
ABSTRACT
Autophagy is a lysosomal degradation system by which cytosolic materials and damaged organelles are broken down into basic components. To explore the physiological role of autophagy in glomerular endothelial cells (GEnCs), we compared the autophagic flux among cells in the kidney under starvation. Inhibition of autophagy by chloroquine administration significantly increased the number of autophagosomes or autolysosomes in GEnCs and proximal tubular cells, but not in podocytes, suggesting that the GEnCs exhibit substantial autophagic activity. Next, we analyzed endothelial and hematopoietic cell-specific atg5-deficient mice (atg5-conditional KO [cKO] mice). Glomeruli of 4-wk-old atg5-cKO mice exhibited slightly distended capillary loops accompanied by an accumulation of reactive oxygen species (ROS). Glomeruli of 8-wk-old atg5-cKO mice showed a lobular pattern with thickening of the capillary loops and mesangial matrix expansion; however, the vasculature of other organs was preserved. The atg5-cKO mice died by 12 wk of age, presumably due to pancytopenia resulting from the defect in their hematopoietic lineages. Therefore, we subjected 4-wk atg5-cKO mice to irradiation followed by bone marrow transplantation from normal littermates. Transplanted mice recapitulated the glomerular phenotypes of the atg5-cKO mice with no obvious histological changes in other organs. Twelve-mo-old transplanted mice developed mesangiolysis and glomerulosclerosis with significant deterioration of kidney function. Administration of N-acetyl-l-cysteine, a ROS scavenger, to atg5-cKO mice rescued the glomerular phenotypes. These data suggest that endothelial autophagy protects glomeruli from oxidative stress and maintains the integrity of glomerular capillaries. Enhancing endothelial autophagy may provide a novel therapeutic approach to minimizing glomerular diseases.
Introduction
Autophagy is one of the major degradation pathways in the cell along with the ubiquitin-proteasome system, which contributes to the removal of damaged organelles and aberrant macromolecules, thereby preventing further injury and cellular dysfunction in various organs and cells,Citation1–Citation3 including kidney.Citation4 Results from in vitro experiments using human umbilical vein endothelial cells (HUVECs) suggest that endothelial cell (EC) autophagy is an essential process for mediating proper endothelial function. It regulates the redox balance and nitric oxide (NO) bioavailability under static and shear-stressed conditions.Citation5 Pretreatment with 3-methyladenine, an autophagy inhibitor, inhibits NOS3/eNOS (nitric oxide synthase 3, endothelial cell) expression, whereas stimulation of autophagy with rapamycin produces the opposite result.Citation5 Similarly, in old mice, treatment with the autophagy-enhancing agent trehalose restores the expression of autophagy markers, rescues NO-mediated endothelium-dependent dilatation by reducing oxidative stress, and normalizes inflammatory cytokine expression.Citation6
By contrast, in vivo evidence for the role of endothelial autophagy is scarce. Autophagy regulates maturation and the secretion of VWF (von Willebrand factor); endothelial-specific deletion of the essential autophagy gene Atg7 in mice results in impaired epinephrine-stimulated VWF release, reduced levels of high molecular weight VWF multimers, and significantly increased bleeding times.Citation7 Another report has demonstrated that the same line of mice exhibits impaired autophagic flux accompanied by marked changes in the EC architecture, loss of endothelial markers, and gain of mesenchymal markers, which are consistent with endothelial-to-mesenchymal transition and consequently result in an increased susceptibility to bleomycin-induced pulmonary fibrosis and collagen accumulation. In vitro experiments also confirmed the above by demonstrating that the loss of Atg7 in HUVECs upregulates TGFB1 (transforming growth factor, beta1) signaling and key profibrotic genes.
A large portion of glomerular diseases, including diabetic nephropathy, results from dysfunction and injury of the microvasculature in kidney.Citation8–Citation10 Studies in various animal models have demonstrated that an impaired availability of NO, which is characteristic of endothelial dysfunction, caused by either the inhibition or a genetic defect of NOS3, accelerates the progression of kidney disease.Citation11 To date, there has been significant scientific effort focused on understanding which factors protect tissues from microvascular injury and end-organ damage.Citation9 We deduced that endothelial autophagy is an endogenous protective system and that impaired autophagy in some settings could jeopardize endothelial integrity and lead to organ dysfunction. The role of endothelial autophagy in kidney remains largely unknown (see Discussion). Based on this background, in this study, we established a new line of EC (and hematopoietic cell)-specific autophagy-deficient (atg5-conditional KO [atg5-cKO]) mice and examined whether autophagy is required for normal vessel architecture in individual organs, with a special focus on kidney. In addition, we investigated whether the phenotypes are recapitulated in mice after bone marrow (BM) transplantation (BMT) from the floxed control mice, which restores autophagy in the hematopoietic cells. Furthermore, we tried to rescue the glomerular phenotype of atg5-cKO mice by using an antioxidant.
Results
Glomerular endothelial cells (GEnCs) exhibit high autophagic activity in vivo
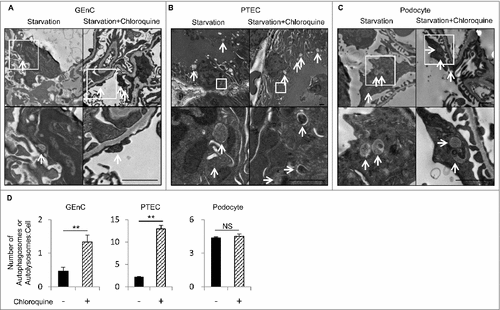
To evaluate in vivo the autophagic flux among the different cell lineages in kidney during the fed or fasted state, the numbers of autophagosomes or autolysosomes were counted on electron micrographs of kidney with or without an administration of chloroquine (50 μg/g body weight), an inhibitor of intralysosomal acidification, 6 h before euthanasia. In the fed mice, the numbers of autophagosomes or autolysosomes in the GEnCs, proximal tubular epithelial cells (PTECs), and podocytes remained almost unchanged before and after chloroquine administration (data not shown). After 24 h of starvation, chloroquine administration significantly increased the number of autophagosomes or autolysosomes in the GEnCs and PTECs (A, B, and D). In contrast, there was a substantial number of autophagosomes or autolysosomes in the podocytes before chloroquine administration, which is in agreement with previous reports,Citation12,Citation13 and the number remained unchanged after chloroquine administration (C and D). These findings demonstrate that the GEnCs exhibit substantial autophagic activity.
Figure 1. GEnCs exhibit high autophagic activity. In vivo evaluation of the autophagic flux among the different cell lineages in the kidney during the fasted state. (A to C) Representative electron micrographs of GEnCs (A), PTECs (B), and podocytes (C) of 8-wk-old WT mice that were subjected to 24 h of starvation with or without chloroquine administration 6 h before euthanasia (n = 3 in each group). Arrows indicate autophagosomes or autolysosomes. The lower panels show a magnification of the indicated areas (white squares) in the upper panels. Bars: 1 μm. (D) Quantification of the number of autophagosomes or autolysosomes per cell among GEnCs, PTECs, or podocytes. For each condition, at least 10 cells were examined in each mouse. Data are provided as the mean ± SD. NS, not significant. Statistically significant differences (**P < 0.01 versus the conditions without chloroquine administration) are indicated.
Generation of EC (and hematopoietic cell)-specific autophagy-deficient mice
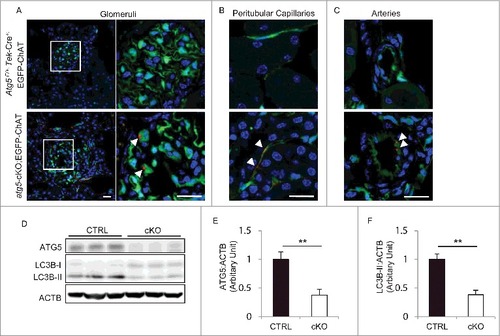
To investigate the function of autophagy in the ECs, we generated EC and hematopoietic cell-specific atg5-deficient mice. We used transgenic mice that expressed Cre recombinase under the promoter of Tek/Tie2 (endothelial-specific receptor tyrosine kinase). Tek-Cre;enhanced green fluorescence protein (EGFP)-chloramphenicol acetyltransferase (ChAT) transgenic mice, in which EGFP can be detected only after Cre-mediated recombination, demonstrated a pan-endothelial-specific pattern of Cre recombinase expression as assessed by immunostaining with PECAM1/CD31 (platelet/endothelial cell adhesion molecule 1), a marker of ECs (Fig. S1). We then crossed the Tek-Cre transgenic mice with mice bearing an Atg5flox allele (Atg5F/F mice). The resulting Atg5-floxed Tek-Cre+ (Atg5F/F ;Tek-Cre+) mice were born at the expected Mendelian frequency (Atg5F/F ;Tek-Cre+: 27%; Atg5F/+ ;Tek-Cre+: 23%; Atg5F/F ;Tek-Cre−: 27%; Atg5F/+ ;Tek-Cre−: 22%; n = 211). To ascertain the autophagy deficiency in atg5F/F ;Tek-Cre+ (atg5-cKO) mice, we performed immunofluorescence analysis for SQSTM1/p62 (sequestosome 1), one of the selective substrates of autophagy, in 8-wk-old atg5-cKO;EGFP-ChAT mice and Atg5F/+ ;Tek-Cre+;EGFP-ChAT control mice. In the atg5-cKO;EGFP-ChAT mice, SQSTM1-positive aggregates were localized in EGFP-positive ECs of principal organs, including kidney, liver, spleen, lung, heart, and aorta, but they were rarely seen in Atg5F/+ ;Tek-Cre+;EGFP-ChAT mice (A to C, Fig. S2). In addition, the level of ATG5 protein in the isolated glomeruli of 4-wk-old atg5-cKO mice was significantly reduced when compared to their Atg5F/F or Atg5F/+ control (referred to as Atg5-CTRL) littermates (62% reduction; D and E). Consequently, the conversion of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3B)-I to MAP1LC3B-II, a phagophore and autophagosome-associated, phosphatidylethanolamine conjugate, was significantly suppressed in the atg5-cKO mice when compared to the Atg5-CTRL mice (62% reduction; D and F).
Figure 2. Evidence of autophagy deficiency in the ECs of atg5-cKO mice. (A to C) SQSTM1 accumulation in kidney ECs of atg5-cKO mice. Representative images of the immunofluorescence analysis using 8-wk-old Atg5F/+ ;Tek-Cre+;EGFP-ChAT (upper) and atg5-cKO;EGFP-ChAT (lower) mice. Sections containing glomeruli (A), peritubular capillaries (B), and renal small arteries (C) were immunostained for SQSTM1 (red) and for 4’, 6-diamidino-2-phenylindole (DAPI; blue) as counterstaining. (A) The right panels show a magnification of the indicated areas (white squares) in the left panels. Arrowheads indicate SQSTM1-positive aggregates localized in GFP-positive ECs of atg5-cKO;EGFP-ChAT mice. Bars: 20 μm. (D) Western blot analysis using isolated glomeruli from 4-wk-old atg5-cKO mice and their Atg5-CTRL littermates. (E and F) Densitometric quantification of the levels of ATG5 protein (E) and the conversion of MAP1LC3B-I to MAP1LC3B-II (F). n = 3 in each group. Data are provided as the mean ± SD. Statistically significant differences (**P < 0.01 versus the Atg5-CTRL mice) are indicated.
GEnCs depend on autophagy for their integrity
Tek is expressed in some hematopoietic lineages as well as in ECs.Citation14 A previous study reports that the absence of autophagy in hematopoietic cells leads to cell death.Citation15 In our model, there was no significant difference in peripheral blood cell counts between atg5-cKO and Atg5-CTRL mice at the age of 3 wk. However, the atg5-cKO mice gradually developed severe pancytopenia (Fig. S3A) and splenomegaly (Fig. S3B and S3C) accompanied by significant weight loss (Fig. S3D) by the age of 6 to 8 wk. We analyzed the proportion of BM hematopoietic cells from 3- and 8-wk-old mice at each developmental stage by flow cytometry using antibodies for KIT/c-KIT (KIT proto-oncogene receptor tyrosine kinase), LY6A/SCA1 (lymphocyte antigen 6 complex, locus A), CD44, and LY76/TER119 (lymphocyte antigen 76) (Fig. S3E and S3F). The overall BM cell count was substantially reduced in the atg5-cKO mice because BM was prominently replaced with fat and fibrous tissues (Fig. S3G). We found that the proportion of hematopoietic stem cells, defined as lineage marker-negative (Lin−) KIT+ LY6A+ cells, remained almost unchanged in 3- and 8-wk-old atg5-cKO mice; in contrast, that of the Lin− KIT+ LY6A− compartment, which contains more mature hematopoietic progenitors, significantly decreased at 8 wk of age (Fig. S3E and S3F). Similarly, the proportion of CD44+ LY76− cells, which contains erythroid progenitor cells, significantly increased, while that of the CD44− LY76+ compartment, which contains more mature erythroid cells, significantly decreased in 8-wk-old atg5-cKO mice. These data suggest the later obstruction of BM cell differentiation into hematopoietic and erythroid progenitors under autophagy deficiency. The atg5-cKO mice all died between the ages of 8 and 12 wk, most likely due to severe hematopoietic injury.
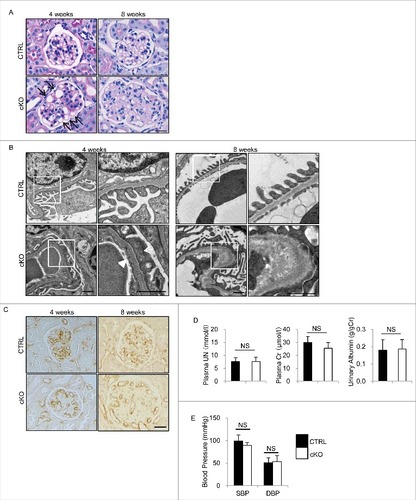
Although periodic-acid Schiff (PAS) and hematoxylin-eosin (HE) staining demonstrated that 4-wk-old atg5-cKO mice showed no obvious change in their principal organs, the glomeruli showed slightly distended capillary loops on light microscopy (A). Furthermore, electron microscopy of 4-wk-old atg5-cKO mice showed segmental loss of glomerular endothelial fenestra accompanied by foot process effacement of podocytes adjacent to the transformed ECs (B). Therefore, we hereafter focused on autophagy in the GEnCs. Glomeruli of 8-wk-old atg5-cKO mice were enlarged and exhibited a lobular pattern with thickening of the capillary loops and mesangial matrix expansion without cell proliferation, but the other organs (liver, spleen, lung, heart, aorta, brain, and retina) remained unchanged (A and S4). Immunohistochemical staining for PECAM1 of the glomerulus of atg5-cKO mice showed distended capillary loops localized to the periphery with loss of complex tuft pattern (C). The detachment of GEnCs was rarely observed at 4 wk of age; however, it became obvious, leading to the thickness of glomerular basement membrane (GBM) by 8 wk of age (B). There were no significant differences in kidney function (assessed by plasma urea nitrogen [UN] and creatinine), urinary albumin to creatinine ratio and blood pressure between 4- or 6-wk-old atg5-cKO and Atg5-CTRL mice, the latter suggesting that glomerular defects may not be derived from hemodynamic change (D and E). We then evaluated peritubular capillaries and tubular function of 8-wk-old atg5-cKO and Atg5-CTRL mice. There were no significant differences in the density of peritubular capillaries and urinary concentrations of glucose and phosphate between 2 groups at 3 or 8 wk of age (Fig. S5A and S5B). Metabolome analysis revealed similar profiles of urinary amino acids, although the levels of some amino acids exhibited slightly higher in Atg5-CTRL mice. (Fig. S5C). Taken together, among the ECs investigated, only GEnCs depend on autophagy for their integrity.
Figure 3. Glomerular abnormalities of atg5-cKO mice. Representative images of PAS staining (A), electron micrographs (B), and immunohistochemical analysis for PECAM1 (C) of glomeruli from 4- and 8-wk-old atg5-cKO and Atg5-CTRL mice (n = 4 to 6 in each group). (A) Distended capillary loops (arrows). (B) Segmental loss of glomerular endothelial fenestra (arrowhead) accompanied by foot process effacement of podocytes (arrow) adjacent to the transformed ECs. The right panels show a magnification of the indicated areas (white squares) in the left panels. Bars: 20 μm (A and C) and 1 μm (B). Plasma UN and creatinine, urinary albumin/creatinine ratio (D), and blood pressure (E) of 4- (D) and 6- (E) wk-old atg5-cKO and Atg5-CTRL mice are shown. n = 6 for plasma UN and creatinine; n = 9 to 14 for urinary albumin/creatinine ratio; and n = 3 or 4 (E) in each group. Data are provided as the mean ± SD. NS, not significant. SBP, systolic blood pressure; DBP, diastolic blood pressure.
Autophagy in the GEnCs protects glomeruli from oxidative stress
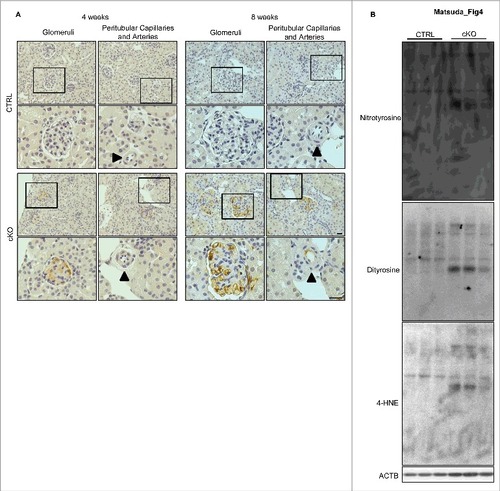
Since autophagy contributes to the alleviation of oxidative stress,Citation16,Citation17 we investigated the effects of autophagy deficiency on the redox status of glomeruli. Immunohistochemical analysis revealed the presence of dityrosine, a marker of protein oxidation, in the glomerular capillaries of 4-wk-old atg5-cKO mice, but not in those of their Atg5-CTRL littermates (A). Of note, we observed no positive staining for dityrosine in the peritubular capillaries, renal small arteries, or other principle organs of atg5-cKO mice (A and S6). In 8-wk-old atg5-cKO mice, we observed more diffuse and intense staining of dityrosine in the glomeruli as well as some positive staining in the peritubular capillaries and renal small arteries, but little staining was seen in the other principle organs (A and S6). Western blot analysis using isolated glomeruli also revealed the accumulation of reactive oxygen species (ROS), including dityrosine, nitrotyrosine, and 4-hydroxy-2-nonenal (4-HNE), in the glomeruli of 8-wk-old atg5-cKO mice, which supports the findings of the immunohistological analysis (B). These findings suggest that autophagy in the GEnCs protects glomeruli from oxidative stress.
Figure 4. Accumulation of ROS in the glomeruli of atg5-cKO mice. (A) Representative images of the immunohistochemical analysis for dityrosine in kidney sections from 4- or 8-wk-old Atg5-CTRL (upper) and atg5-cKO (lower) mice. Small arteries are indicated with arrowheads. The lower panels show a magnification of the indicated areas (black squares) in the upper panels. Bars: 20 μm. (B) Western blot analysis for dityrosine, nitrotyrosine, and 4-HNE using isolated glomeruli homogenates from 8-wk-old atg5-cKO mice and their Atg5-CTRL littermates (n = 3 in each group).
Autophagy in the GEnCs is responsible for the defects in atg5-cKO mice
It has been reported that Tek is expressed in nonendothelial cells, such as hematopoietic stem cells, and it is unclear which component of autophagy deficiency is responsible for the defects observed in the glomerular capillaries of atg5-cKO mice. To investigate this, we subjected 4-wk-old Atg5-CTRL and atg5-cKO mice to irradiation followed by BMT from age-matched Tek-Cre− littermates (Atg5-CTRL recipients and atg5-cKO recipients, respectively). We found no difference in urinary albumin excretion or kidney function, as assessed by plasma UN measurements, between 6-mo-old Atg5-CTRL and atg5-cKO recipients (A and B). However, when compared to Atg5-CTRL recipients, the atg5-cKO recipients showed a significant increase in plasma UN and albuminuria at the age of 12 mo. During the observation periods (up to 12 mo), there was no significant difference in body weight, peripheral blood cell counts, or other biological parameters between the Atg5-CTRL and atg5-cKO recipients (C and D, ). Within 2 mo after BMT, approximately half of the atg5-cKO recipients died, whereas less than 20% of the Atg5-CTRL recipients died (E).
Figure 5. Long-term effects of autophagy deficiency in GEnCs. The long-term effects of autophagy deficiency in GEnCs were investigated in the Atg5-CTRL and atg5-cKO mice rescued by BMT at 4 wk of age (Atg5-CTRL recipients and atg5-cKO recipients, respectively). The blood UN (A), urinary albumin:creatinine ratio (B), body weight (C), peripheral blood cell counts (D), Kaplan-Meier survival curves (E), representative images of PAS staining (F), quantification of the ratio of sclerotic glomeruli per total glomeruli (G), and representative images of Masson-trichrome staining (H), immunostaining for ADGRE1 (I), electron micrographs of glomeruli (J), immunostaining for dityrosine (K), blood pressure (L) of mice at the indicated ages are shown. (A and B) n = 3 for 6-mo-old Atg5-CTRL recipients; n = 22 to 25 for 12-mo-old Atg5-CTRL recipients; n = 4 or 5 for 6-mo-old atg5-cKO recipients; and n = 11 or 12 for 12-mo-old atg5-cKO recipients. (C and D) n = 14 to 16 for Atg5-CTRL recipients; and n = 7 or 8 for atg5-cKO recipients. (G) n = 14 for Atg5-CTRL recipients; and n = 7 for atg5-cKO recipients. (L) n = 5 to 7 for Atg5-CTRL and atg5-cKO recipients, respectively. Data are provided as the mean ± SD. NS, not significant. Statistically significant differences (*P < 0.05, **P < 0.01 versus the Atg5-CTRL recipients) are indicated. (H to K) The right panels show a magnification of the indicated areas (black or white squares) in the left panels. Bars: 20 μm (F, H, I, and K) and 1 μm (J). SBP, systolic blood pressure; DBP, diastolic blood pressure.
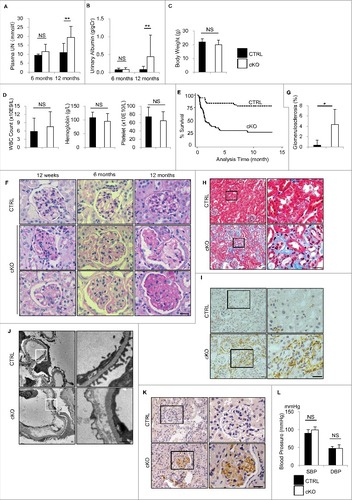
Table 1. Biochemical analysis of blood from endothelial-specific autophagy-deficient mice rescued by BMT
In the glomeruli of 12-qk-old atg5-cKO recipients, PAS staining revealed slight expansion of the mesangial matrix and thickening of the GBM, which recapitulates the phenotype of atg5-cKO mice (F). More remarkable expansion of the mesangial matrix and thickening of the GBM was observed at the ages of 6 and 12 mo (F). In addition, mesangiolysis and glomerulosclerosis were observed in 12-mo-old atg5-cKO recipients (F and G). The distribution of glomerulosclerosis showed no deviation between the superficial cortex and the juxtamedullary zone. In contrast, no obvious histological changes were detected in the other organs of atg5-cKO recipients, including the liver, spleen, lung, heart, aorta, retina, and mesentery (Fig. S7). Masson-trichrome staining demonstrated that 12-mo-old atg5-cKO recipients exhibited severe interstitial fibrosis with massive leukocyte infiltration (H). ADGRE1/F4/80 (adhesion G protein-coupled receptor E1)-positive macrophages were abundant in the interstitium of atg5-cKO recipients, but were almost absent in the interstitium of their Atg5-CTRL recipients (I). Electron microscopy of 12-mo-old atg5-cKO recipients showed the widespread loss of fenestra in the GEnCs with detachment from the GBM and foot process effacement of podocytes (J). In 12-mo-old atg5-cKO recipients, we observed diffuse and intense staining of dityrosine in the glomeruli (K). Similar to before BMT, there were no significant differences in blood pressure between 12-wk-old Atg5-CTRL and atg5-cKO recipients, suggesting that glomerular defects may not be derived from hemodynamic change (L). Although we cannot clearly identify the cause of death in atg5-cKO recipients, considering that the extent of glomerular phenotype in atg5-cKO recipients becomes more severe time-dependently, it is probable that they die of renal failure. These findings indicate that autophagy deficiency in the GEnCs, and not in the hematopoietic cells, is responsible for the defects in atg5-cKO mice.
Endothelial-specific autophagy-deficient mice exhibit GEnC injury
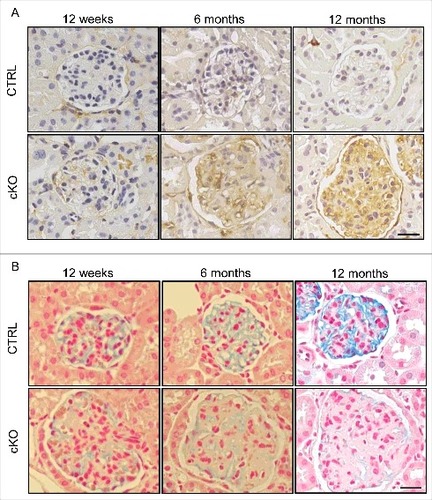
We analyzed the effects of autophagy deficiency on GEnC injury. Immunohistochemical analysis showed the expression of ICAM1 (intercellular adhesion molecule 1) in the glomeruli of 6-mo-old atg5-cKO recipients, but not in the glomeruli of their Atg5-CTRL recipient littermates; this expression of ICAM1 became more prominent in the 12-mo-old atg5-cKO recipients (A). We next examined the glycocalyx of GEnCs, which regulates glomerular microvessel permeability.Citation18,Citation19 We observed a decrease in colloidal iron staining in atg5-cKO recipients when compared to their Atg5-CTRL recipient littermates at the age of 12 wk and 6 mo. This decrease became more prominent in the 12-mo-old atg5-cKO recipients, indicating a marked reduction of the glycocalyx (B). These observations demonstrate the GEnC injury in endothelial-specific autophagy-deficient mice.
Figure 6. Endothelial-specific autophagy-deficient mice exhibit GEnC injury. Representative images of staining for ICAM1 (A) and colloidal iron (B) in the Atg5-CTRL and atg5-cKO mice at the indicated ages after rescue by BMT at 4 wk of age (Atg5-CTRL recipients and atg5-cKO recipients, respectively). Bars: 20 μm.
Antioxidant treatment alleviates the abnormality in endothelial-specific autophagy-deficient mice
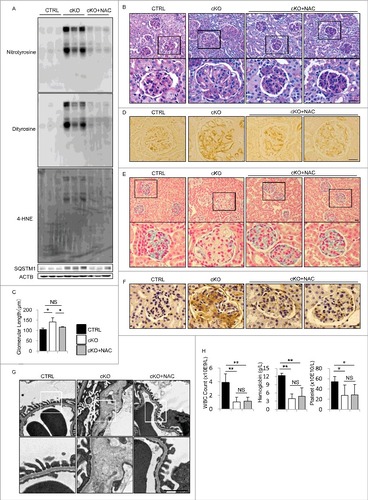
To investigate whether the increased oxidative stress observed in the glomeruli of atg5-cKO mice could lead to the glomerular abnormalities, we treated atg5-cKO mice with an antioxidant, N-acetyl-l-cysteine (NAC), for 8 wk. Administration of NAC via drinking water (5 mg/mL) from birth significantly reduced the accumulation of ROS (as assessed by the protein levels of dityrosine, nitrotyrosine, and 4-HNE) and SQSTM1 in the kidney of atg5-cKO mice, suggesting that the antioxidant alleviates autophagy dependency in atg5-cKO mice (A). PAS staining, the assessment of diameter of glomeruli, and immunohistochemical analysis for PECAM1 revealed that the administration of NAC significantly ameliorated the glomerular pathologies, such as distended capillary loops and glomerular enlargement (B to D). GEnC injury was significantly alleviated, as seen by the restoration of the glycocalyx of GEnCs, a reduction in ICAM1 expression, and amelioration of detachment of GEnCs (E to G). Furthermore, the administration of NAC significantly ameliorated foot process effacement of podocytes (G). Notably, the administration of NAC did not improve pancytopenia, suggesting that the reduced oxidative stress did not restore hematopoietic cell function (H). Collectively, pharmacological intervention with an antioxidant alleviated the abnormalities of endothelial-specific autophagy-deficient mice.
Figure 7. Administration of NAC alleviates the abnormalities of endothelial-specific autophagy-deficient mice. (A) Western blot analysis for nitrotyrosine, dityrosine, 4-HNE, and SQSTM1 using kidney cortex lysates from 8-wk-old atg5-cKO mice, atg5-cKO mice with NAC administration (atg5-cKO+NAC), and their Atg5-CTRL littermates. (B, D to H) Representative images of PAS staining (B), immunohistochemical analysis for PECAM1 (D), colloidal iron staining (E), immunohistochemical analysis for ICAM1 (F), electron micrographs (G), and peripheral blood cell counts (H) in the glomeruli of 8-wk-old atg5-cKO, atg5-cKO+NAC, and Atg5-CTRL mice. In (B), (E), and (G), the lower panels show a magnification of the indicated areas (black or white squares) in the upper panels. (C) For each condition, the diameter of the glomeruli was calculated from at least 20 glomeruli in each mouse. n = 3 (A, B, D to G); 4 (C); and 4 to 7 (H) in each group. Data are provided as the mean ± SD. NS, not significant. Statistically significant differences (*P < 0.05, **P < 0.01 versus the Atg5-CTRL or atg5-cKO+NAC mice) are indicated. Bars: 20 μm (B, D to F) and 1 μm (G).
Discussion
In this study, we demonstrated that endothelial autophagy protects glomeruli from oxidative stress and maintains the integrity of glomerular capillaries.
A prominent finding in our study is that the GEnCs have a substantial capacity to upregulate autophagic activity in response to starvation. Elucidating the autophagic flux in vivo is difficult and is thus usually substituted by an assessment of the number of autophagosomes; however, the number or volume of autophagosomes does not always reflect the autophagic flux, because theoretically and in many cases autophagosomes accumulate due to a block in trafficking to lysosomes without a concomitant increase in autophagosome biogenesis. A high autophagic flux in GEnCs is partially supported by the observation that the ATG5 protein level is significantly reduced in the isolated glomeruli of 4-wk-old atg5-cKO mice when compared to the glomeruli of their Atg5-CTRL littermates, despite the glomeruli contain autophagy-competent cells of other cell lineages (D to F). This unexpected finding led us to explore the role of autophagy in GEnCs.
The glomeruli of 8-wk-old atg5-cKO mice exhibited a lobular pattern with thickening of the capillary loops and mesangial matrix expansion, suggesting that autophagy maintains the integrity of the glomerular capillaries. While preparing this manuscript, Lenoir O. et al. report similar but milder phenotypes of another strain of EC-specific atg5-knockout (Atg5-floxed Cdh5/VE-cadherin [cadherin 5]-Cre+) mice (atg5F/F ;Cdh5-Cre+); the atg5F/F ;Cdh5-Cre+ mice are indiscernible from their control littermates in terms of weight, size, albuminuria, and UN levels up to 20 wk of age.Citation20 The glomeruli from atg5F/F ;Cdh5-Cre+ mice display slightly dilated capillaries with discrete podocyte foot process effacements and the loss of glomerular endothelial fenestrations from 10 wk of age. The differences in the phenotypes between the 2 lines of EC-specific atg5-knockout mice (the atg5F/F ;Cdh5-Cre+ mice display a milder phenotype) may be due to the activity and/or site specificity of Cre recombinase; in CDH5-Cre+ mice, approximately 75% of the GEnCs express Cre recombinase,Citation20 whereas almost all ECs in Tek-Cre+ mice express Cre recombinase (Fig. S1). Alternatively, malnutrition, manifesting as pancytopenia and body weight loss, may make the atg5-cKO mice more dependent on autophagy. In either case, our study has some advantages over the previous study: we clearly demonstrated that GEnCs, and not hematopoietic cells, are responsible for the defects in atg5-cKO mice in a BMT experiment, and we provided some insight into the mechanism of the autophagy dependency of GEnCs (see below).
Recently, the prominent roles of endothelial injury in the initiation or progression of kidney injury have emerged. An increased production of ROS, in addition to a reduced bioavailability of NO, is involved in endothelial injury. NOX (nicotinamide adenine dinucleotide phosphate oxidase) contributes to glomerular ROS production during diabetic nephropathy and aging.Citation21,Citation22 In addition, endothelial mitochondria play a prominent role in the production of ROS; in contrast to cardiac myocytes and PTECs, glycolysis is the major source of adenosine triphosphate (ATP) production in ECs, but it is now recognized that endothelial mitochondria-derived ROS promote inflammation and decrease NO bioavailability.Citation23,Citation24 In this study, western blot analysis for COX4I1 (cytochrome c oxidase subunit 4I1) using isolated glomeruli of 8-wk-old mice revealed a significant decrease in atg5-cKO mice compared with Atg5-CTRL mice, suggesting inadequate autophagy contributes to mitochondrial dysfunction in glomeruli (Fig. S8A). However, it was difficult to observe mitochondria in GEnCs by electron microscopy because of their scanty cytoplasm. By contrast, the mRNA expression of Cyba/p22phox and Ncf1/p47phox, NOX subunits, in whole kidney lysate from 8-wk-old mice was significantly increased in atg5-cKO mice compared with Atg5-CTRL mice (Fig. S8B). These data suggest that ROS derived from NOX rather than mitochondria may be a major culprit in glomerular damage. In any case, ROS can initiate autophagic degradationCitation25,Citation26 and autophagy, in turn reducing oxidative stress through the removal of irreversibly oxidized biomolecules (protein aggregates, DNA, and lipids) and damaged mitochondria.Citation17,Citation27 The observation that the administration of NAC alleviates glomerular defects and reduces the accumulation of SQSTM1 without restoring pancytopenia in atg5-cKO mice clearly suggests that increased oxidative stress is responsible for the glomerular abnormalities and that autophagy in ECs, but not in hematopoietic cells, maintains the integrity of glomerular capillaries by removing and/or restoring oxidized biomolecules and damaged mitochondria.
In this study, we observed effacement of podocyte foot processes adjacent to the transformed ECs even in 4-wk-old atg5-cKO mice. It has been reported that NOS3 deficiency contributes to podocyte injury, highlighting the importance of communication between GEnCs and podocytes.Citation28,Citation29 In this study, western blot analysis has revealed a significant decrease in the ratio of phosphorylated NOS3 (p-NOS3; Ser1177) to total NOS3 in isolated glomeruli of 8-wk-old atg5-cKO mice (Fig. S9). This observation indicates that defective autophagy may contribute to NOS3 inactivation in GEnCs, leading to podocyte injury, which is in good agreement with a recent report.Citation30
The biggest problem that should be solved is why the GEnCs were the only cells among the ECs that were sensitive to autophagy deficiency. Accumulating evidence has suggested that GEnCs are quite unique in their exquisite sensitivity to, or dependence on, the VEGFA (vascular endothelial growth factor A)-KDR/VEGFR2 (kinase insert domain protein receptor) axis; indeed, whole-body deletion of Kdr in adult mice causes marked abnormalities mainly, but not exclusively, in the kidney,Citation31 and glomerular injury is commonly observed in patients receiving antiangiogenic agents that target the VEGFA pathway.Citation32 ROS induce the expression of KDR.Citation33 In another report, ROS derived from NOX enhances KDR autophosphorylation, and subsequent redox signaling is linked to angiogenic responses, such as EC proliferation and migration.Citation34 Induction of podocyte VEGFA overexpression after birth causes massive albuminuria accompanied by glomerular enlargement, and podocyte effacement and fusion in mice.Citation35 In line with these reports, in this study, the protein level of KDR increased in the isolated glomeruli of the atg5-cKO mice (Fig. S10), which may have modulated the glomerular abnormalities in the atg5-cKO mice.
In conclusion, endothelial autophagy protects glomeruli from oxidative stress and maintains the integrity of glomerular capillaries. Enhancing endothelial autophagy may provide a novel therapeutic approach to minimizing glomerular diseases.
Materials and methods
Animals
Tek-Cre transgenic miceCitation36 and CAG-ChAT-EGFP transgenic miceCitation37 were used after backcrossing with C57BL/6N mice more than 10 times. CAG-ChAT-EGFP mice have a loxP-flanked ChAT sequence upstream from EGFP sequence, both under the transcriptional control of a CAG promotor. When bred to mice that express Cre recombinase, the resulting offspring expresses EGFP in Cre expressing tissues. Mice bearing an Atg5flox allele (Atg5F/F mice), in which exon 3 of the Atg5 gene is flanked by 2 loxP sequences, with a C57BL/6N background have been described previously.Citation38 Progeny containing the Atg5flox allele were bred with the Tek-Cre transgenic mice to generate atg5-cKO mice. In NAC treatment experiment, we started to give drinking water containing NAC (5 mg/mL; Sigma-Aldrich, A7250) just from birth, and replaced it every few days. Although the transplacental uptake is controversial, it is certain that they could continuously take NAC for at least 6 wk (between 2 and 8 wk of age). All mice were allowed free access to water and standard mouse chow (Oriental Yeast, MF). All animal experiments were approved by the institutional committee of the Animal Research Committee of Osaka University and were in accordance with the Japanese Animal Protection and Management Law (No. 25).
Blood pressure recordings
Systolic and diastolic blood pressure was measured by tail-cuff plethysmography (HEROTEC, Kyoto, Japan).
Antibodies and reagents
The antibodies and reagents used included the following: antibodies for PECAM1 (Thermo Fisher Scientific, ER-MP12), MAP1LC3B (Cell Signaling Technology [CST], 2775), SQSTM1 (Medical and Biological Laboratory [MBL], PM045), ATG5 (MBL, PM050), ACTB (Sigma-Aldrich, A5316), KIT-APC (BioLegend, 105811), LY6A-PE (BioLegend, 122507), biotinylated lineage markers (PTPRC/CD45R, ITGAM/CD11b, LY6G/Gr-1, CD8A, CD4, and LY76; BioLegend, 103203, 101203, 108403, 100703, 100403, 116203, respectively), CD44-APC (Biolegend, 103011), ADGRE1 (BIO-RAD, MCA497), dityrosine (Japan Institute for the Control of Aging [JaICA], MDT-020P), nitrotyrosine (Abcam, ab7048), 4-HNE (JaICA, MHN-020P), ICAM1 (ReliaTech, 103-M97), COL1A1 (Abcam, ab34710), COX4I1 (MBL, PM063), NOS3 (CST, 32027), p-NOS3 Ser1177 (CST, 9570), Alexa Fluor 555-conjugated secondary antibodies (Molecular Probes, A21434 [anti-rat IgG], A31572 [anti-rabbit IgG]), biotinylated secondary antibodies (Vector Laboratories, BA-1000 [anti-rabbit IgG], BA-2001 [anti-mouse IgG]), and horseradish peroxidase–conjugated secondary antibodies (Dako, P0448 [anti-rabbit IgG], P0447 [anti-mouse IgG]); chloroquine (Sigma-Aldrich, C6628); NAC (as noted above); G418 disulfate (Nacalai tesque, 08973-14); and polymyxin B (Sigma-Aldrich, P1004).
Isolation of glomeruli
The isolation of glomeruli was conducted as described previously.Citation39 In brief, mice were perfused through the heart with magnetic Dynabeads (4.5 μm in diameter; Life Technologies, 14013). Kidneys were minced into small pieces, then digested by collagenase type II (Gibco BRL, 17101-015) and deoxyribonuclease I (Invitrogen, 11533D) in Hank's balanced salt solution (Gibco, 14025–092). The digested tissue was filtered through a 100-μm cell strainer (Thermo Fisher Scientific, 352360) and collected using a magnetic particle concentrator (Invitrogen, 12320D).
Histological analysis
Histological analysis was performed as previously describedCitation40 with some modifications. Mice were transcardially perfused using 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4; composed of 137 mM NaCl [Wako, 191-01665], 2.7 mM KCl [Nacalai tesque, 28514-75], 8 mM Na2HPO4 [Wako, 196-02835], and 1.5 mM KH2PO4 [Wako, 169-04245]). Tissues were postfixed and embedded in paraffin or frozen in OCT compound (Sakura Finetek, 4583). For ICAM1 staining, tissues were fixed in zinc fixative (BD Biosciences, 550523). Paraffin sections were stained with PAS, Masson-trichrome (for kidney), or HE (for other organs). Paraffin-embedded (for PECAM1, dityrosine, ICAM1, and ADGRE1 staining) or fixed-frozen (for PECAM1 and SQSTM1 staining) sections were immunostained with the respective antibodies. Immunohistochemical staining for PECAM1 and dityrosine was performed after antigen retrieval via autoclaving in 0.01 mM citrate buffer (pH 6.0) for 10 min at 120°C and blocking with 1.5% bovine serum albumin (Sigma-Aldrich, A3059) in PBS for 60 min. Immunohistochemical staining for ADGRE1 was performed after enzyme pretreatment using a proteinase K solution (20 μg/ml, Roche, 03115828001) in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) for 3 min at room temperature. Immunofluorescence images were obtained using an Olympus FV1000-D confocal laser scanning microscope (Olympus, Tokyo, Japan). For electron microscopy, kidneys were fixed with 2% glutaraldehyde (Wako, 071–01931) and observed using a Hitachi H-7650 transmission electron microscope (Hitachi, Tokyo, Japan). The procedures for immunohistochemistry in whole-mount-retinasCitation41,Citation42 and -mesenteries,Citation43 and cationic colloidal iron stainingCitation44 have been described in the indicated references.
Western blot analysis
Isolated glomeruli or kidney cortices were lysed in cell lysis buffer (CST, 9803) and a protease inhibitor cocktail, Complete Mini (Roche, 11836170001). The protein concentration was determined using Pierce BCA Protein Assay Reagent (Thermo Fisher Scientific, 23228). Each sample was subjected to immunoblot analysis as described previously.Citation40
Flow cytometry
BM cells were collected by flushing femurs and tibias with PBS containing 1% fetal bovine serum. The suspension was passed through a 70-μm nylon cell strainer (BD Falcon, 352350) to obtain a single-cell suspension. Cells were incubated with biotinylated lineage markers, anti-LY6A-PE, and anti-KIT-APC for the analysis of hematopoietic cell differentiation; with anti-CD44-APC and biotinylated anti-LY76 for analysis of erythroid progenitor differentiation. After 20 min of incubation on ice, the BM cells were washed twice and incubated with streptavidin-488 (Molecular Probes, S32354) for 20 min on ice. Stained cells were resuspended in PBS supplemented with 1% fetal bovine serum and analyzed on a FACSCanto II apparatus (BD Biosciences, San Jose, CA, USA). Multi-parameter data were collected and analyzed using BD FACSDIVA™ (BD Biosciences).
Bone marrow transplantation
Four-wk-old recipient mice (Atg5-CTRL or atg5-cKO) were fasted overnight and lethally irradiated with 7.5 Gy from an X-ray source. BM was collected from age- and sex-matched donors by flushing the femurs and tibias with cold Dulbecco's modified Eagle's medium (Gibco, 08456-36). The BM cells were counted, pelleted by centrifugation, and resuspended in cold PBS just before injection. One h after the irradiation, BM cells (2 × 106/mouse) were injected intravenously into the recipient mice. Transplanted animals were provided with antibiotic-supplemented water (0.1 g G418 disulfate and 100,000 units of polymyxin B/100 ml) for 4 wk postradiation.
Peripheral blood cell counts and biochemical measurements
Peripheral blood cells were counted using Vetscan HM II (Abaxis, Union City, CA, USA). The urinary levels of albumin, glucose, and phosphate were measured using the Microfluoral™ microalbumin test (Progen Biotechnik GmbH, PR2005), Glucose CII Test (Wako), and Phosphor C Test (Wako), and then adjusted to the urinary creatinine level, which was measured using the CRE-EN Kainos kit (Kainos, TKA7500). Urinary amino acids were measured by pressure-assisted capillary electrophoresis-mass spectrometry as previously described.Citation45 The plasma UN concentration was measured using the BUN-test-Wako kit (Wako, 279–36201). Plasma levels of total protein, albumin, globulin, GPT/ALT (glutamic pyruvic transaminase), AMY/amylase, total bilirubin, calcium, and phosphorus were measured using Vetscan VS2 (Abaxis, Union City, CA, USA).
Quantitative RT-PCR analysis
Quantitative RT-PCR analysis was performed as previously described.Citation40 The sequences of primers used were as follows:
Cyba-F 5’-GCACACCGCCATCCACAC-3’
Cyba-R 5’-CCAGCCAACCGAGTCACG–3’
Ncf1-F 5’-CCATCATCCTTCAGACCTATCG-3’
Ncf1-R 5’-CCGCTCTCGCTCTTCTCC–3’
Peritubular capillary density analysis
After immunostaining for PECAM1, each peritubular capillary area was traced and calculated using NIS-Elements Basic Research Microscope Imaging Software (Nikon, Tokyo, Japan). The density was calculated by dividing total cross-section of the peritubular capillaries by the whole area of the field. At least 10 high-power fields (× 1000) were examined for each mouse.
Statistical analysis
All results are expressed as the mean ± standard deviation (SD). Statistical analyses were conducted using JMP software (SAS Institute, Cary, NC). Differences between 2 experimental values were assessed by the Student t test or Wilcoxon signed-rank test as appropriate. Multiple-group comparisons were performed using analysis of variance with post hoc testing using the Tukey-Kramer test. A P < 0.05 was considered to be statistically significant.
Abbreviations
| ACTB | = | actin, beta |
| ADGRE1/F4/80 | = | adhesion G protein-coupled receptor E1 |
| ATG | = | autophagy related |
| ATP | = | adenosine triphosphate |
| BM | = | bone marrow |
| BMT | = | bone marrow transplantation |
| ChAT | = | chloramphenicol acetyltransferase |
| CDH5/VE-cadherin | = | cadherin 5 |
| cKO | = | conditional knockout |
| COL1A1 | = | collagen, type I, alpha 1 |
| COX4I1 | = | cytochrome c oxidase subunit 4I1 |
| CTRL | = | control |
| EC | = | endothelial cell |
| EGFP | = | enhanced green fluorescence protein |
| GBM | = | glomerular basement membrane |
| GEnC | = | glomerular endothelial cell |
| HE | = | hematoxylin-eosin |
| HUVEC | = | human umbilical vein endothelial cell |
| ICAM1 | = | intercellular adhesion molecule 1 |
| KDR/VEGFR2 | = | kinase insert domain protein receptor |
| KIT/c-KIT | = | KIT proto-oncogene receptor tyrosine kinase |
| Lin | = | lineage marker |
| LY6A/SCA1 | = | lymphocyte antigen 6 complex, locus A |
| LY76/TER119 | = | lymphocyte antigen 76 |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| NAC | = | N-acetyl-l-cysteine |
| NO | = | nitric oxide |
| NOS3 | = | nitric oxide synthase 3, endothelial cell |
| NOX | = | nicotinamide adenine dinucleotide phosphate oxidase |
| PAS | = | periodic-acid Schiff |
| PECAM1/CD31 | = | platelet/endothelial cell adhesion molecule 1 |
| PTEC | = | proximal tubular epithelial cell |
| ROS | = | reactive oxygen species |
| SQSTM1/p62 | = | sequestosome 1 |
| TEK/Tie2 | = | endothelial-specific receptor tyrosine kinase |
| TGFB1 | = | transforming growth factor, beta 1 |
| UN | = | urea nitrogen |
| VEGFA | = | vascular endothelial growth factor A |
| VWF | = | von Willebrand factor |
| 4-HNE | = | 4-hydroxy-2-nonenal |
Disclosure of potential conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary_Data.zip
Download Zip (10.4 MB)Acknowledgement
We thank N. Mizushima (University of Tokyo) for the Atg5F/F mice, M. Yanagisawa (Howard Hughes Medical Institute and University of Tsukuba) for the Tek-Cre transgenic mice, J. Miyazaki (Osaka University) for the CAG-ChAT-EGFP mice, and N. Horimoto, K. Endo, and S. Ashitani for technical assistance.
Additional information
Funding
Related Research Data
Reference
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi:10.1038/nature06639. PMID:18305538.
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi:10.1016/j.molcel.2010.09.023. PMID:20965422.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi:10.1016/j.cell.2011.10.026. PMID:22078875.
- Takabatake Y, Kimura T, Takahashi A, Isaka Y. Autophagy and the kidney: health and disease. Nephrol Dial Transplant. 2014;29:1639–47. doi:10.1093/ndt/gft535. PMID:24520117.
- Guo F, Li X, Peng J, Tang Y, Yang Q, Liu L, Wang Z, Jiang Z, Xiao M, Ni C, et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann Biomed Eng. 2014;42:1978–88. doi:10.1007/s10439-014-1033-5. PMID:24838486.
- LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. 2012;590:3305–16. doi:10.1113/jphysiol.2012.229690. PMID:22570377.
- Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs C a, Wu XS, Rovira II, Fergusson MM, Weigert R, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19:1281–7. doi:10.1038/nm.3288. PMID:24056772.
- Cheng H, Harris RC. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc Hematol Disord Drug Targets. 2014;14:22–33. doi:10.2174/1871529X14666140401110841. PMID:24720460.
- Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol. 2015;308:F287–97. doi:10.1152/ajprenal.00533.2014. PMID:25411387.
- Eleftheriadis T, Antoniadi G, Pissas G, Liakopoulos V, Stefanidis I. The renal endothelium in diabetic nephropathy. Ren Fail. 2013;35:592–9. doi:10.3109/0886022X.2013.773836. PMID:23472883.
- Takahashi T, Harris RC. Role of endothelial nitric oxide synthase in diabetic nephropathy: lessons from diabetic eNOS knockout mice. J Diabetes Res. 2014;. 2014:590541. doi:10.1155/2014/590541. PMID:25371905.
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi:10.1091/mbc.E03-09-0704. PMID:14699058.
- Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–96. doi:10.1172/JCI39492. PMID:20200449.
- Constien R, Forde A, Liliensiek B, Gröne HJ, Nawroth P, Hämmerling G, Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi:10.1002/gene.1030. PMID:11353516.
- Mortensen M, Ferguson DJP, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A. 2010;107:832–7. doi:10.1073/pnas.0913170107. PMID:20080761.
- Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol. 2015;35:615–21. doi:10.1007/s10571-015-0166-x. PMID:25722131.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–88. doi:10.1038/cdd.2014.150. PMID:25257172.
- Fridén V, Oveland E, Tenstad O, Ebefors K, Nyström J, Nilsson UA, Haraldsson B. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 2011;79:1322–30. doi:10.1038/ki.2011.58. PMID:21412215.
- Salmon AHJ, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, Bates DO, Peti-Peterdi J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23:1339–50. doi:10.1681/ASN.2012010017. PMID:22797190.
- Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. 2015;11:1130–45. doi:10.1080/15548627.2015.1049799. PMID:26039325.
- Satoh M, Fujimoto S, Haruna Y, Arakawa S, Horike H, Komai N, Sasaki T, Tsujioka K, Makino H, Kashihara N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2005;288:F1144–52. doi:10.1152/ajprenal.00221.2004. PMID:15687247.
- Meyer MR, Barton M, Prossnitz ER. Functional heterogeneity of NADPH oxidase-mediated contractions to endothelin with vascular aging. Life Sci. 2014;118:226–31. doi:10.1016/j.lfs.2013.12.021. PMID:24382462.
- Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112:1171–88. doi:10.1161/CIRCRESAHA.111.300233. PMID:23580773.
- Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011;15:1517–30. doi:10.1089/ars.2010.3642. PMID:21194353.
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi:10.1038/sj.emboj.7601623. PMID:17347651.
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–52. doi:10.1038/cdd.2009.49. PMID:19407826.
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi:10.1016/j.abb.2007.03.034. PMID:17475204.
- Yuen DA, Stead BE, Zhang Y, White KE, Kabir MG, Thai K, Advani SL, Connelly KA, Takano T, Zhu L, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol. 2012;23:1810–23. doi:10.1681/ASN.2011121170. PMID:22997257.
- Ueda S, Ozawa S, Mori K, Asanuma K, Yanagita M, Uchida S, Nakagawa T. ENOS deficiency causes podocyte injury with mitochondrial abnormality. Free Radic Biol Med. 2015;87:181–92. doi:10.1016/j.freeradbiomed.2015.06.028. PMID:26119782.
- Fetterman JL, Holbrook M, Flint N, Feng B, Bretón-Romero R, Linder EA, Berk BD, Duess M-A, Farb MG, Gokce N, et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis. 2016;247:207–17. doi:10.1016/j.atherosclerosis.2016.01.043. PMID:26926601.
- Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–701. doi:10.1681/ASN.2010030295. PMID:20688931.
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–36. doi:10.1056/NEJMoa0707330. PMID:18337603.
- González-Pacheco FR, Deudero JJP, Castellanos MC, Castilla MA, Alvarez-Arroyo MV, Yagüe S, Caramelo C. Mechanisms of endothelial response to oxidative aggression: protective role of autologous VEGF and induction of VEGFR2 by H2O2. Am J Physiol Heart Circ Physiol. 2006;291:H1395–401. doi:10.1152/ajpheart.01277.2005. PMID:16899768.
- Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–35. doi:10.1016/j.cardiores.2006.04.015. PMID:16781692.
- Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, Shen W, Kopp JB, Thomas DB, Tufro A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010;77:989–99. doi:10.1038/ki.2010.64. PMID:20375978.
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi:10.1006/dbio.2000.0106. PMID:11161575.
- Kawamoto S, Niwa H, Tashiro F, Sano S, Kondoh G, Takeda J, Tabayashi K, Miyazaki J. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett. 2000;470:263–8. doi:10.1016/S0014-5793(00)01338-7. PMID:10745079.
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi:10.1038/nature04724. PMID:16625204.
- Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y, Betsholtz C. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161:799–805. doi:10.1016/S0002-9440(10)64239-3. PMID:12213707.
- Yamamoto T, Takabatake Y, Kimura T, Takahashi A, Namba T, Matsuda J, Minami S, Kaimori J-Y, Matsui I, Kitamura H, et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy. 2016;12:801–13. doi:10.1080/15548627.2016.1159376. PMID:26986194.
- Uemura A, Kusuhara S, Wiegand SJ, Yu RT, Nishikawa S. Tlx acts as a proangiogenic switch by regulating extracellular assembly of fibronectin matrices in retinal astrocytes. J Clin Invest. 2006;116:369–77. doi:10.1172/JCI25964. PMID:16424942.
- Fukushima Y, Okada M, Kataoka H, Hirashima M, Yoshida Y, Mann F, Gomi F, Nishida K, Nishikawa S-I, Uemura A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest. 2011;121:1974–85. doi:10.1172/JCI44900. PMID:21505259.
- Yoshida H, Kunisada T, Kusakabe M, Nishikawa S, Nishikawa SI. Distinct stages of melanocyte differentiation revealed by anlaysis of nonuniform pigmentation patterns. Development. 1996;122:1207–14. PMID:8620847.
- Zhang D, Isaka Y, Imamura R, Ichimaru N, Shi Y, Imai E, Tian Y, Ohtsuka A, Takahara S. Glycocalyx damage estimated using colloidal iron staining. Cell Transplant. 2008;17:159–63. doi:10.3727/000000008783907134. PMID:18468245.
- Soga T, Ishikawa T, Igarashi S, Sugawara K, Kakazu Y, Tomita M. Analysis of nucleotides by pressure-assisted capillary electrophoresis-mass spectrometry using silanol mask technique. J Chromatogr A. 2007;1159:125–33. doi:10.1016/j.chroma.2007.05.054. PMID:17543971.