. PMID:25758767.
- Das A, Pushparaj C, Herreros J, Nager M, Vilella R, Portero M, Pamplona R, Matias-Guiu X, Martí RM, Cantí C. T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells. Pigment Cell Melanoma Res. 2013;26:874–85. doi:10.1111/pcmr.12155. PMID:23931340.
- Ardizzone TD, Lu X-H, Dwyer DS. Calcium-independent inhibition of glucose transport in PC-12 and L6 cells by calcium channel antagonists. AJP Cell Physiol. 2002;283:C579–86. doi:10.1152/ajpcell.00451.2001.
- Lemieux B, Percival MD, Falgueyret J-P. Quantitation of the lysosomotropic character of cationic amphiphilic drugs using the fluorescent basic amine Red DND-99. Anal Biochem. 2004;327:247–51. doi:10.1016/j.ab.2004.01.010. PMID:15051542.
- Pushparaj C, Das A, Purroy R, Nàger M, Herreros J, Pamplona R, Cantí C. Voltage-gated calcium channel blockers deregulate macroautophagy in cardiomyocytes. Int J Biochem Cell Biol. 2015;68:166–75. doi:10.1016/j.biocel.2015.09.010. PMID:26429067.
- Shcheglovitov A, Zhelay T, Vitko Y, Osipenko V, Perez-Reyes E, Kostyuk P, Shuba Y. Contrasting the effects of nifedipine on subtypes of endogenous and recombinant T-type Ca2+ channels. Biochem Pharmacol. 2005;69:841–54. doi:10.1016/j.bcp.2004.11.024. PMID:15710361.
- Tian X, Gala U, Zhang Y, Shang W, Nagarkar Jaiswal S, di Ronza A, Jaiswal M, Yamamoto S, Sandoval H, Duraine L, et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015;13:e1002103. doi:10.1371/journal.pbio.1002103. PMID:25811491.
- Marchant JS, Patel S. Two-pore channels at the intersection of endolysosomal membrane traffic. Biochem Soc Trans. 2015;43:434–41. doi:10.1042/BST20140303. PMID:26009187.
- Morgan AJ, Davis LC, Ruas M, Galione A. TPC: the NAADP discovery channel? Biochem Soc Trans 2015;43:384–9. doi:10.1042/BST20140300. PMID:26009180.
- Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–9. doi:10.1083/jcb.200904073. PMID:19620632.
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang K-T, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi:10.1038/nature08030. PMID:19387438.
- Pereira GJS, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor-Young D, Churchill G, et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem. 2011;286:27875–81. doi:10.1074/jbc.C110.216580. PMID:21610076.
- Alegre-Abarrategui J, Christian H, Lufino MMP, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–34. doi:10.1093/hmg/ddp346. PMID:19640926.
- Pereira GJS, Antonioli M, Hirata H, Ureshino RP, Nascimento AR, Bincoletto C, Vescovo T, Piacentini M, Maria Fimia G, Smaili SS. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget. 2017;8:12730–40. PMID:28055974.
- Lu Y, Hao B-X, Graeff R, Wong CWM, Wu W-T, Yue J. Two pore channel 2 (TPC2) inhibits autophagosomal-lysosomal fusion by alkalinizing lysosomal pH. J Biol Chem. 2013;288:24247–63. doi:10.1074/jbc.M113.484253. PMID:23836916.
- Lin P-H, Duann P, Komazaki S, Park KH, Li H, Sun M, Sermersheim M, Gumpper K, Parrington J, Galione A, et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J Biol Chem. 2015;290:3377–89. doi:10.1074/jbc.M114.608471. PMID:25480788.
- Garcia-Rua V, Feijoo-Bandin S, Rodriguez-Penas D, Mosquera-Leal A, Abu-Assi E, Beiras A, Maria Seoane L, Lear P, Parrington J, Portoles M, et al. Endolysosomal two-pore channels regulate autophagy in cardiomyocytes. J Physiol. 2016;594:3061–77. doi:10.1113/JP271332. PMID:26757341.
- Pedersen SF, Owsianik G, Nilius B. TRP channels: An overview. Cell Calcium. 2005;38:233–52. doi:10.1016/j.ceca.2005.06.028. PMID:16098585.
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–47. doi:10.1146/annurev.physiol.68.040204.100431. PMID:16460286.
- Cheng X, Shen D, Samie M, Xu H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010;584:2013–21. doi:10.1016/j.febslet.2009.12.056. PMID:20074572.
- LaPlante JM, Falardeau J, Sun M, Kanazirska M, Brown EM, Slaugenhaupt SA, Vassilev PM. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 2002;532:183–7. doi:10.1016/S0014-5793(02)03670-0. PMID:12459486.
- Dong X-P, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–6. doi:10.1038/nature07311. PMID:18794901.
- Bach G. Mucolipidosis type IV. Mol Genet Metab. 2001;73:197–203. doi:10.1006/mgme.2001.3195. PMID:11461186.
- Altarescu G, Sun M, Moore DF, Smith JA, Wiggs EA, Solomon BI, Patronas NJ, Frei KP, Gupta S, Kaneski CR, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59:306–13. doi:10.1212/WNL.59.3.306. PMID:12182165.
- Curcio-Morelli C, Charles FA, Micsenyi MC, Cao Y, Venugopal B, Browning MF, Dobrenis K, Cotman SL, Walkley SU, Slaugenhaupt SA. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol Dis. 2010;40:370–7. doi:10.1016/j.nbd.2010.06.010. PMID:20600908.
- Jennings JJ, Zhu J-H, Rbaibi Y, Luo X, Chu CT, Kiselyov K. Mitochondrial aberrations in mucolipidosis Type IV. J Biol Chem. 2006;281:39041–50. doi:10.1074/jbc.M607982200. PMID:17056595.
- Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–301. doi:10.1074/jbc.M508211200. PMID:16361256.
- Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008;17:2723–37. doi:10.1093/hmg/ddn174. PMID:18550655.
- Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, Laplante JM, Dice JF, Slaugenhaupt SA. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219:344–53. doi:10.1002/jcp.21676. PMID:19117012.
- Sun T, Wang X, Lu Q, Ren H, Zhang H. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy. 2011;7:1308–15. doi:10.4161/auto.7.11.17759. PMID:21997367.
- Wong C-O, Li R, Montell C, Venkatachalam K. Drosophila TRPML is required for TORC1 activation. Curr Biol. 2012;22:1616–21. doi:10.1016/j.cub.2012.06.055. PMID:22863314.
- Onyenwoke RU, Sexton JZ, Yan F, Díaz MCH, Forsberg LJ, Major MB, Brenman JE. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem J. 2015;470(3):331–42. doi:10.1042/BJ20150219. PMID:26195823.
- Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A. 2015;112:E1373–81. doi:10.1073/pnas.1419669112. PMID:25733853.
- Zeevi DA, Lev S, Frumkin A, Minke B, Bach G. Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy. J Cell Sci. 2010;123:3112–24. doi:10.1242/jcs.067330. PMID:20736310.
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–99. doi:10.1038/ncb3114. PMID:25720963.
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. doi:10.1038/ncomms12109. PMID:27357649.
- Di Palma F, Belyantseva IA, Kim HJ, Vogt TF, Kachar B, Noben-Trauth K. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc Natl Acad Sci U S A. 2002;99:14994–9. doi:10.1073/pnas.222425399. PMID:12403827.
- Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I, Muallem S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009;10:1157–67. doi:10.1111/j.1600-0854.2009.00924.x. PMID:19522758.
- Choi S, Kim HJ. The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem Biophys Res Commun. 2014;443:56–61. doi:10.1016/j.bbrc.2013.11.044. PMID:24269818.
- Zeevi DA, Frumkin A, Offen-Glasner V, Kogot-Levin A, Bach G. A potentially dynamic lysosomal role for the endogenous TRPML proteins. J Pathol. 2009;219:153–62. doi:10.1002/path.2587. PMID:19557826.
- Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal. 2009;2:ra23.
- Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–25. doi:10.1113/jphysiol.2010.201855. PMID:21135052.
- Wyrsch P, Blenn C, Bader J, Althaus FR. Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose) polymerases and Ca(2+). Mol Cell Biol. 2012;32:3541–53. doi:10.1128/MCB.00437-12. PMID:22751932.
- Wang Q, Guo W, Hao B, Shi X, Lu Y, Wong CWM, Ma VWS, Yip TTC, Au JSK, Hao Q, et al. Mechanistic study of TRPM2-Ca(2+)-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy. 2016;12:1340–54. doi:10.1080/15548627.2016.1187365. PMID:27245989.
- Hall DP, Cost NG, Hegde S, Kellner E, Mikhaylova O, Stratton Y, Ehmer B, Abplanalp WA, Pandey R, Biesiada J, et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell. 2014;26:738–53. doi:10.1016/j.ccell.2014.09.015. PMID:25517751.
- Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–7. doi:10.1126/science.1058519. PMID:11161216.
- Schmitz C, Perraud A-L, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi:10.1016/S0092-8674(03)00556-7. PMID:12887921.
- Ryazanova L V, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A, Ryazanov AG. TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun. 2010;1:109. doi:10.1038/ncomms1108. PMID:21045827.
- Visser D, Middelbeek J, van Leeuwen FN, Jalink K. Function and regulation of the channel-kinase TRPM7 in health and disease. Eur J Cell Biol. 2014;93:455–65. doi:10.1016/j.ejcb.2014.07.001. PMID:25073440.
- Oh HG, Chun YS, Park C-S, Kim T-W, Park MK, Chung S. Regulation of basal autophagy by transient receptor potential melastatin 7 (TRPM7) channel. Biochem Biophys Res Commun. 2015;463:7–12. doi:10.1016/j.bbrc.2015.05.007. PMID:25983327.
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–24. doi:10.1038/39807. PMID:9349813.
- Farfariello V, Amantini C, Santoni G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J Leukoc Biol. 2012;92:421–31. doi:10.1189/jlb.0312123. PMID:22753949.
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi:10.1038/sj.emboj.7601623. PMID:17347651.
- Li B-H, Yin Y-W, Liu Y, Pi Y, Guo L, Cao X-J, Gao C-Y, Zhang L-L, Li J-C. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Dis. 2014;5:e1182. doi:10.1038/cddis.2014.146. PMID:24743737.
- Lu S, Xu D. Cold stress accentuates pressure overload-induced cardiac hypertrophy and contractile dysfunction: role of TRPV1/AMPK-mediated autophagy. Biochem Biophys Res Commun. 2013;442:8–15. doi:10.1016/j.bbrc.2013.10.128. PMID:24211590.
- Li Q, Li L, Wang F, Chen J, Zhao Y, Wang P, Nilius B, Liu D, Zhu Z. Dietary capsaicin prevents nonalcoholic fatty liver disease through transient receptor potential vanilloid 1-mediated peroxisome proliferator-activated receptor δ activation. Pfluügers Arch Eur J Physiol. 2013;465:1303–16. doi:10.1007/s00424-013-1274-4.
- Chien C-S, Ma K-H, Lee H-S, Liu P-S, Li Y-H, Huang Y-S, Chueh S-H. Dual effect of capsaicin on cell death in human osteosarcoma G292 cells. Eur J Pharmacol. 2013;718:350–60. doi:10.1016/j.ejphar.2013.08.011. PMID:24012930.
- Liu A-J, Wang S-H, Hou S-Y, Lin C-J, Chiu W-T, Hsiao S-H, Chen T-H, Shih C-M. Evodiamine Induces Transient Receptor Potential Vanilloid-1-Mediated Protective Autophagy in U87-MG Astrocytes. Evid Based Complement Alternat Med. 2013;2013:354840. doi:10.1155/2013/354840. PMID:24454492.
- Ahn S, Park J, An I, Jung SJ, Hwang J. Transient receptor potential cation channel V1 (TRPV1) is degraded by starvation- and glucocorticoid-mediated autophagy. Mol Cells. 2014;37:257–63. doi:10.14348/molcells.2014.2384. PMID:24658385.
- Nabissi M, Morelli MB, Amantini C, Liberati S, Santoni M, Ricci-Vitiani L, Pallini R, Santoni G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int J cancer. 2015;137:1855–69. doi:10.1002/ijc.29573. PMID:25903924.
- Zhan L, Yang Y, Ma T-T, Huang C, Meng X-M, Zhang L, Li J. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol Cell Biochem. 2015;402:9–22. doi:10.1007/s11010-014-2298-6. PMID:25600591.
- Sukumaran P, Sun Y, Vyas M, Singh BB. TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 2015;6:e1674. doi:10.1038/cddis.2015.7. PMID:25741599.
- Azimi I, Milevskiy MJG, Kaemmerer E, Turner D, Yapa KTDS, Brown MA, Thompson EW, Roberts-Thomson SJ, Monteith GR. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J Cell Sci. 2017;130:2292–305. doi:10.1242/jcs.196659. PMID:28559303.
- Zhang L, Dai F, Cui L, Jing H, Fan P, Tan X, Guo Y, Zhou G. Novel role for TRPC4 in regulation of macroautophagy by a small molecule in vascular endothelial cells. Biochim Biophys Acta. 2015;1853:377–87. doi:10.1016/j.bbamcr.2014.10.030. PMID:25476892.
- Orhon I, Dupont N, Zaidan M, Boitez V, Burtin M, Schmitt A, Capiod T, Viau A, Beau I, Wolfgang Kuehn E, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016;18:657–67. doi:10.1038/ncb3360. PMID:27214279.
- Delmas P, Padilla F, Osorio N, Coste B, Raoux M, Crest M. Polycystins, calcium signaling, and human diseases. Biochem Biophys Res Commun. 2004;322:1374–83. doi:10.1016/j.bbrc.2004.08.044. PMID:15336986.
- Cebotaru V, Cebotaru L, Kim H, Chiaravalli M, Boletta A, Qian F, Guggino WB. Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem. 2014;289:6404–14. doi:10.1074/jbc.M113.501205. PMID:24459142.
- Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95:1383–436. doi:10.1152/physrev.00020.2014. PMID:26400989.
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi:10.1038/nature05122. PMID:16921383.
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi:10.1038/nature04147. PMID:16208375.
- Abdelmohsen K, Srikantan S, Tominaga K, Kang M-J, Yaniv Y, Martindale JL, Yang X, Park S-S, Becker KG, Subramanian M, et al. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol Cell Biol. 2012;32:2530–48. doi:10.1128/MCB.00510-12. PMID:22547681.
- Tang B-D, Xia X, Lv X-F, Yu B-X, Yuan J-N, Mai X-Y, Shang J-Y, Zhou J-G, Liang S-J, Pang R-P. Inhibition of Orai1-mediated Ca 2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J Cell Mol Med. 2017;21:904–15. doi:10.1111/jcmm.13029. PMID:27878958.
- Chen Y-W, Chen Y-F, Chen Y-T, Chiu W-T, Shen M-R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci Rep. 2016;6:22142. doi:10.1038/srep22142. PMID:26917047.
- Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2016;55:818–31. doi:10.1002/mc.22324. PMID:25917875.
- Singh A, Hildebrand ME, Garcia E, Snutch TP. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br J Pharmacol. 2010;160:1464–75. doi:10.1111/j.1476-5381.2010.00786.x. PMID:20590636.
- Kondratskyi A, Yassine M, Slomianny C, Kondratska K, Gordienko D, Dewailly E, Lehen'kyi V, Skryma R, Prevarskaya N. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis. 2014;5:e1193. doi:10.1038/cddis.2014.156. PMID:24763050.
- Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol. 2011;31:3546–56. doi:10.1128/MCB.05114-11. PMID:21730292.
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–45. doi:10.1128/MCB.05124-11. PMID:21670147.
- Yang J, Yu J, Li D, Yu S, Ke J, Wang L, Wang Y, Qiu Y, Gao X, Zhang J, et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 2017;13:82–98. doi:10.1080/15548627.2016.1245261. PMID:27791458.
- Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi:10.1186/gb-2003-4-3-207. PMID:12620097.
- Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14:461–71. doi:10.1038/nrn3529. PMID:23783197.
- Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–67. doi:10.1152/physrev.00007.2002. PMID:12087134.
- Cochet-Bissuel M, Lory P, Monteil A. The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci. 2014;8:132. doi:10.3389/fncel.2014.00132. PMID:24904279.
- Sun X, Cao Y-B, Hu L-F, Yang Y-P, Li J, Wang F, Liu C-F. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagic degradation. Brain Res. 2011;1396:77–87. doi:10.1016/j.brainres.2011.04.011. PMID:21529788.
- Cang C, Zhou Y, Navarro B, Seo Y-J, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell. 2013;152:778–90. doi:10.1016/j.cell.2013.01.023. PMID:23394946.
- Shieh CC, Coghlan M, Sullivan JP, Gopalakrishnan M. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol Rev. 2000;52:557–94. PMID:11121510.
- Klein B, Wörndl K, Lütz-Meindl U, Kerschbaum HH. Perturbation of intracellular K(+) homeostasis with valinomycin promotes cell death by mitochondrial swelling and autophagic processes. Apoptosis. 2011;16:1101–17. doi:10.1007/s10495-011-0642-9. PMID:21877215.
- Kim S-H, Choi Y-J, Kim K-Y, Yu S-N, Seo Y-K, Chun S-S, Noh K-T, Suh J-T, Ahn S-C. Salinomycin simultaneously induces apoptosis and autophagy through generation of reactive oxygen species in osteosarcoma U2OS cells. Biochem Biophys Res Commun. 2016;473:607–13. doi:10.1016/j.bbrc.2016.03.132. PMID:27033598.
- Yu K-Y, Wang Y-P, Wang L-H, Jian Y, Zhao X-D, Chen J-W, Murao K, Zhu W, Dong L, Wang G-Q, et al. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res Cardiol. 2014;109:416. doi:10.1007/s00395-014-0416-y. PMID:24847907.
- Perez-Neut M, Haar L, Rao V, Santha S, Lansu K, Rana B, Jones WK, Gentile S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget. 2016;7:21991–2004. doi:10.18632/oncotarget.7831. PMID:26942884.
- Cang C, Aranda K, Seo Y, Gasnier B, Ren D. TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function. Cell. 2015;162:1101–12. doi:10.1016/j.cell.2015.08.002. PMID:26317472.
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–47. doi:10.1046/j.1365-201X.2003.01060.x. PMID:12558550.
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol 2010; 72:95–121. doi:10.1146/annurev-physiol-021909-135811. PMID:19827947.
- Zhong J, Kong X, Zhang H, Yu C, Xu Y, Kang J, Yu H, Yi H, Yang X, Sun L. Inhibition of CLIC4 enhances autophagy and triggers mitochondrial and ER stress-induced apoptosis in human glioma U251 cells under starvation. PLoS One. 2012;7:e39378. doi:10.1371/journal.pone.0039378. PMID:22761775.
- Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–75. doi:10.1038/ncb2090. PMID:20711182.
- Xia D, Qu L, Li G, Hongdu B, Xu C, Lin X, Lou Y, He Q, Ma D, Chen Y. MARCH2 regulates autophagy by promoting CFTR ubiquitination and degradation and PIK3CA-AKT-MTOR signaling. Autophagy. 2016;12:1614–30. doi:10.1080/15548627.2016.1192752. PMID:27308891.
- Wang L, Shen M, Guo X, Wang B, Xia Y, Wang N, Zhang Q, Jia L, Wang X. Volume-sensitive outwardly rectifying chloride channel blockers protect against high glucose-induced apoptosis of cardiomyocytes via autophagy activation. Sci Rep. 2017;7:44265. doi:10.1038/srep44265. PMID:28300155.
- Wartosch L, Fuhrmann JC, Schweizer M, Stauber T, Jentsch TJ. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009;23:4056–68. doi:10.1096/fj.09-130880. PMID:19661288.
- Graves AR, Curran PK, Smith CL, Mindell JA. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–92. doi:10.1038/nature06907. PMID:18449189.
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A, et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–96. doi:10.1016/S0896-6273(01)00189-1. PMID:11182090.
- Su J, Xu Y, Zhou L, Yu H-M, Kang J-S, Liu N, Quan C-S, Sun L-K. Suppression of chloride channel 3 expression facilitates sensitivity of human glioma U251 cells to cisplatin through concomitant inhibition of Akt and autophagy. Anat Rec (Hoboken). 2013;296:595–603. doi:10.1002/ar.22665. PMID:23408563.
- Westermann B. The mitochondria-plasma membrane contact site. Curr Opin Cell Biol. 2015;35:1–6. doi:10.1016/j.ceb.2015.03.001. PMID:25801776.
- Okeke E, Dingsdale H, Parker T, Voronina S, Tepikin A V. Endoplasmic reticulum-plasma membrane junctions: structure, function and dynamics. J Physiol. 2016;594:2837–47. doi:10.1113/JP271142. PMID:26939537.
- Nascimbeni AC, Giordano F, Dupont N, Grasso D, Vaccaro MI, Codogno P, Morel E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017;:e201797006.
- Xu H, Ren D. Lysosomal Physiology. Annu Rev Physiol. 2015;77:57–80. doi:10.1146/annurev-physiol-021014-071649. PMID:25668017.
- Dimroth P, Kaim G, Matthey U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol. 2000;203:51–9. PMID:10600673.
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein D. Autophagosome Precursor Maturation Requires Homotypic Fusion. Cell. 2011;146:303–17. doi:10.1016/j.cell.2011.06.023. PMID:21784250.
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse Autophagosome Membrane Sources Coalesce in Recycling Endosomes. Cell. 2013;154:1285–99. doi:10.1016/j.cell.2013.08.044. PMID:24034251.