
ABSTRACT
M. tuberculosis causes an enormous worldwide burden of disease. Its success depends upon subverting the antimicrobial capacity of macrophages. We have known for decades that M. tuberculosis impairs phagosomal trafficking to avoid lysosomal degradation, but the mechanism is unclear. Recent work has described a phagolysosomal pathway called LC3-associated phagocytosis (LAP), in which LC3 associates with microbe-containing phagosomes. Macrophage pathogen recognition receptors (PRRs) initiate LAP, and NADPH oxidase and RUBCN/RUBICON are required for LAP. We discovered that CpsA, an exported M. tuberculosis virulence factor, blocks LAP by interfering with recruitment of CYBB/NOX2 (cytochrome b-245, beta polypeptide) to the mycobacterial phagosome. In macrophages and in mice, M. tuberculosis mutants lacking cpsA are successfully cleared by NADPH oxidase and the ensuing LC3-associated lysosomal trafficking pathway. CpsA belongs to the LytR-CpsA-Psr family, which is found widely in Gram-positive bacilli. This family is known for its enzymatic role in cell wall assembly. However, our data suggest that CpsA inhibits CYBB oxidase independently of a cell wall function. Thus, CpsA may have evolved from an enzyme involved in cell wall integrity to an indispensable virulence factor that M. tuberculosis uses to evade the innate immune response.
LAP is distinct from canonical macroautophagy/autophagy. Whereas both depend on some of the same autophagy-related (ATG) proteins, they have unique requirements. During autophagy of invading microbes (called xenophagy), microorganisms are captured by an LC3-decorated, double-membrane transient organelle, the phagophore. The autophagy initiation complex is involved in generating this double-membrane compartment. In LAP, LC3 is conjugated to a single-membrane phagosome in a process that does not depend on the autophagy initiation complex. LAP is initiated by signaling through pathogen recognition receptors such as TLR2 (toll-like receptor 2) and C-type lectin receptors (CLRs), which promote NADPH oxidase recruitment to the phagosome. NADPH oxidase generates reactive oxygen species (ROS), which are required for LAP. RUBCN, which is also required for LAP, stabilizes the NADPH oxidase. Thus, LAP is distinguished from xenophagy based on its dependence on NADPH oxidase and RUBCN, whereas xenophagy depends on the autophagy initiation complex.
Neither of these lysosomal trafficking pathways effectively controls M. tuberculosis. Whereas several studies have explored how M. tuberculosis inhibits xenophagy, none address how it blocks LAP. It is puzzling that M. tuberculosis does not robustly activate LAP, because the tubercle bacilli have numerous TLR2 and CLR ligands. Yet, in mice, neither the NADPH oxidase nor ATG proteins make a substantial contribution to M. tuberculosis control. One interpretation is that the NADPH oxidase and LC3 trafficking are not important in M. tuberculosis pathogenesis. However, both are clearly important in host defense against other microbes. Our study suggests an alternative explanation: there is no difference in the burden of M. tuberculosis in mice deficient in CYBB and LC3-trafficking because M. tuberculosis effectively inactivates these host defenses.
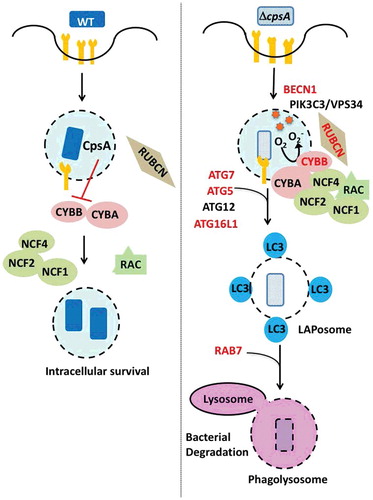
We hypothesized that CpsA might inhibit xenophagy after we found that it interacted with xenophagy receptor proteins, CALCOCO2/NDP52 and TAX1BP1, in a yeast 2-hybrid screen. Consistent with our initial idea, when we compared the ∆cpsA mutant to the wild-type (WT) control, the mutant exhibited dramatically enhanced LC3-associated lysosomal trafficking. Moreover, whereas the ∆cpsA mutant grows normally in broth culture, it is highly attenuated in macrophages. The intracellular growth defect of the ∆cpsA mutant is restored in macrophages deficient in BECN1, ATG5, ATG7 or ATG16L1; these factors are required for both LAP and xenophagy. When we tested unique pathway components, we were surprised to find that the intracellular growth defect is also rescued in macrophages lacking RUBCN or CYBB. In contrast, there is little rescue in macrophages lacking xenophagy-specific factors (ATG14, ULK1, and MB21D1/cGAS). These findings suggested that the ∆cpsA mutant is cleared by LAP, not xenophagy. Consistent with the genetic data, we found that CpsA blocked CYBB recruitment and ROS generation on phagosomes harboring WT M. tuberculosis, thereby blocking downstream LAP-mediated killing ().
Figure 5. CpsA inhibits phagosomal recruitment of NADPH oxidase and prevents LAP. WT M. tuberculosis (left) exports CpsA, which prevents recruitment of the NADPH oxidase (which is composed of subunits CYBA/p22phox, CYBB/gp91phox/NOX2, NCF4/p40phox, NCF1/p47phox, NCF2/p67phox). The NADPH oxidase is activated by GTP-bound RAC1-RAC2 and is stabilized by RUBCN. When CpsA is lacking (right), the NADPH oxidase is recruited to the phagosome, which results in ROS production. Subsequently LC3 is recruited to the phagosomal membrane through the ATG conjugation system (ATG5, ATG7, ATG12, ATG16L1, and others). The LAPosome matures and is delivered to the lysosome, which depends upon RAB7. Proteins in red are those that are required for macrophages to kill the ∆cpsA mutant.
Our results in vivo were consistent with our findings in macrophages. The ∆cpsA mutant is attenuated early in infection when there is only an innate immune response. As we found in vitro, the growth defect of the ∆cpsA mutant is partially rescued in mice lacking the NADPH oxidase CYBB (cybb/nox2−/−) or ATG5 and ATG7 in their myeloid cells. In contrast, xenophagy-specific mouse mutants (Atg14 cKO, Mb21d1/cGAS KO) fail to rescue. Together, our data demonstrate that CpsA is a significant mediator of the resistance of M. tuberculosis to NADPH oxidase and LC3-trafficking in vivo.
The next step was to determine how CpsA works in this context. CpsA harbors 2 domains: a LytR-CpsA-Psr (LCP) domain and a LytR domain. The LCP domain is widely conserved in Gram-positive bacteria and thought to transfer cell wall polysaccharides from lipid bound precursors to peptidoglycan. One straightforward idea that we considered is that a cell wall defect in the ∆cpsA mutant might expose more pathogen-associated molecular patterns that drive LAP. However, our data do not support this. First, the cell wall phenotypes of the ∆cpsA mutant are subtle at best. Moreover, PRR signaling in macrophages is not increased in response to the mutant relative to WT bacilli. To test whether CpsA inhibits LAP based upon an activity independent of cell wall metabolism, we examined whether it could alter trafficking of fungal zymosan. Normally zymosan recruits the NADPH oxidase and activates LAP, but when we transfect macrophages with CpsA, CYBB is no longer recruited. Thus, CpsA is sufficient to inhibit LAP, consistent with the idea that it impairs trafficking independently of any mycobacterial cell wall function.
Ongoing work is aimed at defining how CpsA modulates NADPH oxidase recruitment and identifying other effectors that inhibit LAP and xenophagy. These mechanisms could be treatment targets. To this end, it will be important to determine whether catalytic activity of CpsA is required for it to block the NADPH oxidase. Another outstanding question is whether CpsA actually binds CALCOCO2 or TAX1BP1 during infection and interferes with their functions. While CALCOCO2 and TAX1BP1 are involved in xenophagy, they have not yet been implicated in LAP. Whereas our data show that LAP makes the major contribution to bacterial killing, it is possible that CpsA inhibits both LAP and xenophagy. Numerous host-directed therapies under investigation activate LC3 pathways, but we do not yet know whether they activate xenophagy or LAP. A more refined understanding of these LC3-trafficking pathways during M. tuberculosis infection may reveal differences in their antimicrobial capacities and identify opportunities for combination therapy.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.