
ABSTRACT
TRAF6 (TNF receptor associated factor 6) plays a pivotal role in NFKB activation and macroautphagy/autophagy activation induced by TLR4 (toll like receptor 4) signaling. The objective of this study was to determine the functional role of PRDX1 (peroxiredoxin 1) in NFKB activation and autophagy activation. PRDX1 interacted with the ring finger domain of TRAF6 and inhibited its ubiquitin-ligase activity. The inhibition on TRAF6 ubiquitin-ligase activity by PRDX1 induced the suppression of ubiquitination of an evolutionarily conserved signaling intermediate in Toll pathways (ECSIT) essential for NFKB activation and BECN1 (beclin 1) required for autophagy activation. An inhibitory effect of PRDX1 on TRAF6 was clearly evidenced in PRDX1-knockdown (PRDX1KD) THP-1, PRDX1KD MDA-MB-231, and PRDX1KD SK-HEP-1 cells. PRDX1KD THP-1 cells showed increases of NFKB activation, pro-inflammatory cytokine production, NFKB-dependent gene expression induced by TLR4 stimulation, and resistance against Salmonella typhimurium infection. Additionally, migration and invasion abilities of PRDX1KD MDA-MB-231 and PRDX1KD SK-HEP-1 cancer cells were significantly enhanced compared to those of control cancer cells. Taken together, these results suggest that PRDX1 negatively regulates TLR4 signaling for NFKB activation and autophagy functions such as bactericidal activity, cancer cell migration, and cancer cell invasion by inhibiting TRAF6 ubiquitin-ligase activity.
Abbreviations: 3-MA: 3-methyladenine; BECN1: beclin 1; CHUK/IKKA: conserved helix-loop-helix ubiquitous kinase; ECSIT: ECSIT signalling integrator; ELISA: enzyme-linked immunosorbent assay; NFKB: nuclear factor kappa-light-chain-enhancer of activated B cells; IB: immunoblotting; IKBKB/IKKB: inhibitor of nuclear factor kappa B kinase subunit beta; IL1B: interleukin 1 beta; IL6: interleukin 6; IP: immunoprecipitation; LPS: lipopolysaccharide; MAP1LC3/LC3: microtuble associated protein 1 light chain 3; MAP3K7/TAK1: mitogen-activated protein kinase kinase kinase 7; MAPK14/p38: mitogen-activated protein kinase 14; mROS: mitochondrial reactive oxygen species; PRDX1: peroxiredoxin 1; PRDX6: peroxiredoxin 6; RELA/p65: RELA proto-oncogene, NF-kB subunit; TRAF6 TNF: receptor associated factor 6.
Introduction
TRAF6 (TNF receptor associated factor 6), a member of the TNF receptor associated factor (TRAF) protein family, is essential for the regulation of toll like receptor (TLR)-mediated signaling [Citation1]. TLRs play pivotal roles in the initiation of innate immunity and induction of adaptive immune responses by recognizing distinct pathogen-associated molecular patterns of pathogens [Citation1–Citation3]. Upon TLR stimulation, MYD88 (myeloid differentiation primary response 88) recruits IRAK1 (interleukin 1 receptor associated kinase 1) and IRAK4 (interleukin 1 receptor associated kinase 4). IRAK1 is then phosphorylated, resulting in recruitment of TRAF6, an E3 ubiquitin ligase and a scaffold protein. TRAF6 is then autoubiquitinated through Lys63(K63)-linked ubiquitin chains [Citation4–Citation6]. Ubiquitinated TRAF6 activates a complex containing MAP3K7/TAK1 (mitogen-activated protein kinase kinase kinase 7), TAB1 (TGF-beta activated kinase 1 [MAP3K7] binding protein 1), and TAB2 (TGF-beta activated kinase 1 [MAP3K7] binding protein 2) [Citation7–Citation9]. This complex then induces phosphorylation of the IKK (inhibitor of IκB kinase) complex, leading to activation of NFKB (nuclear factor kappa-light-chain-enhancer of activated B cells) and induction of expression of genes encoding inflammatory cytokines.
TRAF6 ubiquitin-ligase activity plays a pivotal role in the initiation and function of cellular autophagy. It has been recently reported that TRAF6 controls TLR4 (toll like receptor 4)-induced autophagy via ubiquitination of BECN1 (beclin 1) [Citation10]. TLR4 signaling triggers K63-linked ubiquitination of BECN1 through a mechanism that depends on TRAF6 [Citation10]. Ubiquitination of BECN1 can amplify TLR-induced autophagy [Citation10]. In addition, it has been reported that TLR4 and TLR3 activation can induce autophagy via TICAM1 (toll like receptor adaptor molecule 1) in lung cancer cells, which in turn promotes ubiquitination of TRAF6 that is essential for TLR4- and TLR3-triggered increases of multiple cytokines to enhance cell migration and invasion, including IL6 (interleukin 6), CCL2 (C-C motif chemokine ligand 2), CCL20 (C-C motif chemokine ligand 20), VEGFA (vascular endothelial growth factor A), and MMP2 (matrix metallopeptidase 2) [Citation11]. These results strongly demonstrate that TRAF6 ubiquitin-ligase activity might be essential for the activation of NFKB and the regulation of cellular autophagy activation induced by TLR stimulation.
Peroxiredoxins (PRDXs) are antioxidant proteins that constitute the potent defense system to maintain redox balance by converting hydrogen peroxide to water [Citation12–Citation14]. So far, 6 different PRDXs (PRDX1 to PRDX6) in intracellular compartments have been reported [Citation12–Citation14]. Among them, PRDX1 (peroxiredoxin 1) has been identified as a 23-kDa stress-induced macrophage redox protein with multiple functions [Citation12,Citation13]. Extracellular PRDX1 plays a pivotal role in inflammation regulation [Citation15,Citation16]. It can act as a molecular chaperone with the ability to modulate actions of numerous molecules [Citation17,Citation18] or as a regulator of transcription [Citation19]. Moreover, PRDX1 expression is correlated with reactive oxygen species and the incidence of malignant disease [Citation20]. PRDX1 knockout mice have shown malignancies in the intestine, lymphomas, and sarcomas with a high frequency, suggesting that PRDX1 as a tumor suppressor might play a role in cancer development and progression [Citation18,Citation20,Citation21]. However, little is known about the functional role of PRDX1 in NFKB activation or autophagy activation.
Therefore, the objective of this study was to determine the functional role of PRDX1 in NFKB activation and autophagy activation. Our results showed that PRDX1 interacted with the ring finger domain of TRAF6 and inhibited its ubiquitin-ligase activity. The inhibition of TRAF6 ubiquitin-ligase activity attenuated the ubiquitination of ECSIT (ECSIT signaling integrator) protein essential for NFKB activation and BECN1 protein required for autophagy activation after TLR4 stimulation. The inhibitory effect of PRDX1 was clearly evidenced in PRDX1KD THP-1 cells. Moreover, PRDX1KD MDA-MB-231 and PRDX1KD SK-HEP-1 cancer cells showed increased cellular migration and invasion, suggesting that PRDX1 could negatively regulate tumor progression. Together, our data demonstrate a novel regulatory mechanism and function of PRDX1 in TLR4-mediated signaling and autophagy activation through inhibiting TRAF6 ubiquitin-ligase activity.
Results
PRDX1 negatively regulates the activation of NFKB induced by TLR4 stimulation
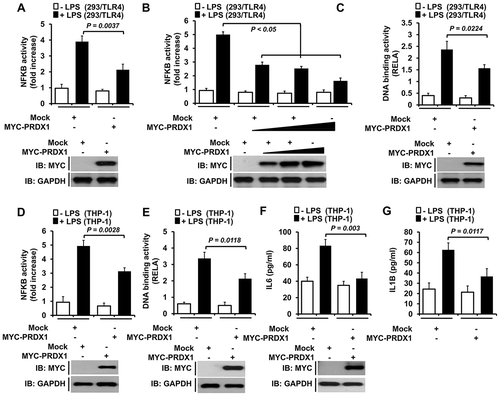
Recent reports have suggested that PRDX1 has several different functions in TLR4-mediated signaling. It is a ligand for TLR4, a modulator of intercellular signaling, and a transcriptional regulator [Citation12,Citation13,Citation16,Citation19]. However, little is known about the molecular mechanisms by which PRDX1 is implicated in TLR4-mediated signaling. To explore this issue, we examined whether PRDX1 was associated with activation of NFKB induced by TLR4 stimulation. NFKB reporter activity in response to lipopolysaccharide (LPS) stimulation in 293/TLR4 cells overexpressing PRDX1 was significantly attenuated compared to that in mock transfected cells ()). The attenuation of NFKB activity was clearly evidenced in a dose-dependent manner and in RELA/p65 (RELA proto-oncogene, NF-kB subunit) DNA-binding assay (,)). To confirm these results, human monocytic THP-1 cells were transfected with mock or MYC-PRDX1 vector and NFKB activities were then measured by using luciferase reporter assay and RELA DNA-binding assay. Consistently, NFKB activities were significantly attenuated in cells transfected with MYC-PRDX1(,), Fig. S1,). Moreover, levels of pro-inflammatory cytokines such as IL6 and IL1B (interleukin 1 beta) produced in response to LPS stimulation were markedly decreased in MYC-PRDX1-overexpressed cells (,)), suggesting that PRDX1 negatively regulated the activation of NFKB induced by TLR4 stimulation.
Figure 1. PRDX1 overexpression inhibits NFKB activation induced by TLR4. (A) 293/TLR4 cells were transfected with vector control (mock) or MYC-PRDX1 vector together with pBIIx-luc and Renilla luciferase vector, untreated or treated with LPS (200 ng/ml) for 6 h, and then subjected to luciferase activity assay. Results are expressed as fold-induction in luciferase activity relative to that in untreated cells. All error bars represent mean ±SEM from triplicate samples. Western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control. (B) 293/TLR4 cells were transfected with mock or different concentrations of MYC-PRDX1 vector. Luciferase reporter assay was then performed as described in (A). Results are expressed as fold-induction in luciferase activity relative to that in untreated cells. All error bars represent ±SEM of the mean from triplicate samples. Western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control. (C) 293/TLR4 cells were transfected with mock or MYC-PRDX1 vector, untreated or treated with LPS (200 ng/ml) for 6 h, and then analyzed for RELA DNA-binding activity. All error bars represent mean ±SEM of the mean from triplicate samples. IB western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control. (D) THP-1 cells were transfected with a mock or MYC-PRDX1 vector, untreated or treated with LPS (200 ng/ml) for 6 h, and then luciferase reporter assay was performed. Results are expressed as the fold-induction in luciferase activity relative to that in untreated cells. All error bars represent mean ±SEM from triplicate samples. Western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control. (E) THP-1 cells were transfected with mock or MYC-PRDX1 vector, untreated or treated with LPS (200 ng/ml) for 6 h, and then analyzed for RELA DNA-binding activity. All error bars represent mean ±SEM from triplicate samples. Western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control. (F and G) THP-1 cells were transfected with mock or MYC-PRDX1 vector, untreated or treated with LPS (200 ng/ml) for 9 h, and then subjected to enzyme-linked immunosorbent assay (ELISA) to determine levels IL6 (F) and IL1B (G) produced. All error bars represent mean ±SEM from triplicate samples. Western blot analysis was used to measure the expression of transiently transfected MYC-PRDX1 (lower panel). The expression of GAPDH was used as a loading control.
Enhanced TLR4-mediated signaling in PRDX1-knockdown THP-1 cells
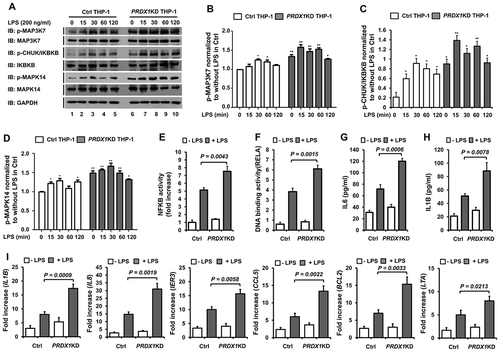
To verify the negative regulation of TLR4-mediated signaling by PRDX1, PRDX1-knockdown (PRDX1KD) THP-1 cells were generated by shRNA lentivirus particles targeting endogenous PRDX1. Control (Ctrl) THP-1 cells were also generated by using control shRNA lentivirus particles (Fig. S2). Ctrl and PRDX1KD THP-1 cells were treated with LPS for different time periods as indicated in ) and activation of TLR4-mediated signaling was then examined. LPS stimulation induced the phosphorylation of MAP3K7, leading to activation of CHUK/IKKA (conserved helix-loop-helix ubiquitous kinase), IKBKB/IKKB (inhibitor of nuclear factor kappa B kinase subunit beta), and MAPK14/p38 (mitogen-activated protein kinase 14) in Ctrl THP-1 cells. Such activations were markedly enhanced in PRDX1KD THP-1 cells ()). Levels of phosphorylated MAP3K7, CHUK/IKBKB, and MAPK14 were significantly higher in PRDX1KD THP-1 cells (-)). Furthermore, NFKB activity induced by LPS treatment was markedly higher in PRDX1KD THP-1 cells than that in Ctrl THP-1 cells (,)). Based on these results, we further examined whether NFKB activation by TLR4 stimulation in PRDX1KD THP-1 cells could lead to the production of pro-inflammatory cytokines IL6 and IL1B and the expression of NFKB-dependent genes. Upon TLR4 stimulation, IL6 and IL1B production was significantly enhanced in Ctrl THP-1 cells (,h)). Interestingly, their levels were significantly elevated in PRDX1KD THP-1 cells compared to those in Ctrl THP-1 cells (,h)). To verify the ELISA results, intracellular cytokine staining assay was performed in Ctrl and PRDX1KD THP-1 cells (Fig. S3). Upon LPS stimulation, levels of IL1B and IL6 were significantly higher in PRDX1KD THP-1 cells than those in Ctrl THP-1 cells (Fig. S3). We next evaluated expression levels of 6 different NFKB-dependent genes (IL1B, IL8, IER3 [immediate early response 3],CCL5 [C-C motif chemokine ligand 5],BCL2 [BCL2, apoptosis regulator], and LTA [lymphotoxin alpha]) containing specific KB-binding DNA sequences. These genes were significantly upregulated in LPS-treated PRDX1KD THP-1 cells compared to those in LPS-treated Ctrl THP-1 cells ()). Taken together, these results strongly suggest that PRDX1 negatively regulates TLR4-mediated signaling for NFKB activation.
Figure 2. PRDX1-knockdown THP-1 cells enhance TLR4-mediated signals. (A) Control (Ctrl) and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) for different time periods and then subjected to western blot analyses using anti-phospho-MAP3K7, anti-MAP3K7, anti-phospho-CHUK/IKBKB, anti-IKBKB, anti-phospho-MAPK14, anti-MAPK14, or anti-GAPDH antibody as a loading control. (B-D) Band intensity of phospho-MAP3K7 (B), phospho-CHUK/IKBKB (C), and phospho-MAPK14(D) was analyzed with Image J program (bottom). Data shown are averages from a minimum of 3 independent experiments (± SEM). * p < 0.05, ** p < 0.01 when compared to Ctrl THP-1 (without LPS). (E) Ctrl and PRDX1KD THP-1 cells were transfected with pBIIx-luc and Renilla luciferase vector, untreated or treated with LPS (200 ng/ml) for 6 h, and then analyzed for luciferase activity. Results are expressed as fold-induction in luciferase activity relative to that in untreated cells. All error bars represent mean ±SEM from triplicate samples. (F) Ctrl and PRDX1KD THP-1 cells were untreated or treated with LPS (200 ng/ml) for 6 h and then analyzed for RELA DNA-binding activity. All error bars represent mean ±SEM from triplicate samples. (G and H) Ctrl and PRDX1KD THP-1 cells were untreated or treated with LPS (200 ng/ml) for 9 h and used for enzyme-linked immunosorbent assay (ELISA) to determine levels of IL6 (G) and IL1B (H) produced. All error bars represent mean ±SEM from triplicate samples. (I) Control (Ctrl) and PRDX1KD THP-1 cells were untreated or treated with LPS (200 ng/ml) for 6 h. Total RNA was extracted and quantitative RT-PCR analysis was performed using gene-specific primers for IL1B, IL8, IER3, CCL5, BCL2, and LTA. All error bars represent mean ±SEM from triplicate samples.
PRDX1 inhibits TRAF6 ubiquitin-ligase activity
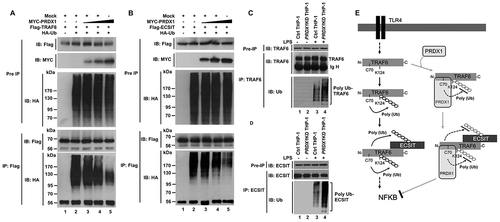
We next explored the molecular mechanism by which PRDX1 negatively regulated TLR4-mediated signaling. Our recent studies have revealed that PRDX6 (peroxiredoxin 6) interrupts the formation of TRAF6-ECSIT complex which plays a key role in the activation of NFKB induced by TLR4 stimulation through molecular interaction between TRAF6 and PRDX6 [Citation22–Citation25]. Therefore, we hypothesized that PRDX1 also could affect the formation of TRAF6-ECSIT complex. An immunoprecipitation (IP) assay in HEK293T cells revealed that MYC-PRDX1 proteins were precipitated with Flag-TRAF6 protein (), lane 4), whereas no significant interaction between PRDX1 and ECSIT was found (Fig. S4). Endogenous PRDX1 proteins in THP-1 cells were also significantly pulled down with TRAF6 proteins (), lane 2), indicating that PRDX1 could interact with TRAF6, but not with ECSIT. To figure out the PRDX1 interaction site on TRAF6, HEK293T cells were transfected with Flag-TRAF6 wild type (WT), Flag-TRAF6 truncation mutants, or MYC-PRDX1 vector as indicated in ), and then IP assay was performed using anti-Flag antibody. MYC-PRDX1 proteins were markedly precipitated with Flag-TRAF6 WT only (), lane 7), but not with Flag-TRAF6 truncation mutants (), lane 8–10), indicating that PRDX1 could interact with the ring finger domain of TRAF6. To verify the above result, HEK293T cells were transfected with Flag-TRAF6 [1–110] or MYC-PRDX1 vector as indicated in ), and then IP assay was performed using anti-Flag antibody. As expected, MYC-PRDX1 proteins were markedly precipitated with Flag-TRAF6 [1–110] (), lane 2). Consistent with a previous report [Citation24], MYC-ECSIT proteins interacted with Flag-TRAF6 WT, Flag-TRAF6 [110–522], and Flag-TRAF6 [260–522] (), lane 7, 8, and 9), whereas a relatively weak interaction with Flag-TRAF6 [349–522] could be seen (), lane 10), indicating that the C-terminal region of TRAF6 comprising coiled coil and TRAF-C domains might be required for ECSIT binding ()). These results suggest that PRDX1 and ECSIT proteins interact with the ring finger domain and the C-terminal region of TRAF6, respectively, as depicted in ).
Figure 3. PRDX1 interacts with TRAF6. (A) HEK293T cells were transfected with mock, Flag-TRAF6, or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by immunoblotting (IB) using anti-Flag or anti-MYC antibody. (B) THP-1 cells were treated with LPS (200 ng/ml) for 60 min. Immunoprecipitation assay was performed using lysates with IgG antibody and anti-TRAF6 antibody. IB assay was then performed using anti-TRAF6 and anti-PRDX1 antibodies. (C) HEK293T cells were transfected with mock, Flag-TRAF6 wild type (WT), Flag-TRAF6 truncated mutants, or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody. (D) HEK293T cells were transfected with mock, Flag-TRAF6 [1–110], or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody. (E) HEK293T cells were transfected with Mock, Flag-TRAF6 wild type (WT), Flag-TRAF6 truncated mutants or MYC-ECSIT as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody, and detected with anti-mouse Ig HRP TrueBlot antibody. (F) Model of how PRDX1 and ECSIT interact with TRAF6. See text for details.
![Figure 3. PRDX1 interacts with TRAF6. (A) HEK293T cells were transfected with mock, Flag-TRAF6, or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by immunoblotting (IB) using anti-Flag or anti-MYC antibody. (B) THP-1 cells were treated with LPS (200 ng/ml) for 60 min. Immunoprecipitation assay was performed using lysates with IgG antibody and anti-TRAF6 antibody. IB assay was then performed using anti-TRAF6 and anti-PRDX1 antibodies. (C) HEK293T cells were transfected with mock, Flag-TRAF6 wild type (WT), Flag-TRAF6 truncated mutants, or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody. (D) HEK293T cells were transfected with mock, Flag-TRAF6 [1–110], or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody. (E) HEK293T cells were transfected with Mock, Flag-TRAF6 wild type (WT), Flag-TRAF6 truncated mutants or MYC-ECSIT as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-MYC antibody, and detected with anti-mouse Ig HRP TrueBlot antibody. (F) Model of how PRDX1 and ECSIT interact with TRAF6. See text for details.](/cms/asset/ea9bf57c-6f49-4757-831b-036450c0c905/kaup_a_1474995_f0003_b.gif)
TRAF6 catalyzes its own Lys63-linked ubiquitination which depends on its ring finger domain [Citation26]. Since PRDX1 interacted with the ring finger domain of TRAF6 ()), we next examined whether PRDX1-TRAF6 interaction might affect TRAF6 ubiquitin-ligase activity. Flag-TRAF6 and HA-Ub were expressed in HEK293T cells along with different concentrations of MYC-PRDX1 and IP assay was performed using anti-Flag antibody. Ubiquitination of TRAF6 was strongly increased in the absence of MYC-PRDX1 (), lane 2), whereas significant decreases in ubiquitination of TRAF6 were observed in the presence of MYC-PRDX1 (), lane 3–5), indicating that PRDX1 inhibited the ubiquitination of TRAF6. The interaction between TRAF6 and ECSIT leads to ubiquitination of ECSIT that is crucial for the activation of NFKB in response to TLR4 stimulation [Citation22–Citation24]. Therefore, we further examined whether PRDX1 could affect TRAF6-induced ubiquitination of ECSIT. As shown in ), ubiquitination of Flag-ECSIT was significantly enhanced in the absence of MYC-PRDX1 (), lane 2), whereas marked decreases in ubiquitination of Flag-ECSIT were observed in the presence of MYC-PRDX1(), lane 3–5). To verify the results, Ctrl and PRDX1KD THP-1 cells were treated with or without LPS, and then the ubiquitination of TRAF6 or ECSIT was evaluated by anti-Ub antibody after an immunoprecipitation assay was performed using anti-TRAF6 ()) or anti-ECSIT antibody ()). As expected, the ubiquitination of TRAF6 or ECSIT in response to LPS stimulation was seen in Ctrl THP-1 cells (,)) and significant increases could be seen in PRDX1KD THP-1 cells (,)). Taken together, these results demonstrate that, upon TLR stimulation, TRAF6 is auto-ubiquitinated at its K124 residue with simultaneous ubiquitination of ECSIT protein (), left) [Citation22–Citation26]. The complex is further associated with TAB1 and TAB2 proteins to facilitate the activation of MAP3K7, leading to the activation of NFKB [Citation24,Citation26,Citation27]. However, the interaction between PRDX1 and TRAF6 might lead to inhibition of TRAF6 ubiquitin-ligase activity by masking the C70 residue of TRAF6 which inhibits the ubiquitination of TRAF6 and ECSIT, leading to inhibition of NFKB activation (), right).
Figure 4. PRDX1 inhibits ubiquitination of TRAF6 and ECSIT. (A) HEK293T cells were transfected with Mock, Flag-TRAF6, HA-Ub, or different concentrations of MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-HA antibody. (B) HEK293T cells were transfected with Mock, Flag-ECSIT, HA-Ub, or different concentrations of MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-HA antibody. (C and D) Ctrl and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) for 60min. Immunoprecipitation assay was performed using lysates with anti-TRAF6 (C) or anti-ECSIT antibody (D). IB assay was performed using anti-TRAF6, anti-ECSIT, and anti-Ub antibodies. (E) Model showing how PRDX1 inhibits ubiquitination of TRAF6 and ECSIT. Upon TLR4 stimulation, TRAF6 is auto-ubiquitinated. It simultaneously ubiquitinates its associated proteins including ECSIT. The TRAF6-ECSIT complex induces activation of TLR4 downstream molecules such as MAP3K7 and IKKs, leading to activation of NFKB (left). In contrast, the interaction of PRDX1 with the ring finger domain of TRAF6 inhibits TRAF6 ubiquitin ligase activity, resulting in inhibition of ubiquitination of TRAF6 and ECSIT which leads to inhibition of NFKB activation (right).
PRDX1 inhibits autophagy activation through inhibition of BECN1 ubiquitination
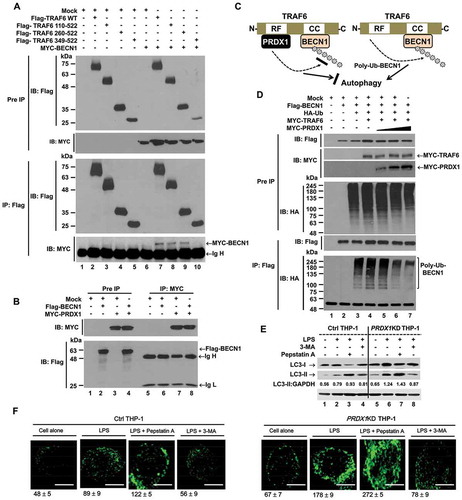
A recent report has shown that TRAF6-mediated ubiquitination of BECN1 is critical for TLR4-triggered autophagy in macrophages [Citation10]. Since PRDX1 inhibited ubiquitin-ligase activity of TRAF6, we examined whether PRDX1 was functionally associated with TRAF6-mediated autophagy activation. Consistent with a previous report, Flag-TRAF6 WT interacted with MYC-BECN1 (), lane 7) and 2 Flag-TRAF6 truncation mutants, Flag-TRAF6 [110–522] and Flag-TRAF6 [260–522], significantly interacted with MYC-BECN1 (), lanes 8 and 9) whereas no significant interaction could be seen in Flag-TRAF6 [349–522] (), lane 10). No significant interaction between Flag-BECN1 and MYC-PRDX1 was observed ()). These results indicate that BECN1 interacts with the coiled-coil domain of TRAF6, but not with PRDX1 as depicted in ). Since PRDX1 inhibited TRAF6 ubiquitin-ligase activity through its interaction with the ring finger domain of TRAF6, we examined whether PRDX1 could inhibit TRAF6-mediated ubiquitination of BECN1. As expected, ubiquitination of BECN1 was markedly enhanced in the presence of TRAF6 (), lane 3 vs. lane 4). However, ubiquitination of BECN1 was gradually attenuated in the presence of PRDX1 (), lane 4 vs. lane 5–7), strongly suggesting that PRDX1 could attenuate the ubiquitination of BECN1 through inhibiting TRAF6 ubiquitin-ligase activity. To verify the functional role of PRDX1 in autophagy activation in macrophages, Ctrl and PRDX1KD THP-1 cells treated with or without LPS in the presence or absence of 3-methyladenine (3-MA), an inhibitor of phosphatidylinositol 3-kinases, and pepstatin A, a lysosomal protease inhibitor, for 6 h, and then the ratio between amounts of MAP1LC3/LC3-II (microtubule associated protein 1 light chain 3) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was monitored by western blotting. Upon TLR4 stimulation, levels of LC3-II were markedly elevated in PRDX1KD THP-1 cells compared to those in Ctrl THP-1 cells (), lane 2 vs lane 6). As expected, significant accumulation of LC3-II protein levels could be detected in treatment of the pepstatin A in both cells (), lane 3 vs lane 7), whereas marked attenuations could be seen in treatment of 3-MA (), lane 4 vs lane 8). Upon TLR4 stimulation, moreover, the number of autophagosomes in PRDX1KD THP-1 cells was significantly higher than that in Ctrl THP-1 cells (), Ctrl treated LPS vs. PRDX1KD treated LPS). In addition, the number of autophagosomes was significantly increased in both cells treated with pepstatin A, whereas marked attenuations could be seen in treatment of 3-MA ()). These results demonstrate that TRAF6 facilitates autophagy activation via ubiquitination of BECN1 (), right), whereas the interaction of PRDX1 with the ring finger domain of TRAF6 induces attenuation of BECN1 ubiquitination, leading to inhibition of autophagosome formation (), left).
Figure 5. PRDX1 is negatively involved in autophagy activation through inhibiting BECN1 ubiquitination. (A) HEK293T cells were transfected with Mock, Flag-TRAF6 wild type (WT), Flag-TRAF6 truncated mutants, or MYC-BECN1 as indicated. At 38 h post transfection, transfected cells were extracted, immunoprecipitated with anti-Flag antibody, and then subjected to IB assay using anti-Flag or anti-MYC antibody. (B) HEK293T cells were transfected with Mock, Flag-BECN1, or MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-MYC antibody followed by IB using anti-Flag or anti-MYC antibody. (C) Model showing of how PRDX1-TRAF6 interaction inhibits the ubiquitination of BECN1. See text for details. (D) HEK293T cells were transfected with Mock, Flag-BECN1, HA-Ub, MYC-TRAF6, or different concentrations of MYC-PRDX1 as indicated. At 38 h post transfection, transfected cells were extracted and cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by IB using anti-Flag or anti-HA antibody. (E) Control (Ctrl) and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) in the presence or absence of pepstatin A (10 μM) and 3-MA (5 mM) for 6 h as indicated. These cells were lysed and subjected to SDS-PAGE followed by immunoblotting with LC3-I/-II or GAPDH antibody. Band intensity was quantified using ImageJ software. Quantitative data were calculated from 3 independent experiments. Data are shown as mean ±SEM. (F) Control (Ctrl) and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) in the presence or absence of pepstatin A (10 μM) and 3-MA (5 mM) for 6 h as indicated, and then fixed. Immunofluorescence assay was performed with anti-LC3 antibody. Digital images were captured with confocal microscopy and the number of LC3-puncta was scored. Quantification represents the mean ±SEM of puncta per cell (n = 5) from 3 independent experiments. Scale bar: 10 μm.
PRDX1 negatively regulates bactericidal activity and cancer progression
Autophagy is critically associated with bacterial elimination in infectious diseases [Citation28] and cancer progression [Citation29–Citation34]. TRAF6 is functionally implicated in bactericidal activity through regulation of mitochondrial reactive oxygen species (mROS) [Citation25,Citation35] and autophagy activation through ubiquitination of BECN1 [Citation10]. We therefore investigated whether PRDX1 was functionally associated with bactericidal activity and cancer progression through inhibition of TRAF6 ubiquitin-ligase activity. Previous reports have shown that TRAF6-mediated ubiquitination of ECSIT is critical for mROS production associated with bactericidal activity [Citation25,Citation35]. Consistent with previous results, mROS level in PRDX1KD THP-1 cells was significantly higher than that in Ctrl THP-1 cells (Fig. S5), suggesting that PRDX1 could negatively regulate mROS through inhibiting TRAF6-mediated ubiquitination of ECSIT. To determine the functional role of PRDX1 in bactericidal activity, Ctrl or PRDX1KD THP-1 cells were infected with the facultative intracellular pathogen Salmonella typhimurium and survival of the bacterium was then measured. The number of colonies in Ctrl THP-1 cells was significantly increased in a time-dependent manner. However, it was significantly decreased in PRDX1KD THP-1 cells ()). In addition, the survival of S. typhimurium was significantly attenuated in PRDX1KD THP-1 cells compared to that in Ctrl THP-1 cells ()). These results suggest that PRDX1 is negatively implicated in the bactericidal activity by the regulation of mROS production, which might be functionally due to the inhibition of TRAF6-mediated ubiquitination of ECSIT protein.
Figure 6. PRDX1 negatively regulates bactericidal activity and cancer progression in vitro. (A and B) Control (Ctrl) and PRDX1KD THP-1 cells were infected with Salmonella wild type (14028s strain) at a multiplicity of infection of 10 bacteria/cell as described in Methods. Cells were lysed with 0.5% deoxycholate in Dulbecco’s PBS. Bacteria were diluted (x 50) and plated onto LB agar. The number of colonies was counted and presented (A). The number of colonies at T = 0 was presented as an average of both cell lines (control and PRDX1KD cells). Percentage survival was obtained by dividing the number of bacteria recovered after 6 h (T6) or 12 h (T12) by the number of bacteria present at time 0 (T0) and multiplying by 100 (B). All error bars represent mean ±SEM of 3 independent experiments. * p < 0.05. (C) Ctrl and PRDX1KD MDA-MB-231 cells were seeded into 12-well cell culture plates, cultured in DMEM supplemented with 10% FBS, and allowed to grow to near confluence. Confluent monolayers were carefully wounded with a sterile yellow Gilson-pipette tip and cellular debris was gently washed away with culture medium, and then treated with vehicle (DMSO,<0.2% in culture medium) or 3-MA (5 mM) for different time periods as indicated. Scale bar: 500 μm. (D) Ctrl and PRDX1KD SK-HEP-1cells were seeded into 12-well cell culture plates, cultured in DMEM supplemented with 10% FBS, and allowed to grow to near confluence. Confluent monolayers were carefully wounded with a sterile yellow Gilson-pipette tip and cellular debris was gently washed away with culture medium, and then treated with vehicle (DMSO,<0.2%) or 3-MA (5 mM) for different time periods as indicated. Scale bar: 500 μm. (E) Ctrl and PRDX1KD MDA-MB-231 cells were suspended in DMEM containing vehicle or 3-MA (5 mM) and added to the top chambers of the transwells in 24-well plates. After an overnight incubation, cells were fixed and stained with crystal violet. Scale bar: 50 μm. Number of cell migration was counted and results are presented as mean ±SEM of 3 independent experiments. (F) Ctrl and PRDX1KD SK-HEP-1cells were suspended in DMEM containing vehicle or 3-MA (5 mM) and added to the top chambers of the transwells in 24-well plates. After an overnight incubation, cells were fixed and stained with crystal violet. Scale bar: 50 μm. Number of cell migration was counted and results are presented as mean ±SEM of 3 independent experiments.
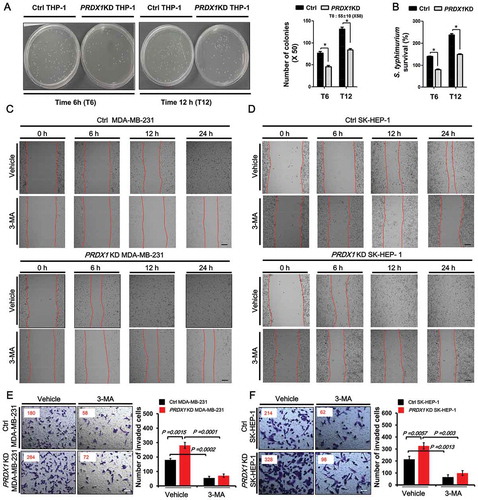
Next, we examined whether PRDX1-induced inhibition of autophagy activation was associated with cancer progression. First, we generated PRDX1KD MDA-MB-231 and PRDX1KD SK-HEP-1 cancer cells (Fig. S6). We then performed wound healing and transwell invasion assays to assess the capacity of cancer migration and invasion, respectively. The migration of PRDX1KD MDA-MB-231 and PRDX1KD SK-HEP-1 cells treated with vehicle was increased in a time-dependent manner, as compared with those of Ctrl cells (,)). As expected, the treatment of 3-MA, an autophagy inhibitor, induced the marked inhibition of migration in both cells (,), vehicle vs. 3-MA). Additionally, transwell invasion assay revealed significant increases of cellular invasive ability in PRDX1KD MDA-MB-231 and PRDX1KD SK-HEP-1 cells compared to Ctrl MDA-MB-231 and Ctrl SK-HEP-1 cells (,)), whereas the marked attenuation could be detected in the treatment of 3-MA (,), vehicle vs. 3-MA). These results suggest that PRDX1 is negatively implicated in cancer progression through inhibition of autophagy activation.
Discussion
In this study, we provided 2 insights into the regulation of PRDX1 in TLR4-induced activation of NFKB and autophagy activation. Our biochemical study revealed that PRDX1 interacted with the ring finger domain of TRAF6, which critically affected TRAF6 ubiquitin-ligase activity, resulting in attenuation of ubiquitination of TRAF6, ECSIT, and BECN1. Inhibition of TRAF6 and ECSIT ubiquitination led to inhibition of TLR4-induced activation of NFKB while inhibition of BECN1 ubiquitination led to inhibition of TLR4-induced autophagy activation. Moreover, we found that PRDX1-knockdown MDA-MB-231 and SK-HEP-1 cancer cells showed increases in the capacity of cellular migration and invasion, indicating that PRDX1 was negatively implicated in cancer progression through inhibition of autophagy activation.
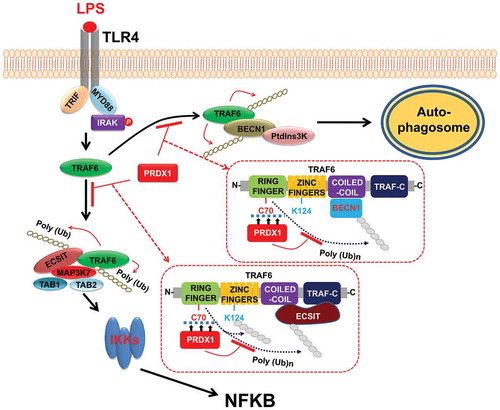
Protein ubiquitination as a posttranslational modification is important for signaling activation and protein trafficking, thereby regulating various biological processes [Citation36,Citation37]. In TLR-mediated signaling, TRAF6 as an E3 ubiquitin ligases plays pivotal roles in the activation of NFKB [Citation4–Citation6]. TRAF6 conjugates K63-linked ubiquitin chains to itself and other proteins and activates ubiquitin-dependent kinase MAP3K7, which in turn activates IKK and MAPKs such as MAPK8/JNK (mitogen-activated protein kinase 8) and MAPK14. The C70 residue of TRAF6 ring finger domain is critical for its ubiquitin-ligase activity [Citation26]. The TRAF6C70A mutant resulted in defective E3 ubiquitin-ligase activity. Interestingly, we found that PRDX1 interacted with the ring finger domain of TRAF6, resulting in the attenuation of ubiquitination of TRAF6 and ECSIT proteins and inhibition of NFKB activation induced by TLR4 stimulation. These results strongly suggest that PRDX1 negatively regulates TLR4-induced activation of NFKB via inhibiting TRAF6 ubiquitin-ligase activity. TRAF6 is also involved in TLR-induced autophagy activation [Citation10]. It has been reported that TLR4 signaling triggers K63-linked ubiquitination of BECN1 through a mechanism that depends on TRAF6. Consistently, we found that TRAF6 induced ubiquitination of BECN1. However, BECN1 ubiquitination was significantly attenuated in the presence of PRDX1. Moreover, our data showed that TLR4-induced autophagy activation was significantly enhanced in PRDX1KD THP-1 cells. These results suggest that TRAF6 functions as an E3 ligase to ubiquitinate BECN1 whereas PRDX1 inhibits the ubiquitination of BECN1 through inhibiting TRAF6 ubiquitin-ligase activity to control the extent of autophagy elicited by TLR4 signaling ().
Figure 7. Negative regulation of PRDX1 in TLR4-induced NFKB activation and autophagy activation by inhibiting TRAF6 ubiquitin-ligase activity. Upon TLR4 stimulation, TRAF6 E3 ligase activity is enhanced and BECN1 is ubiquitinated. Ubiquitinated BECN1 facilitates the oligomerization of BECN1 and the activation of the phosphatidylinositols 3-kinase, leading to formation of autophagosomes. The engagement of TLR4 also triggers TRAF6 auto-ubiquitination and its associated proteins, including ECSIT. The TRAF6-ECSIT complex induces activations of TLR4 downstream molecules such as MAP3K7 and IKKs, leading to the activation of NFKB. PRDX1 interacts with the ring finger domain of TRAF6 which inhibits TRAF6 ubiquitin-ligase activity. The inhibition of TRAF6 ubiquitin-ligase activity results in inhibition of TRAF6-mediated ubiquitination required for NFKB activation and autophagy activation.
PRDXs are a family of antioxidant enzymes also known as scavengers of peroxide in mammalian cells [Citation12,Citation13]. Previous reports have shown that overexpression of PRDX1 is associated with cancer development [Citation21,Citation29–Citation34]. In addition, PRDX1 expression is correlated with reactive oxygen species and the incidence of malignant disease [Citation21]. PRDX1-deficiency in mouse model exhibited hemolytic anemia and multiple tumors, including mammary carcinomas [Citation18,Citation34,Citation38]. The tumor suppressive effects of PRDX1 were closely associated with a reduction of MYC transcriptional activity [Citation38] or of PTEN (phosphatase and tensin homolog)/AKT1 (AKT serine/threonine kinase 1) activity [Citation18]. In lung cancer, additionally, the PRDX1-induced tumor suppressant effect is mediated by the modulation of the ROS-mediated activation of the KRAS-MAPK/ERK pathway [Citation39]. Moreover, PRDX1 promotes the reactivation of PTPRJ/DEP-1 (protein tyrosine phosphatase, receptor type J), a tumor suppressor that counteracts the action of the transforming kinase, FLT3 (fms related tyrosine kinase 3)-internal tandem duplication (ITD), in human acute myeloid leukemia (AML) [Citation40]. Increasing evidence shows that autophagy is critically implicated in cancer development and progression [Citation20,Citation41–Citation45]. Although cellular and molecular mechanisms by which autophagy is regulated in cancer cell migration and invasion remain unclear, a recent report has shown that induction of autophagy contributes to TLR4- and TLR3-triggered progression of lung cancer cells through promoting TRAF6 ubiquitination [Citation11]. We found that PRDX1-knockdown human breast MDA-MB-231 cells and human hepatic adenocarcinoma SK-HEP-1 cells possessed enhanced cell migration and invasion properties. Although the precise molecular and cellular mechanisms by which PRDX1 is involved in cancer progression through autophagy activation are currently unknown, our data suggest that PRDX1 might be involved in cancer cell migration and invasion through inhibiting TRAF6-mediated autophagy activation.
In conclusion, in TLR4-induced NFKB activation and autophagy, PRDX1 regulates TRAF6 ubiquitin-ligase activity as depicted in . Upon TLR4 stimulation, TRAF6 is auto-ubiquitinated. It also simultaneously ubiquitinates its associated proteins including ECSIT. The TRAF6-ECSIT complex then induces activation of TLR4 downstream molecules such as MAP3K7 and IKKs, leading to activation of NFKB [Citation22,Citation24,Citation27]. TRAF6 also ubiquitinates BECN1. Ubiquitinated BECN1 then induces autophagy [Citation10]. However, the interaction between PRDX1 and the ring finger domain of TRAF6 inhibits TRAF6 ubiquitin-ligase activity, resulting in inhibition of TRAF6-mediated ubiquitination required for NFKB activation and autophagy activation. Our current results will contribute to our understanding of cancer development and progression in various cancers.
Materials and methods
Cell lines
HEK293T human embryonic kidney cells were purchased from the American Type Culture Collection (ATCC, CRL-11,268) and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific,11,965,092). HEK293 cells expressing human TLR4 (293/TLR4) were purchased from InvivoGen (293-htlr4a) and maintained per the manufacturer’s protocol. THP-1 human monocytic cells were purchased from ATCC (TIB-202) and maintained in RPMI 1640 medium (Thermo Fisher Scientific,11,875,093) containing 10% fetal bovine serum (FBS; Hyclone, 11,306,060), 2 mM L-glutamine (GIBCO, A2916801), 100 units/ml penicillin (GIBCO, 15,140,122), 100μg/ml streptomycin (GIBCO, 15,140,122), and 5 × 10−5 M β-mercaptoethanol (GIBCO, 21,985,023). Human breast carcinoma cell line MDA-MB-231 (HTB-26) and human hepatic adenocarcinoma cell line SK-HEP-1 cells (HTB-52) were obtained from ATCC and maintained in DMEM supplemented with 10% FBS. 3-Methyladenine (3-MA, M9281), and pepstatin A (P4265) were purchased from Sigma. Stock solutions were prepared in dimethyl sulfoxide (DMSO; Sigma, 472,301). The final concentration of DMSO in culture medium was <0.2% volume.
Plasmids
The following plasmids were used: Flag-tagged PRDX1 (Addgene, 38,086; Markus Landthaler lab), Flag-tagged TRAF6 (Addgene, 21,624; John Kyriakis), and Flag-tagged BECN1 (Addgene, 24,388; Qing Zhong). MYC-tagged ECSIT, HA-tagged Ub and Flag-tagged ECSIT were kindly provided by Jae-Hyuck Shim (University of Massachusetts Medical School, USA). The constructs coding for full-length PRDX1 and BECN1 with the MYC tag were cloned into the pCMV-3Tag-7 vector (Agilent technologies, 240,202) using Flag-tagged PRDX1 and Flag-tagged BECN1, respectively, as a template. Flag-tagged TRAF6 truncated mutants were generated as described previously [Citation25].
Generation of PRDX1-knockdown cells
Lentivirus containing small hairpin RNA (shRNA) targeting human PRDX1 (sc-36,177-V) and control shRNA lentivirus(sc-108,080) were purchased from Santa Cruz Biotechnology. Cells were cultured in 24-well cell culture plates (2 × 104 cells per well) and infected with control shRNA lentivirus or PRDX1 shRNA lentivirus according to the manufacturer’s protocol. Control (Ctrl) or PRDX1-knockdown (PRDX1KD) cells were selected in puromycin (4–8 μg/ml; Sigma, P8833) -containing medium and cultured as described previously [Citation23–Citation25].
Luciferase reporter assay
Luciferase reporter assay was performed as described previously [Citation23–Citation25,Citation46]. Briefly, cells were transiently transfected with or without appropriate vectors indicated in each experiment using Lipofectamine 2000 (Thermo Fisher Scientific, 11,668,019) or Neon® Transfection System (Thermo Fisher Scientific, MPK5000). After 24 h, cells were transiently transfected with NFKB-dependent reporter construct pBIIx-luc and Renilla luciferase vector (a kind gift from Sankar Ghosh, Columbia University, USA). Cells were treated with or without lipopolysaccharide (LPS, 200 ng/ml; Sigma, serotype 0128:B12) for 6 h. Luciferase activity was then measured using dual-luciferase assay kit (Promega, E1910).
Measurement of proinflammatory cytokines and RELA dna-binding assay
Cells were treated with or without LPS (200 ng/ml) for 9 h. Supernatants were then harvested. Levels of IL1B and IL6 in supernatants were measured using Human IL1B (R&D Systems, HSLB00C) and IL6 ELISA kits (R&D Systems, D6050) per the manufacturer’s protocol. For RELA/p65 DNA-binding assay, cells were transiently transfected with or without appropriate vectors indicated in each experiment using Lipofectamine 2000 or Neon® Transfection System. Cells were then treated with or without LPS (200 ng/ml) for 6 h. Nuclear proteins were then prepared using CelLytic NuCLEAR extraction kit (Sigma, NXTRACT) per the manufacturer’s protocol. Activity of transcription factor RELA was determined using Trans AM NFKB transcription factor assay kit (Active Motif North America, 40,596) per the manufacturer’s instructions [Citation24,Citation46].
Western blotting and immunoprecipitation (IP) assays
Western blotting and immunoprecipitation (IP) assays were performed as described previously [Citation23–Citation25,Citation46]. Briefly, HEK293T cells were transiently transfected with or without appropriate vectors indicated in each experiment using Lipofectamine 2000. At 38 h after transfection, transfected cells were harvested and cell lysates were immunoprecipitated with either anti-Flag (Sigma-Aldrich, F-3165) or anti-MYC (Cell Signaling Technology, 2276) antibody. Immunoprecipitated complexes were separated by 6–10% SDS-PAGE and probed with anti-HA (Sigma-Aldrich, H3663), anti-MYC, or anti-Flag antibody. Ctrl and PRDX1KD THP-1 cells treated with or without 3-MA (5 mM) and pepstatin A (10 μM) were stimulated with LPS (200 ng/ml) for 6 h. Whole cell lysates were subjected for immunoblot analysis of LC3A/B (Cell Signaling Technology, 4108S) and GAPDH (Cell Signaling Technology, 2118S) as a loading control. For endogenous immunoprecipitation assay, THP-1 cells were treated with LPS (200 ng/ml) for 60 min. Immunoprecipitation assay was performed using cell lysates with IgG (Santa Cruz Biotechnology, sc-3888) antibody and anti-TRAF6 (Santa Cruz Biotechnology, sc-7221) antibody. Immunoblot (IB) assay was performed with anti-TRAF6 and anti-PRDX1 (Abcam, ab41906) antibodies. Ctrl and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) for 60 min. Immunoprecipitation assay was performed using anti-TRAF6 or anti-ECSIT (Abcam, ab21288) antibody. IB assay was performed with anti-TRAF6, anti-ECSIT, and anti-Ub (Abcam, ab7780) antibodies. Ctrl and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) for different time periods. Cells were lysed and cell lysates were separated by 6–10% SDS-PAGE and probed with the following antibodies: phospho-MAP3K7/TAK1 (Cell Signaling Technology, 9339), MAP3K7 (Cell Signaling Technology, 4505), phospho-MAPK14/p38 (Cell Signaling Technology, 9211), MAPK14 (Cell Signaling Technology, 9212), phospho-CHUK/IKKA, IKBKB/IKKB (Cell Signaling Technology, 2697), IKBKB/IKKB (Cell Signaling Technology, 2684), LC3A/B (Cell Signaling Technology, 4108S), and GAPDH (Cell Signaling Technology, 2118S). Anti-Mouse Ig HRP TrueBlot antibodies was purchased from Rockland Immunochemicals Inc. (18–8817-30).
Quantitative real-time polymerase chain reaction (QRT-PCR)
Ctrl and PRDX1KD THP-1 cells were treated with or without LPS (200 ng/ml) for 6 hr. Total RNA was isolated and cDNA was synthesized using the RNeasy Mini Kit (Qiagen, 74,104) and the QuantiTect Reverse Transcription kit (Qiagen, 205,311) per the manufacturer’s protocol. Primers hIL1B (PPH00171C), hIL8 (PPH00568A), hIER3 (PPH10008E), hCCL5 (PPH00703A), hBCL2 (PPH00079B), and hLTA (PPH00337E) were purchased from Qiagen. qRT-PCR was performed using Rotor-Gene Q (Qiagen, 9,001,550) per the manufacturer’s protocol.
Immunofluorescence microscopy
Cells were grown on glass coverslips overnight, fixed with 4% paraformaldehyde (Sigma, P-6148), and treated with 0.2% Triton X-100 (Sigma, T9284) to permeabilize for 30 min on ice. Immunofluorescence microscopy assay for detection of LC3 puncta was performed as described previously [Citation11]. Slides were mounted in VECTASHIELD mounting medium (Vector Laboratories, H-1000) and examined under a LSM 710 laser-scanning confocal microscope (Carl Zeiss,Jena, Germany).
Salmonella infection assay
Salmonella infection was performed as described previously [Citation25,Citation47]. Briefly, 5 × 105 control (Ctrl) or PRDX1KD THP-1 cells were cultured in fresh RPMI 1640 complete medium without antibiotics in the presence of phorbol 12-myristate 13-acetate (PMA, 20 ng/ml; Sigma, 16,561–29-8) for 24 hand seeded into culture wells at a concentration of 7 × 105 cells/ml. The next day, non-adherent cells were removed and medium was replaced with fresh complete medium without antibiotics. Cells were infected with Salmonella typhimurium wild type (14028s strain; a kind gift from Dongwoo Shin, Sungkyunkwan University, Korea) at a multiplicity of infection of 10 bacteria/cell. Culture plates were centrifuged at 200 × g for 5 min and incubated at 37°C for 30min to allow phagocytosis to occur. The medium was then replaced with fresh medium containing gentamicin (20 μg/ml; Sigma, G1272) and incubated for different time periods. Total cell population in the well was harvested. An aliquot of the harvested cell population was centrifuged and macrophages were lysed by 0.5% deoxycholate (Sigma, D6750) in Dulbecco’s phosphate-buffered saline (Sigma, D8537).
Wound-healing and transwell migration assay
A wound-healing assay was performed as described previously [Citation11]. Briefly, cells were seeded into 12-well plates and grown to confluence. Cell monolayer was gently scratched with a sterile yellow Gilson-pipette tip to form a wide gap of approximately 400 μm. Cells were then rinsed with culture medium to remove floating cells and debris. Cells were treated with vehicle (DMSO,<0.2% in DMEM culture medium) or 3-MA (5 mM), and images were captured after 0 h, 6 h, 12 h, or 24 h. For migration assay, transwell inserts (8-μm pore; Corning, 3422) were placed into wells. The cells (5 × 104 cells/well) were suspended in DMEM containing vehicle or 3-MA (5 mM) and added to the top chambers of the transwells in 24-well plates, and DMEM with 10% FBS was added to the bottom chambers. After an overnight incubation, the cells that remained in the top chamber (non-migrated) were removed, and the cells in the bottom chamber (migrated) were fixed and stained with crystal violet to visualize the nuclei. All experiments were conducted in triplicate and repeated twice.
Statistical analysis
In vitro data are presented as mean ±SEM of the mean from triplicate samples. Statistical differences were analyzed by ANOVA or Student’s t-test using GraphPad Prism5.0 (GraphPad Software, San Diego, CA, USA).
Supplemental Material
Download MS Word (215.1 KB)Acknowledgments
We would like to thank Hyehwa Forum members for their helpful discussion.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;8:675–680.
- Kopp EB, Medzhitov R. The Toll-receptor family and control of innate immunity. CurrOpin Immunol. 1999;1:13–18.
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;3:179–190.
- Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
- Kaisho T, Akira S. Toll-like receptors and their signaling mechanism in innate immunity. Acta OdontolScand. 2001;3:124–130.
- Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;5:816–825.
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, et al. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256.
- Takaesu G, Kishida S, Hiyama A, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;4:649–658.
- Qian Y, Commane M, Ninomiya-Tsuji J, et al. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFkappa B. J BiolChem. 2001;276:41661–41667.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42.
- Zhan Z, Xie X, Cao H, et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257–268.
- Ishii T, Yamada M, Sato H, et al. Cloning and characterization of a 23-kDa stress-induced mouse peritoneal macrophage protein. J BiolChem. 1993;268:18633–18636.
- Yamaguchi M, Sato H, Bannai S. Induction of stress proteins in mouse peritoneal macrophages by oxidized low-density lipoprotein. BiochemBiophys Res Commun. 1993;193:1198–1201.
- Wood ZA, Schröder E, Robin Harris J, et al. Structure, mechanism andregulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40.
- Shichita T, Hasegawa E, Kimura A, et al. Peroxiredoxin family proteins are keyinitiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917.
- Riddell JR, Wang XY, Minderman H, et al. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol. 2010;18:1022–1030.
- Jang HH, Lee KO, Chi YH, et al. Twoenzymes in one; two yeast peroxiredoxins display oxidative stress-dependentswitching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635.
- Cao J, Schulte J, Knight A, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKTactivity. EMBO J. 2009;28:1505–1517.
- Wang X, He S, Sun JM, et al. Selective association of peroxiredoxin 1 with genomic DNA and COX-2 upstream promoter elements in estrogen receptor negative breast cancer cells. MolBiol Cell. 2010;21:2987–2995.
- Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancertherapy. Expert Rev Mol Med. 2009;11:e36.
- Hoshino I, Matsubara H, Akutsu Y, et al. Tumor suppressor Prdx1 is a prognostic factor inesophageal squamous cell carcinoma patients. Oncol Rep. 2007;4:867–871.
- Kopp E, Medzhitov R, Carothers J, et al. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signaltransduction pathway. Genes Dev. 1999;13:2059–2071.
- Wi S M, Park J, Shim JH, et al. Ubiquitination of ECSIT is crucialfor the activation of p65/p50 NF-κBs in Toll-like receptor 4 signaling. MolBiol Cell. 2015;26:151–160.
- Wi SM, Moon G, Kim J, et al. TAK1-ECSIT-TRAF6complex plays a key role in the TLR4 signal to activate NF-κB. J BiolChem. 2014;289:35205–35214.
- Min Y, Wi SM, Shin D, et al. Peroxiredoxin-6 negatively regulates bactericidal activity and NF-κB activity by interrupting TRAF6-ECSIT complex. Front Cell Infect Microbiol. 2017;7:94.
- Lamothe B, Besse A, Campos AD, et al. Site-specificLys-63-linked tumor necrosis factor receptor-associated factor 6auto-ubiquitination is a critical determinant of I kappa B kinase activation. JBiolChem. 2007;282:4102–4112.
- Kishida S, Sanjo H, Akira S, et al. TAK1-bindingprotein 2 facilitates ubiquitination of TRAF6 and assembly of TRAF6 with IKK inthe IL-1 signaling pathway. Genes Cells. 2005;10:447–454.
- Yuk JM, Yoshimori T, Jo EK. Autophagy and bacterial infectious diseases. ExpMol Med. 2012;44:99–108.
- Yanagawa T, Ishikawa T, Ishii T, et al. Peroxiredoxin I expressionin human thyroid tumors. Cancer Lett. 1999;145:127–132.
- Yanagawa T, Iwasa S, Ishii T, et al. Peroxiredoxin I expressionin oral cancer: a potential new tumor marker. Cancer Lett. 2000;156:27–35.
- Quan C, Cha EJ, Lee HL, et al. Enhanced expression of peroxiredoxin I and VI correlates withdevelopment, recurrence and progression of human bladdercancer. J Urol. 2006;175:1512–1516.
- Odreman F, Vindigni M, Gonzales ML, et al. Proteomic studieson low- and high-grade human brain astrocytomas. J ProteomeRes. 2005;4:698–708.
- Park JH, Kim YS, Lee HL, et al. Expression of peroxiredoxinand thioredoxin in human lung cancer and paired normal lung. Respirology. 2006;11:269–275.
- Neumann CA, Krause DS, Carman CV, et al. Essential role forthe peroxiredoxin Prdx1 in erythrocyte antioxidant defence andtumour suppression. Nature. 2003;424:561–565.
- West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophagebactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480.
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin inendocytosis and signaling. Science. 2007;315:201–205.
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533.
- Egler RA, Fernandes E, Rothermund K, et al. Regulation of reactive oxygen species, DNA damage,and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050.
- Park YH, Kim SU, Lee BK, et al. Prx I suppresses K-ras-driven lungtumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxid RedoxSignal. 2013;19:482–496.
- Godfrey R, Arora D, Bauer R, et al. Cell transformation byFLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumorsuppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood. 2012;119:4499–4511.
- Mah LY, Ryan KM. Autophagy and cancer. Cold Spring HarbPerspectBiol. 2012;4:a008821.
- Botti J, Djavaheri-Mergny M, Pilatte Y, et al. Autophagy signaling and the cogwheelsof cancer. Autophagy. 2006;2:67–73.
- Hippert MM, O’Toole PS, Thorburn A. Autophagyin cancer: good, bad, or both? CancerRes. 2006;66:9349–9351.
- Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747.
- White E, DiPaola RS. The double-edgedsword of autophagy modulation in cancer. ClinCancer Res. 2009;15:5308–5316.
- Min Y, Wi SM, Kang JA, et al. Cereblon negatively regulates TLR4 signaling through the attenuation of ubiquitination of TRAF6. Cell Death Dis. 2016;7:e2313.
- Valle E, Guiney DG. Characterization of Salmonella-induced celldeath in human macrophage-like THP-1 cells. InfectImmun. 2005;73:2835–2840.