
ABSTRACT
Autophagy is required for cellular homeostasis and can determine cell viability in response to stress. It is established that MTOR is a master regulator of starvation-induced macroautophagy/autophagy, but recent studies have also implicated an essential role for the MAPK8/cJun NH2-terminal kinase 1 signal transduction pathway. We found that MAPK8/JNK1 and MAPK9/JNK2 were not required for autophagy caused by starvation or MTOR inhibition in murine fibroblasts and epithelial cells. These data demonstrate that MAPK8/9 has no required role in starvation-induced autophagy. We conclude that the role of MAPK8/9 in autophagy may be context-dependent and more complex than previously considered.
Abbreviations: AKT: thymoma viral proto-oncogene;ALB: albumin; ATG4: autophagy related 4; BCL2: B cell leukemia/lymphoma 2; BECN1: beclin 1, autophagy related; BNIP3: BCL2/adenovirus E1B interacting protein 3; CQ: chloroquine diphosphate; DMEM: Dulbecco’s modified Eagle’s medium; EDTA: ethylenediaminetetraacetic acid; EBSS: Earle’s balanced salt solution; FBS: fetal bovine serum; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; HRAS: Harvey rat sarcoma virus oncogene; IgG: Immunoglobulin G; MAPK3/ERK1: mitogen-activated protein kinase 3; MAPK8/JNK1: mitogen-activated protein kinase 8; MAPK9/JNK2: mitogen-activated protein kinase 9; MAPK10/JNK3: mitogen-activated protein kinase 10; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MEFs: mouse embryonic fibroblasts; MTOR: mechanistic target of rapamycin kinase; RPS6KB1/p70: ribosomal protein S6 kinase, polypeptide 1; PPARA: peroxisome proliferator activated receptor alpha; SEM: standard error of the mean; SQSTM1/p62: sequestosome 1; TORC1: target of rapamycin complex 1; TORC2: target of rapamycin complex 2; TRP53: transforming related protein 53; TUBA: tubulin alpha; UV: ultraviolet; WT: wild-type
Introduction
Autophagy is a cellular mechanism that enables recycling of cytoplasmic contents and plays an important role in cellular homeostasis [Citation1,Citation2]. Macroautophagy/autophagy is initiated with the formation of the phagophore, a double-membrane structure that surrounds a portion of cytoplasm [Citation3]. These structures progress to the development of autophagosomes that fuse with lysosomes to form autolysosomes that release nutrients to the cytoplasm [Citation3]. A major regulator of autophagy is established to be the MTOR (mechanistic target of rapamycin kinase) pathway that can sense cellular energy and amino acid levels [Citation4]. TORC1 (target of rapamycin complex 1) is activated when cells are in a nutrient rich environment, which leads to inhibition of autophagy [Citation4]. However, suppression of TORC1 activity in response to low energy balance or amino acid starvation causes increased autophagy [Citation4].
It was recently reported that MAPK8/JNK1 (mitogen-activated protein kinase 8), but not MAPK9/JNK2 (mitogen-activated protein kinase 9), is required for the induction of autophagy by starvation [Citation5]. This mechanism of MAPK8 signaling is mediated by BCL2 (B cell leukemia/lymphoma 2) phosphorylation (on Thr69, Ser70, and Ser87) that disrupts the interaction of BCL2 with BECN1 (beclin 1, autophagy related) and initiates BECN1-dependent autophagy [Citation5]. Subsequent studies have provided strong support for this role of MAPK8 in autophagy [Citation6–Citation19], although contributing roles for MAPK9 [Citation20–Citation23] and alternative potential functions of MAPK8 related to autophagy have also been reported [Citation24–Citation29].
The MAPK8-promoted autophagy pathway raises several questions. First, what is the mechanistic relationship between the MAPK8 and MTOR pathways in the regulation of autophagy by starvation? Second, since MAPK8 exhibits functional redundancy as a BCL2 kinase [Citation20,Citation30–Citation34], what mechanism accounts for the requirement of MAPK8 for starvation-induced autophagy? The purpose of this study was to examine these two questions in the context of starvation-induced autophagy. We show that the role of MAPK8/9 in autophagy may be context-dependent and substantially more complex than previously considered.
Results
MTOR-regulated autophagy does not require MAPK8/9
It is established that MTOR is a master regulator of starvation-induced autophagy [Citation4]. The requirement of MAPK8 for starvation-induced autophagy [Citation5] may therefore reflect a role for MAPK8 upstream or down-stream of MTOR. Indeed, previous studies have demonstrated that MAPK8/9 can be activated in response to TORC1 signaling [Citation35] and that TORC1 activation can require MAPK8/9 signaling [Citation28]. Alternatively, MAPK8/9 and TORC1 may function in parallel pathways that control autophagy.
To test whether MAPK8/9 plays a role in autophagy induction downstream of MTOR, we examined the effect of torin 1, a small molecule inhibitor of MTOR. Control studies demonstrated that torin 1 prevented the phosphorylation of the MTOR substrate RPS6KB1/p70 (ribosomal protein S6 kinase, polypeptide 1) on Thr389 in both wild-type (WT) and mapk8−/- mapk9−/- immortalized mouse embryonic fibroblasts (MEFs) (Figure. 1(a) and S1A). To assess the effect of torin 1 on autophagy, we initially examined the formation of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) puncta in the cytoplasm by fluorescence microscopy. We found that torin 1 caused LC3B puncta in both WT and mapk8−/- mapk9−/- immortalized MEFs (). This observation suggested that MAPK8/9 deficiency caused no major change in autophagy as a result of MTOR inhibition. This conclusion was confirmed by measurement of autophagic flux by monitoring the formation of phosphatidylethanolamine-conjugated LC3B-II in response to MTOR inhibition in the presence of a lysosomal inhibitor [Citation36] ( and S1B) and reduced accumulation of the autophagic substrate SQSTM1/p62 (sequestosome 1) ( and S1C).
Figure 1. Autophagy caused by MTOR inhibition does not require MAPK8/9 in immortalized MEFs. (a) The amount of RPS6KB1, p-Thr389 RPS6KB1, and TUBA (tubulin alpha) in WT and mapk8−/- mapk9−/- immortalized MEFs after incubation without or with 250 nM torin 1 (2 or 4 h) was examined by immunoblot analysis. (b) WT and mapk8−/- mapk9−/- immortalized MEFs were transduced with a lentivirus vector that expresses GFP-LC3B. Puncta formation following incubation of the cells with 250 nM torin 1 (2 and 4 h) was examined by fluorescence microscopy. Scale bar: 30 µm. (c) LC3B and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression by WT and mapk8−/- mapk9−/- immortalized MEFs after incubation (2 h) without or with 250 nM torin 1 in the absence or presence of 25 µM chloroquine (CQ) was examined by immunoblot analysis. The LC3B-II:GAPDH ratios were normalized to the mean of WT control condition (first lane). The ‘Change in MAP1LC3B-II’ was calculated by subtracting MAP1LC3B-II:GAPDH (media+ CQ condition) from MAP1LC3B-II:GAPDH (torin 1+ CQ condition). The data presented represent the mean ± SEM; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Two-way ANOVA was used for the analysis of LC3B-II expression and Student’s t-test is used for the flux analysis. (d) The amount of SQSTM1 and TUBA in WT and mapk8−/- mapk9−/- immortalized MEFs after incubation with 250 nM torin 1 (2 or 4 h) was examined by immunoblot analysis. The SQSTM1:TUBA ratio was quantified and normalized to SQSTM1 expression in WT cells treated without torin 1 (mean ± SEM; n = 3 independent experiments; **, p < 0.01 (two-way ANOVA). (e) RPS6KB1, p-Thr389 RPS6KB1, AKT, p-Thr308 AKT, p-Ser473 AKT, and GAPDH expression by WT and mapk8−/- mapk9−/- immortalized MEFs after incubation (2 or 4 h) without or with 200 nM rapamycin was examined by immunoblot analysis. (f) LC3B and TUBA expression by WT and mapk8−/- mapk9−/- immortalized MEFs after incubation (2 h) without or with 200 nM rapamycin in the presence or absence of 25 µM chloroquine (CQ) was examined by immunoblot analysis. The LC3B-II:TUBA ratios were normalized to the mean of WT control (first lane). The data presented represent the mean ± SEM; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001. Two-way ANOVA was used for the analysis of LC3B-II expression and Student’s t-test was used for the flux analysis
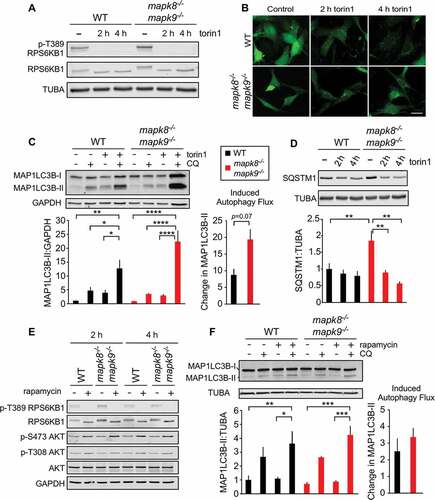
Torin 1 is an active site-directed inhibitor of MTOR and therefore inhibits both MTOR complexes TORC1 and TORC2 (target of rapamycin complex 2) [Citation37]. To test the role of TORC1, we used a low dose of rapamycin to selectively block TORC1 [Citation37]. Control studies demonstrated that rapamycin inhibited phosphorylation of the TORC1 substrate RPS6KB1 at Thr389, but not the TORC2 substrate AKT (thymoma viral proto-oncogene) at target site Ser473 (). We found that treatment of WT and mapk8−/- mapk9−/- immortalized MEFs with rapamycin caused a similar increase in autophagic flux that was measured by monitoring the accumulation of LC3B-II in the presence of a lysosomal inhibitor [Citation36] ( and S1D).
Collectively, these data demonstrate that MAPK8/9 are not required for autophagy induced in response to inhibition of TORC1 (by rapamycin) or inhibition of TORC1 and TORC2 (by torin 1).
Starvation-induced autophagy does not require MAPK8/9
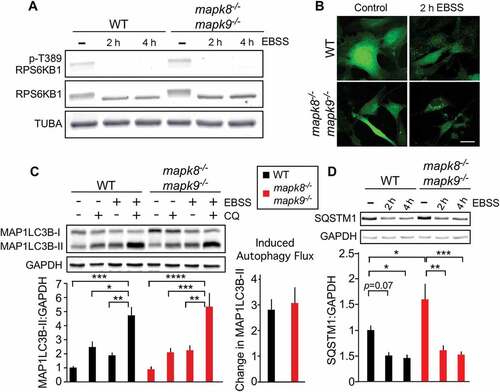
Our analysis of the effects of MTOR on autophagy in immortalized MEFs demonstrated that MAPK8/9 does not regulate autophagy downstream of MTOR (). It was therefore possible that MAPK8/9 functions upstream of MTOR in the regulation of autophagy by starvation [Citation28]. To test this hypothesis, we examined TORC1 signaling in response to starvation. We found that starvation prevented the phosphorylation of the TORC1 substrate RPS6KB1 at Thr389 in both WT and mapk8−/- mapk9−/- immortalized MEFs (Figure. 2(a) and S2A). MAPK8/9 are therefore not required for suppression of TORC1 signaling by starvation. Studies of autophagy demonstrated similar LC3B puncta formation following starvation of WT and mapk8−/- mapk9−/- immortalized MEFs (). Similarly, no differences in autophagic flux ( and S2B) or accumulation of the autophagic substrate SQSTM1 ( and S2C) were detected between WT and mapk8−/- mapk9−/- immortalized MEFs. This analysis suggests that MAPK8/9 in immortalized MEFs play no required role in starvation-induced autophagy.
Figure 2. Autophagy caused by starvation does not require MAPK8/9 in immortalized MEFs. (a) RPS6KB1, p-Thr389 RPS6KB1, and TUBA expression by WT and mapk8−/- mapk9−/- immortalized MEFs after incubation with EBSS containing 5mM glucose (2 or 4 h) was examined by immunoblot analysis. (b) WT and mapk8−/- mapk9−/- immortalized MEFs were transduced with a lentivirus vector that expresses GFP-LC3B. Puncta formation following incubation with EBSS containing 5 mM glucose (2 h) was examined by fluorescence microscopy. Scale bar: 30 µm. (c) LC3B and GAPDH in WT and mapk8−/- mapk9−/- immortalized MEFs after incubation (2 h) in medium or with EBSS containing 5 mM glucose in the presence or absence of 25 µM chloroquine (CQ) was examined by immunoblot analysis. The LC3B-II:GAPDH ratios were normalized to the average of WT control condition (first lane). The data presented represent the mean ± SEM; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***; p < 0.001; ****; p < 0.0001. Two-way ANOVA was used for the analysis of LC3B-II expression and Student’s t-test was used for the flux analysis. (d) The amount of SQSTM1 and GAPDH in WT and mapk8−/- mapk9−/- immortalized MEFs after incubation with EBSS containing 5 mM glucose (2 or 4 h) was examined by immunoblot analysis. The SQSTM1:GAPDH ratio was quantified and normalized to WT cells treated without EBSS (mean ± SEM; n = 3 independent experiments); *, p < 0.05; **, p < 0.01; ***; p < 0.001 (two-way ANOVA)
MAPK8/9 activation and MTOR inhibition
Our inability to detect a difference in autophagy between WT and mapk8−/- mapk9−/- immortalized MEFs was not anticipated. One explanation for this finding is that MAPK8/9 are not regulated under the conditions that we employed for these experiments. Indeed, we found that MTOR inhibition caused by starvation (Figure. 3(a) and S3A) or treatment with torin 1 ( and S3B) under the conditions of the autophagy assays did not lead to MAPK8/9 activation. It is therefore possible that in a different cell type, or under different culture conditions, MAPK8/9 could be activated and thus may contribute to the regulation of autophagy during starvation or torin 1 treatment.
Figure 3. Effect of starvation and MTOR inhibition on MAPK8/9 activation is not sufficient to cause autophagy. (a and b) MAPK8/9 activation in immortalized MEFs was examined by immunoblot analysis of p-Thr183/Tyr185 MAPK8/9, MAPK8/9, and GAPDH in cells after incubation (2 or 4 h) with EBSS containing 5 mM glucose (a) or 250 nM torin 1 (b). Lanes 1 and 2 represent positive and negative controls: lysates of WT MEFs exposed to 60 J/m2 UV and mapk8−/- mapk9−/- MEFs, respectively. (c) WT and mapk8−/- mapk9−/- immortalized MEFs were exposed to UV radiation (60 J/m2) and cell extracts were prepared at 45 min post-irradiation. The expression of LC3B, p-Thr183/Tyr185 MAPK8/9, MAPK8/9, and GAPDH was examined by immunoblot analysis
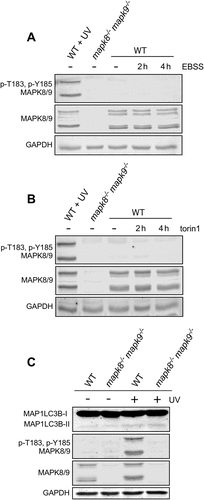
To test whether MAPK8/9 activation is sufficient for the induction of autophagy, we examined the effect of MAPK8/9 activation on the conversion of LC3B-I to LC3B-II by immunoblot analysis using WT and mapk8−/- mapk9−/- immortalized MEFs. We found that activated MAPK8/9 caused no change in LC3B-II formation ( and S3C). These data suggest that MAPK8/9 activation is not sufficient for autophagy and that MAPK8/9-promoted autophagy likely involves interactions between MAPK8/9 and other pro-autophagic signaling pathways.
MAPK8/9 is not required for starvation or torin 1-induced autophagy in primary MEFs
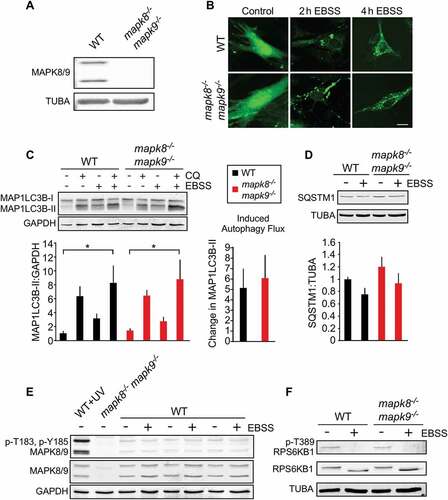
Our initial studies of immortalized MEFs may be compromised by the loss of TRP53 (transforming related protein 53) function in these cells, which may change autophagic responses [Citation38]. We therefore repeated our studies using early passage primary WT and mapk8−/- mapk9−/- MEFs (Figure 4(a)). The primary MEFs exhibited reduced autophagy flux compared with immortalized MEFs. We found that MAPK8/9 deficiency caused no change in LC3B puncta formation in response to starvation (). Similarly, MAPK8/9-deficiency caused no change in starvation-induced autophagic flux ( and S4A) or accumulation of the autophagic substrate SQSTM1 ( and S4B). Control studies demonstrated that starvation under these conditions did not activate MAPK8/9 (), but did prevent phosphorylation of the TORC1 substrate RPS6KB1 at Thr389 ( and S4C).
Figure 4. Autophagy caused by starvation does not require MAPK8/9 in primary MEFs. (a) (Z)-4-Hydroxytamoxifen-treated primary Rosa-CreERT (WT) MEFs and Rosa-CreERT Mapk8LoxP/LoxP mapk9−/- MEFs were examined by immunoblot analysis by probing with antibodies to MAPK8/9 and TUBA. (b) WT and mapk8−/- mapk9−/- primary MEFs were transduced with a lentivirus vector that expresses GFP-LC3B. Puncta formation following incubation with EBSS containing 5 mM glucose (2 and 4 h) was examined by fluorescence microscopy. Scale bar: 25 µm. (c) LC3B and GAPDH expression by WT and mapk8−/- mapk9−/- primary MEFs after incubation (2 h) in medium or with EBSS containing 5 mM glucose in the presence or absence of 25 µM chloroquine (CQ) was examined by immunoblot analysis. The LC3B-II:GAPDH ratios were normalized to the mean of WT control (first lane). The data presented represent the mean ± SEM; n = 3 independent experiments; *, p < 0.05. Two-way ANOVA was used for the analysis of LC3B-II expression and Student’s t-test was used for the flux analysis. (d) The amount of SQSTM1 and TUBA in WT and mapk8−/- mapk9−/- primary MEFs after incubation with EBSS containing 5 mM glucose (2 h) was examined by immunoblot analysis. The SQSTM1:TUBA ratio was quantified and normalized to SQSTM1 expression in WT non-starved cells (mean ± SEM; n = 3 independent experiments). (e) MAPK8/9 activation in WT and mapk8−/- mapk9−/- primary MEFs was examined by immunoblot analysis of p-Thr183/Tyr185 MAPK8/9, MAPK8/9, and GAPDH after incubation (2 h) with media or EBSS containing 5 mM glucose. WT MEFs exposed to UV (60 J/m2) and mapk8−/- mapk9−/- represent positive and negative controls. (f) RPS6KB1, p-Thr389 RPS6KB1, and TUBA in WT and mapk8−/- mapk9−/- primary MEFs after incubation (2 h) with EBSS containing 5 mM glucose was examined by immunoblot analysis
Studies of torin 1-induced autophagy demonstrated similar inhibition of the phosphorylation of the TORC1 substrate RPS6KB1 at Thr389 in primary WT and mapk8−/- mapk9−/- MEFs (Figure S5A) and similar torin 1-induced LC3B puncta formation (Fig. S5B). A small decrease in autophagic flux (Fig. S5C) and modestly increased accumulation of the autophagic substrate SQSTM1 (Fig. S5D) was detected in mapk8−/- mapk9−/- primary MEFs compared with WT primary MEFs.
Together, these data indicate that MAPK8/9 play no required role in starvation-induced autophagy in primary MEFs.
Requirement of MAPK8 and MAPK9 for starvation-induced autophagy in primary MEFs
It is possible that our analysis of compound MAPK8/9-deficiency in MEFs is compromised by compensatory mechanisms that are engaged in these cells. Indeed, the role of MAPK8/9 in starvation-induced autophagy was first established in studies using mapk8−/- and mapk9−/- primary MEFs [Citation5]. We therefore examined the effects of starvation on WT, mapk8−/-, and mapk9−/- primary MEFs. Starvation prevented the phosphorylation of the TORC1 substrate RPS6KB1 at Thr389 in these cells (Fig. S6A). Studies of WT, mapk8−/-, and mapk9−/- primary MEFs identified similar LC3B puncta formation (Fig. S6B), accumulation of the autophagic substrate SQSTM1 (Figure S6C), and autophagic flux (Fig. S6D). These data demonstrate that MAPK8-deficiency and MAPK9-deficiency (Fig. S6) cause no major defects in starvation-induced autophagy.
Requirement of MAPK8/9 for starvation-induced autophagy in primary epithelial cells
Our analysis of primary and immortalized MEFs suggests that MAPK8/9 play no required role in starvation-induced autophagy ( and ). However, studies of a different cell type may lead to a different conclusion. We therefore examined starvation-induced autophagic flux in primary kidney epithelial cells. We found that MAPK8/9 deficiency caused no difference in autophagic flux in these epithelial cells (Fig. S7A) and no difference in the accumulation of the autophagic substrate SQSTM1 (Fig. S7B) in response to starvation. These data demonstrate that MAPK8/9 have no required role in primary kidney epithelial cells for starvation-induced autophagy.
Requirement of MAPK8/9 for autophagy in response to Ras and hypoxia
If MAPK8/9 are not required for autophagy in response to starvation or MTOR inhibition, it is possible that MAPK8/9 may be required for autophagy in response to other stimuli. We therefore examined control trp53−/- MEFs (-Ras) and Hras (Harvey rat sarcoma virus oncogene)-transformed (+ Ras) trp53−/- MEFs without (WT) and with ablation of the Mapk8 and Mapk9 genes (Fig. S8A). Autophagic flux studies demonstrated no significant effects of MAPK8/9 deficiency (Fig. S8B). These data indicate that MAPK8/9 are not required for autophagy in control or Hras-transformed trp53−/- MEFs.
We also studied control and Hras-transformed trp53−/- MEFs without (WT) and with ablation of the Mapk8 and Mapk9 genes under hypoxia (1.5% O2) conditions (Fig. S9A). Autophagic flux studies demonstrated no significant effects of MAPK8/9 deficiency (Fig. S9B). These data indicate that MAPK8/9 are not required for autophagy in control or Hras-transformed trp53−/- MEFs under hypoxia conditions.
MAPK8/9-regulated autophagy in primary hepatocytes
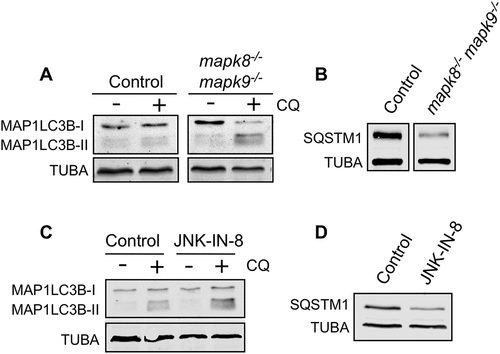
Autophagy plays a major role in systemic metabolic homeostasis [Citation39] and hepatic lipid metabolism [Citation40]. Moreover, hepatocytes have been reported to exhibit MAPK8/9-dependent autophagy [Citation6]. We therefore examined the requirement of MAPK8/9 for autophagy using primary hepatocytes prepared from Albumin-Cre−/+ (Alb-Cre−/+) control mice and Alb-cre−/+ Mapk8LoxP/LoxP Mapk9LoxP/LoxP mice. We found that MAPK8/9-deficient hepatocytes exhibited increased accumulation of LC3B-II following lysosomal inhibition and the reduced accumulation of the autophagic substrate SQSTM1 in MAPK8/9-deficient hepatocytes compared with control hepatocytes (Figure 5(a,b), S10A and B). These observations suggest that MAPK8/9 may inhibit autophagy in hepatocytes.
To confirm the conclusion that MAPK8/9 suppresses autophagic flux in primary hepatocytes, we examined wild-type (WT) hepatocytes treated with JNK-IN-8, a potent and selective small molecule inhibitor of MAPK8/9 [Citation41]. We found that treatment with JNK-IN-8 caused increased accumulation of LC3B-II following lysosomal inhibition ( and S10C) and reduced accumulation of the autophagic substrate SQSTM1 ( and S10D). These data indicate that pharmacological inhibition of MAPK8/9 causes increased autophagic flux.
Figure 5. MAPK8/9 suppresses autophagy in primary hepatocytes. (a) LC3B and TUBA in WT and mapk8−/- mapk9−/- primary hepatocytes after incubation (6 h) with medium or EBSS containing 5 mM glucose in the presence or absence of 25 µM chloroquine (CQ) was examined by immunoblot analysis. (b) The amount of SQSTM1 and TUBA in WT and mapk8−/- mapk9−/- primary hepatocytes was examined by immunoblot analysis. (c and d) WT primary hepatocytes were incubated (6 h) without or with 2 µM JNK-IN-8. LC3B and TUBA in WT primary hepatocytes in the presence or absence of 25 µM CQ was examined by immunoblot analysis (c). The amount of SQSTM1 and TUBA was examined by immunoblot analysis (d)
The increased autophagy caused by MAPK8/9 loss-of-function in hepatocytes compared with WT hepatocytes was unexpected. This increased autophagy may reflect the established role of MAPK8/9 to strongly suppress the transcriptional activity of the PPARA (peroxisome proliferator activated receptor alpha) nuclear receptor [Citation42] that can promote autophagy through increased expression of autophagic genes [Citation43]. This role of MAPK8/9 to suppress basal autophagy in hepatocytes is similar to the established function of MAPK8, MAPK9, and MAPK10/JNK3 (mitogen-activated protein kinase 10) in neurons to suppress basal autophagy by repression of autophagic gene expression [Citation27].
Discussion
Studies of the genetically tractable organism Drosophila melanogaster demonstrate that the basket gene, which encodes a MAPK8/9 ortholog, can increase autophagy [Citation44]. It is established that Basket in Drosophila promotes autophagy by causing increased expression of autophagy proteins [Citation44]. This mechanism is also found in mammals [Citation45–Citation51]. Thus, MAPK8/9 may promote autophagy by increasing the expression of ATG4 (autophagy related 4) [Citation49], ATG5 [Citation45], BECN1 [Citation48], BNIP3 (BCL2/adenovirus E1B interacting protein 3) [Citation47,Citation50], LC3B [Citation46,Citation51], and SQSTM1 [Citation46]. MAPK8/9-regulated gene expression therefore represents an evolutionarily conserved mechanism of autophagic regulation. The specific physiological effects of MAPK8/9 on the expression of autophagic proteins likely reflects the combinatorial actions of MAPK8/9-regulated transcription factors together with other transcription factors that are activated by different signal transduction pathways. MAPK8/9 may therefore play an essential role in mammalian autophagy in some specific physiological or pathological contexts.
Recently, MAPK8 was proposed to promote starvation-induced autophagy in MEFs by a non-transcriptional mechanism [Citation5]. It was reported that mapk8−/- MEFs, but not mapk9−/- MEFs, are resistant to starvation-induced autophagy [Citation5]. The proposed mechanism was mediated by phosphorylation of BCL2 on Thr69, Ser70, and Ser87 by MAPK8, disruption of BCL2-BECN1 complexes, and the initiation of BECN1-dependent autophagy [Citation5]. This mechanism implies that MAPK8 is essential for BCL2 phosphorylation [Citation5]. However, other studies indicate that MAPK8 may only serve a redundant role in BCL2 phosphorylation [Citation20,Citation30–Citation34]. The essential non-transcriptional role of MAPK8 in starvation-induced autophagy [Citation5] is therefore likely to be mediated by BCL2 phosphorylation only in specific cellular contexts.
Possible roles of MAPK8 in starvation-induced autophagy include functional interactions with TORC1, a master regulator of starvation-induced autophagy [Citation4]. Indeed, previous studies have demonstrated that MAPK8/9 can be activated in response to TORC1 signaling [Citation35] and that TORC1 activation can require MAPK8/9 signaling [Citation28]. However, we found no requirement for MAPK8/9 upstream () or downstream () of TORC1 during starvation-induced autophagy. The essential function of MAPK8/9 must therefore play a role in a pathway that is parallel to TORC1. These observations led us to test whether MAPK8/9 is required for starvation-induced autophagy. Our analysis demonstrates that MAPK8/9 is not required for starvation-induced autophagy in mouse fibroblasts or epithelial cells ( and S7).
Many reported studies support the conclusion that MAPK8/9 plays an essential role in the promotion of autophagy [Citation5–Citation23]. In contrast, our analysis does not indicate a major role for MAPK8/9 in starvation-induced autophagy. It is likely that small differences in cell culture conditions and starvation conditions may contribute to discrepancies between our analysis () and previously published reports of the role of MAPK8/9 in MEFs [Citation5]. The use of the small molecule SP600125, that inhibits MAPK8/9 and many other protein kinases [Citation52], to draw conclusions concerning the specific role of MAPK8/9 may also contribute to conclusions [Citation8,Citation10,Citation12–Citation19,Citation22,Citation51] that contrast with those drawn from our analysis of MAPK8/9 knockout cells. Finally, some studies that have identified a role for MAPK8/9 do not focus on starvation-induced autophagy. One example is the requirement of MAPK8/9 for oncolytic adenovirus-mediated autophagy [Citation20]. Clearly our conclusions concerning the lack of a major role for MAPK8/9 in autophagy are restricted to the starvation, transformation, and hypoxia paradigms that we studied. Moreover, it should be noted that we found that starvation or an MTOR inhibitor under the conditions of our autophagy assays did not lead to MAPK8/9 activation. It is therefore possible that in a different cell type, or under different culture conditions, MAPK8/9 could be activated and thus may contribute to the regulation of autophagy.
In conclusion, our analysis demonstrates that there is no required role for MAPK8/9 in starvation-induced autophagy. However, this signaling pathway can regulate autophagy by a conserved mechanism that leads to regulated expression of autophagy proteins in Drosophila [Citation44] and mammals [Citation45–Citation51]. MAPK8/9-regulated gene expression may lead to increased autophagy, but different paradigms of MAPK8/9-regulated gene expression could lead to reduced autophagy. For example, MAPK8/9-regulated gene expression suppresses basal autophagy in neurons [Citation27]. A similar mechanism may account for MAPK8/9-mediated repression of autophagy in hepatocytes (). It is also possible that MAPK8/9 may promote autophagy by a non-transcriptional mechanism mediated by BCL2 phosphorylation in some specific physiological or pathological conditions. Collectively, our analysis shows that the role of MAPK8/9 in autophagy may be context-dependent and substantially more complex than previously considered.
Materials and methods
Cell culture
Primary MEFs were prepared from embryonic day 13.5 WT, mapk8−/-, mapk9−/-, Rosa-CreERT and Rosa-CreERT Mapk8LoxP/LoxP mapk9−/- mice and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, 11,960,077) supplemented with 10% bovine growth serum (BGS; Thermo Fisher Scientific, SH30541.03), 1% penicillin/streptomycin (Thermo Fisher Scientific, 15,140,122) plus 1% L-glutamine (Thermo Fisher Scientific, 25,030,081) [Citation53]. CreERT primary MEFs were treated (24 h) with 1 µM (Z)-4-hydroxytamoxifen (Millipore Sigma, H7904) in culture media at passage #1. Autophagy experiments were conducted on MEFs between passages #2 and #3.
Immortalized WT and mapk8−/- mapk9−/- MEFs have been described previously [Citation54]. We have also previously described trp53−/- MEFs, trp53−/- mapk8−/- mapk9−/- MEFs, Hras-transformed trp53−/- MEFs, and Hras transformed trp53−/- mapk8−/- mapk9−/- MEFs [Citation55]. These cells were cultured in DMEM supplemented with 10% BGS, 1% penicillin/streptomycin, plus 1% L-glutamine.
Primary kidney epithelial cells were isolated from ROSA-CreERT mice or ROSA-CreERT Mapk8LoxP/LoxP mapk9−/- mice. Kidneys were digested (2 h) with 0.1% collagenase A (Millipore Sigma, 11,088,793,001) plus 0.1% trypsin (Thermo Fisher Scientific, 15,090,046) in DMEM with 150 mM NaCl [Citation56]. The cells were maintained in DMEM/F-12 media (Thermo Fisher Scientific, 11,039–047) containing 10% BGS, plus 150 mM urea and 150 mM NaCl. The cells were treated (24 h) with 1 µM (Z)-4-hydroxytamoxifen and employed for autophagy studies after 3 d.
Primary hepatocytes were isolated from WT (Alb-Cre−/+) and MAPK8/9-deficient (Alb-Cre−/+ Mapk8LoxP/LoxP Mapk9LoxP/LoxP) mice [Citation42] using a modified 2-step perfusion method [Citation57] with Hanks’ Balanced Salt Solution (HBSS; Thermo Fisher Scientific, 14,175) and Liver Digest Medium (Thermo Fisher Scientific, 17,703,034). Cells were seeded on plates pre-coated (1 h) with collagen I (Thermo Fisher Scientific, A1048301) in DMEM supplemented with 10% BGS, 2 mM sodium pyruvate (Thermo Fisher, 11,360,070), 1 μM dexamethasone (Millipore Sigma, D4902), 100 nM insulin (Millipore Sigma, I0516) plus 2% penicillin/streptomycin. After attachment (2 h), the medium was removed and the hepatocytes were incubated (22 h) in maintenance medium (DMEM (4.5 g/L glucose) supplemented with 10% BGS, 0.2% bovine serum albumin (Millipore Sigma, A8806), 2 mM sodium pyruvate, 2% penicillin/streptomycin, 0.1 μM dexamethasone, and 1 nM insulin. Autophagy studies were performed within 48 h. Pharmacological studies of MAPK8/9 inhibition were performed by treating hepatocytes with 2 µM JNK-IN-8 (Millipore Sigma, 420,150) or dimethyl sulfoxide (DMSO; Millipore Sigma, D2650).
Autophagy studies
Autophagy promoted by amino acid starvation was examined using cells (70% confluent) washed 3 times with modified Earle’s Balanced Salt Solution (EBSS; 117.24 mM NaCl, 26.19 mM NaHCO3, 5.33 mM KCl, 0.816 mM MgCl2, 1.01 mM NaH2PO4, 1.8 mM CaCl2, 10.92 mM HEPES, pH 7.4, 0.224 mM sodium pyruvate) and then incubated with EBSS containing 5 mM glucose. Autophagy was also examined in cells treated with pharmacological inhibitors of MTOR (250 nM torin 1; Tocris, 4247) and TORC1 (200 nM rapamycin; Millipore Sigma, R8781) or under hypoxia (1.5% O2) conditions. Autophagic flux was examined by measuring the LC3B-II:GAPDH ratio or LC3B-II:TUBA (tubulin alpha) ratio by immunoblot analysis, normalization of the data to the control condition (without autophagy induction or CQ), and calculation of the increased LC3B-II:GAPDH (or TUBA) ratio caused by treatment of the cells with 25 µM chloroquine diphosphate (CQ; Millipore Sigma, 25,745) to inhibit lysosomal protein degradation [Citation36].
Immunoblot analysis
Cell extracts were prepared using Triton lysis buffer (20 mM Tris at pH 7.4, 1% Triton X-100 [Millipore Sigma, X100], 10% glycerol [Millipore Sigma, G7793], 137 mM NaCl, 2 mM EDTA [Millipore Sigma, EDS], 25 mM β-glycerophosphate [Millipore Sigma, G9422], 1 mM sodium orthovanadate [Millipore Sigma, S6508], 1 mM phenylmethylsulfonyl fluoride [Millipore Sigma, P7626], 10 µg/mL aprotinin [Millipore Sigma, A6106] plus leupeptin [Millipore Sigma, L2884]). Extracts (20–40 μg of protein) were examined by protein immunoblot analysis by probing with antibodies described in antibodies section. Immunocomplexes were detected using IRDye-conjugated secondary antibodies to mouse IgG (LI-COR Biosciences, 925–68,070) and rabbit IgG (LICOR Biosciences, 925–32,211) and quantified by using the Odyssey infrared imaging system (LI-COR Biosciences).
Antibodies
We obtained antibodies to AKT (Cell Signaling Technology, 9272), p-Thr308 AKT (Cell Signaling Technology, 5106), p-Ser473 AKT (Cell Signaling Technology, 9271), GAPDH (Santa Cruz Biotechnology, sc-25,778), LC3B (Cell Signaling Technology, 2775), MAPK8/9 (R&D Systems, AF1387), p-Thr183/Tyr185 MAPK8/9 (Cell Signaling Technology, 9255 [] and Cell Signaling Technology, 4668 [Figs. S8 and S9]), RPS6KB1 (Cell Signaling Technology, 9202), p-Thr389 RPS6KB1 (Cell Signaling Technology, 9206), SQSTM1 (Progen, GP62-C), and TUBA (Millipore Sigma, T5168), from the indicated suppliers.
LC3B puncta formation
Cells were transduced with a lentiviral vector that expresses GFP-LC3B according to the manufacturer’s instructions (LentiBrite GFP-LC3 Lentiviral Biosensor; Millipore Sigma, 17–10,193). The cells were plated on a glass culture dish and incubated with culture media, EBSS containing 5mM glucose, or culture media supplemented with 250 nM torin 1. Live cell images were acquired at 0, 2, and 4 h using Leica TCS SP2 confocal microscope and Leica Confocal Software.
Statistical analysis
Data are presented as the mean and standard error (SEM). Statistical analysis was performed using two-tailed Student’s t-test for pair-wise comparisons and two-way ANOVA for multiple group comparisons as indicated at the figure legends.
1466013.zip
Download Zip (7 MB)Acknowledgments
We would like to thank Tetsuro Ishii for providing sqstm1−/- MEFs for SQSTM1 antibody validation studies, members of the Davis Laboratory for scientific discussions, and Kathy Gemme for expert administrative assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplementary data can be accessed here.
Additional information
Funding
Related Research Data
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. PMID:15068787.
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. PMID:15102438.
- Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem. 2000;69:303–342. PMID:10966461.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. PMID:28388417.
- Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. PMID:18570871.
- Niso-Santano M, Malik SA, Pietrocola F, et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015;34:1025–1041. PMID:25586377.
- Pattingre S, Bauvy C, Carpentier S, et al. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. PMID:19029119.
- Siddiqui MA, Malathi K. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J Biol Chem. 2012;287:43651–43664. PMID:23109342.
- Sun ZL, Dong JL, Wu J. Juglanin induces apoptosis and autophagy in human breast cancer progression via ROS/JNK promotion. Biomed Pharmacother. 2017;85:303–312. PMID:27899257.
- Vasilevskaya IA, Selvakumaran M, Roberts D, et al. JNK1 inhibition attenuates hypoxia-induced autophagy and sensitizes to chemotherapy. Mol Cancer Res. 2016;14:753–763. PMID:27216154.
- Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. PMID:18769111.
- Yan H, Gao Y, Zhang Y. Inhibition of JNK suppresses autophagy and attenuates insulin resistance in a rat model of nonalcoholic fatty liver disease. Mol Med Rep. 2017;15:180–186. PMID:27909723.
- Zhu X, Zhou M, Liu G, et al. Autophagy activated by the c-Jun N-terminal kinase-mediated pathway protects human prostate cancer PC3 cells from celecoxib-induced apoptosis. Exp Ther Med. 2017;13:2348–2354. PubMed PMID: 28565848.
- Chen YY, Sun LQ, Wang BA, et al. Palmitate induces autophagy in pancreatic beta-cells via endoplasmic reticulum stress and its downstream JNK pathway. Int J Mol Med. 2013;32:1401–1406. PMID:24142192.
- Granato M, Santarelli R, Lotti LV, et al. JNK and macroautophagy activation by bortezomib has a pro-survival effect in primary effusion lymphoma cells. PLoS One. 2013;8:e75965. PMID:24086672.
- He W, Wang Q, Srinivasan B, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33:3004–3013. PMID:23831571.
- Jin HO, Hong SE, Park JA, et al. Inhibition of JNK-mediated autophagy enhances NSCLC cell sensitivity to mTORC1/2 inhibitors. Sci Rep. 2016;6:28945. PMID:27358039.
- Liu E, Lopez Corcino Y, Portillo JA, et al. Identification of signaling pathways by which CD40 stimulates autophagy and antimicrobial activity against toxoplasma gondii in macrophages. Infect Immun. 2016;84:2616–2626. PMID:27354443.
- Yang J, Yao S. JNK-Bcl-2/Bcl-xL-Bax/Bak pathway mediates the crosstalk between matrine-induced autophagy and apoptosis via interplay with Beclin 1. Int J Mol Sci. 2015;16:25744–25758. PMID:26516844.
- Klein SR, Piya S, Lu Z, et al. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene. 2015;34:5295–5301. PMID:25619840.
- Zhang Q, Kuang H, Chen C, et al. The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat Immunol. 2015;16:458–466. PMID:25799126.
- Bock BC, Tagscherer KE, Fassl A, et al. The PEA-15 protein regulates autophagy via activation of JNK. J Biol Chem. 2010;285:21644–21654. PMID:20452983.
- Lin Z, Liu T, Kamp DW, et al. AKT/mTOR and c-Jun N-terminal kinase signaling pathways are required for chrysotile asbestos-induced autophagy. Free Radic Biol Med. 2014;72:296–307. PMID:24735948.
- He JD, Wang Z, Li SP, et al. Vitexin suppresses autophagy to induce apoptosis in hepatocellular carcinoma via activation of the JNK signaling pathway. Oncotarget. 2016;7:84520–84532. PMID:27588401.
- Long Z, Chen B, Liu Q, et al. The reverse-mode NCX1 activity inhibitor KB-R7943 promotes prostate cancer cell death by activating the JNK pathway and blocking autophagic flux. Oncotarget. 2016;7:42059–42070. PMID:27275542.
- Palumbo C, De Luca A, Rosato N, et al. c-Jun N-terminal kinase activation by nitrobenzoxadiazoles leads to late-stage autophagy inhibition. J Transl Med. 2016;14:37. PMID:26847645.
- Xu P, Das M, Reilly J, et al. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–322. PMID:21325132.
- Basu S, Rajakaruna S, Reyes B, et al. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy. 2014;10:1193–1211. PMID:24813396.
- Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29:2070–2082. PMID:20101227.
- Tournier C, Dong C, Turner TK, et al. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. PMID:11390361.
- Du L, Lyle CS, Chambers TC. Characterization of vinblastine-induced Bcl-xL and Bcl-2 phosphorylation: evidence for a novel protein kinase and a coordinated phosphorylation/dephosphorylation cycle associated with apoptosis induction. Oncogene. 2005;24:107–117. PMID:15531923.
- Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–656. PMID:19917720.
- He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. PMID:22258505.
- Mellor HR, Rouschop KM, Wigfield SM, et al. Synchronised phosphorylation of BNIP3, Bcl-2 and Bcl-xL in response to microtubule-active drugs is JNK-independent and requires a mitotic kinase. Biochem Pharmacol. 2010;79:1562–1572. PMID:20100468.
- Lee JH, Budanov AV, Park EJ, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–1228. PMID:20203043.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. PMID:26799652.
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. PMID:19383975.
- Starobinets H, Cancer: DJ. A suppression switch. Nature. 2013;504:225–226. PMID:24305045.
- Lim YM, Lim H, Hur KY, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun. 2014;5:4934. PMID:25255859.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. PMID:19339967.
- Zhang T, Inesta-Vaquera F, Niepel M, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–154. PMID:22284361.
- Vernia S, Cavanagh-Kyros J, Garcia-Haro L, et al. The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20:512–525. PMID:25043817.
- Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. PMID:25383539.
- Wu H, Wang MC, Bohmann D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech Dev. 2009;126:624–637. PMID:19540338.
- Byun JY, Yoon CH, An S, et al. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis. 2009;30:1880–1888. PMID:19783847.
- Artal-Martinez de Narvajas A, Ts G, Js Z, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol. 2013;33:3983–3993. PMID:23918802.
- Chaanine AH, Jeong D, Liang L, et al. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265. PMID:22297293.
- Li DD, Wang LL, Deng R, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–898. PMID:19060920.
- Li Y, Luo Q, Yuan L, et al. JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol Appl Pharmacol. 2012;263:21–31. PMID:22668848.
- Moriyama M, Moriyama H, Uda J, et al. BNIP3 upregulation via stimulation of ERK and JNK activity is required for the protection of keratinocytes from UVB-induced apoptosis. Cell Death Dis. 2017;8:e2576. PMID:28151469.
- Sun T, Li D, Wang L, et al. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J Transl Med. 2011;9:161. PMID:21943220.
- Bain J, McLauchlan H, Elliott M, et al. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. PMID:12534346.
- Das M, Jiang F, Sluss HK, et al. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci U S A. 2007;104:15759–15764. PubMed PMID: 17893331.
- Iordanov MS, Wong J, Newton DL, et al. Differential requirement for the stress-activated protein kinase/c-Jun NH(2)-terminal kinase in RNAdamage-induced apoptosis in primary and in immortalized fibroblasts. Mol Cell Biol Res Commun. 2000;4:122–128. PMID:11170843.
- Kennedy NJ, Sluss HK, Jones SN, et al. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes Dev. 2003;1:629–637. PMID:12629045.
- Follit JA, San Agustin JT, Xu F, et al. The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS Genet. 2008;4:e1000315. PMID:19112494.
- Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. PMID: 177845.