
ABSTRACT
Microvascular barrier dysfunction is the central pathophysiological feature of acute lung injury (ALI). RAB26 is a newly identified small GTPase involved in the regulation of endothelial cell (EC) permeability. However, the mechanism behind this protection has not been clearly elucidated. Here we found that RAB26 promoted the integrity of adherens junctions (AJs) in a macroautophagy/autophagy-dependent manner in ALI. RAB26 is frequently downregulated in mouse lungs after LPS treatment. Mice lacking Rab26 exhibited phosphorylated SRC expression and increased CDH5/VE-cadherin phosphorylation, leading to AJ destruction. rab26-null mice showed further aggravation of the effects of endotoxin insult on lung vascular permeability and water content. Depletion of RAB26 resulted in upregulation of phosphorylated SRC, enhancement of CDH5 phosphorylation, and aggravation of CDH5 internalization, thereby weakening AJ integrity and endothelial barrier function in human pulmonary microvascular endothelial cells (HPMECs). RAB26 overexpression caused active interaction between SRC and the autophagy marker LC3-II and promoted degradation of phosphorylated SRC. Furthermore, RAB26 was involved in a direct and activation-dependent manner in autophagy induction through interaction with ATG16L1 in its GTP-bound form. These findings demonstrate that RAB26 exerts a protective effect on endothelial cell (EC) permeability, which is in part dependent on autophagic targeting of active SRC, and the resultant CDH5 dephosphorylation maintains AJ stabilization. Thus, RAB26-mediated autophagic targeting of phosphorylated SRC can maintain barrier integrity when flux through the RAB26-SRC pathway is protected. These findings suggest that activation of RAB26-SRC signaling provides a new therapeutic opportunity to prevent vascular leakage in ALI.
Abbreviations: AJs: adherens junctions; ALI: acute lung injury; ARDS: acute respiratory distress syndrome; ATG5: autophagy related 5; ATG12: autophagy related 12; ATG 16L1: autophagy related 16 like; 1 BALF: bronchoalveolar lavage fluidCQ: chloroquine; Ctrl: control; EC: endothelial cell; GFP: green fluorescent protein; HA-tagged; RAB26WT: HA-tagged wild-type; RAB26 HA-tagged; RAB26QL: HA-tagged; RAB26Q123LHA-tagged; RAB26NI: HA-tagged; RAB26N177IHPMECs: human pulmonary microvascular endothelial cells; H&E: hematoxylin & eosin; IgG: immunoglobulin; GIF: immunofluorescence; IP: immunoprecipitationi;. p.: intraperitoneal; LPS: lipopolysaccharide; PBS: phosphate-buffered salinesi; RNA: small interfering;RNASQSTM1/p62, sequestosome; 1TBS: Tris-buffered saline; VEGF: vascular endothelial growth factor; WB: western blot; WT: wild-type
Introduction
Acute respiratory distress syndrome (ARDS) is a major cause of acute respiratory failure, and its primary pathogenic factors are dysregulation of inflammation and disruption of alveolar barriers [Citation1]. Microvascular barrier destabilization is a cardinal pathophysiological mechanism in ALI and ARDS, which is linked to fluid accumulation and leukocyte extravasation into the alveolar space [Citation2]. Although considerable progress has been made in the past few decades in understanding the mechanisms responsible for ALI, the morbidity and mortality from ARDS remains high [Citation3–Citation5]. Thus, it is necessary to further explore the underlying mechanism of ALI.
RAB26 belongs to the RAS-like family of small GTPases, which orchestrates vesicle-mediated protein transport [Citation6]. RAB26 was first discovered in the rat pancreas [Citation7,Citation8] and related to the regulation of AMY (amylase) release from parotid acinar cells [Citation9]. Previous studies found that RAB26 acted as a BHLHA15/MIST1 target and regulated lysosomal trafficking and autophagic function in the modulation of exocrine granule maturation [Citation10–Citation13]. In addition, RAB26 had been clearly shown to direct synaptic vesicles for degradation by coupling with phagophore assembly sites [Citation14]. Intriguingly, recent evidence had confirmed that Lc3 overexpression attenuates acute lung injury in septic mice [Citation15]. These studies revealed that RAB26 may play important roles in ALI. In addition, RAB26 interacts with ADRA2A (adrenoceptor alpha 2A) and coordinates transport of ADRA2A from the Golgi to the cell surface [Citation6]. During the inflammatory response, ADRA2A-mediated endothelial adherens junction stabilization prevents neutrophil extravasation [Citation16]. Based on these studies, we speculate that RAB26 may play a positive role in ALI. RAB26 may help counteract ALI.
CDH5 is a dominant factor in the stability of inter-EC AJs, which are critical for maintaining endothelial barrier integrity [Citation17,Citation18]. One important factor that determines the integrity of endothelial cell-to-cell AJs is the adhesive activity and dimer disassembly of CDH5 [Citation19]. It has become apparent that post-translational modification of CDH5 has considerable importance in the induction of endothelial permeability [Citation20]. Published reports demonstrate that several permeability-inducing agents, for example, VEGF, F2/thrombin, histamine and TNF (tumor necrosis factor), can induce tyrosine phosphorylation of CDH5 [Citation21–Citation23]. Phosphorylated CDH5 is endocytosed from AJs through clathrin-coated vesicles, resulting in CDH5 dimer disassembly, which increases endothelial permeability. The tyrosine kinase SRC is required for tyrosine phosphorylation of CDH5 [Citation24]. Recent studies reveal that the complex between active SRC and LC3 is associated with SRC-selective autophagy [Citation25].
In this study, we found that RAB26 can maintain EC integrity by regulating CDH5 phosphorylation, which is a result of the targeted autophagic degradation of SRC via direct and activation-dependent interaction with ATG16L1.
Results
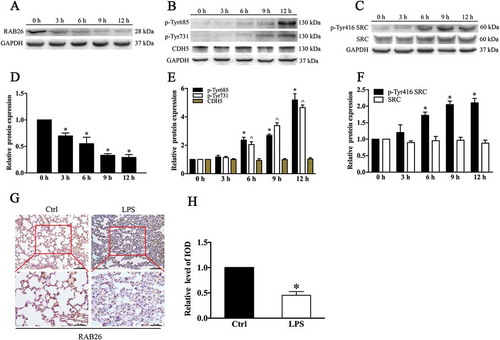
Sublethal endotoxin insult is frequently followed by RAB26 downregulation and CDH5 phosphorylation in mouse lungs
To investigate the RAB26 protein expression after endotoxin challenge in mice, we employed a lipopolysaccharide (LPS)-induced lung injury mouse model. Mice were challenged with intraperitoneal (i.p.) LPS for various time intervals (0, 3, 6, 9 and 12 h). Hematoxylin and eosin (HE) staining of lung sections at 6 h and 12 h after LPS injection clearly verified establishment of lung injury in the animal model (Fig. S1). Compared with the expression in control mice, RAB26 protein expression was decreased in the lungs of LPS-treated mice in a time-dependent manner (). Moreover, histological analyses showed that the RAB26 protein level was decreased in the lung tissue 6 h after LPS treatment ()). To detect the effect of endotoxin on AJs, we examined the phosphorylation of specific tyrosine residues in CDH5, the dominant factor in the stability of inter-EC AJs. Immunoblot analysis showed that LPS treatment increased the level of CDH5 phosphorylation at Y685 and Y731 and did not affect total CDH5 expression (). We also observed that the steady-state levels of phosphorylated SRC were elevated by LPS stimulation, although there were no significant changes in the total SRC expression levels (). These results were consistent with the previous finding that phosphorylated SRC in cells is the primary functional form of the protein and makes up a minor fraction of total SRC [Citation26]. The results validated the previous findings that LPS can increase CDH5 phosphorylation by increasing SRC activity. These data demonstrate that LPS exposure downregulated RAB26 expression, and that RAB26 levels were negatively correlated with CDH5 phosphorylation.
Figure 1. RAB26 is frequently downregulated in mouse lungs and accompanied by phosphorylation of CDH5 after sublethal endotoxin insult. C57BL/6 mice were challenged with LPS (15 mg/kg) for various periods of time (0, 3, 6, 9 and 12 h). (a-c) Dissected mice lungs were homogenized, and western blotting (WB) was performed to determine the expression of RAB26, SRC, p-Tyr416 SRC, and CDH5 and the phosphorylation of the indicated tyrosine residues in CDH5. GAPDH was used as the standard for verifying equivalent loading. (g) Representative immunohistochemical images indicating RAB26 protein expression in mouse lungs with or without LPS administration. The images were performed with 5 mouse lungs in each of the 2 groups. (h) Semiquantitative analysis of the integrated optical density (IOD) of RAB26 expression. * p < 0.01, compared with the control (n = 3) (d) (e) (f) Quantitative analysis of the indicated proteins. * p < 0.01 and ^p < 0.01 versus the 0 h group (n = 3).
Deletion of Rab26 in mice aggravates lps-induced ALI
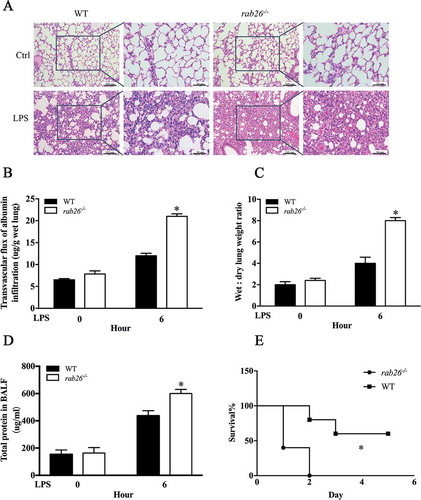
To explore the function of Rab26 in regulation of vascular EC permeability in vivo, we generated rab26 knockout mice. The first exon of Rab26 was selected as the TALEN target site (Fig. S2A). The mice were genotyped by sequence analysis to ensure that homozygous individuals were obtained (Fig. S2B). The rab26 knockout mice were verified by western blotting, which clearly demonstrated that RAB26 expression was eliminated in the lungs (Fig. S2C). In this experiment, CDH5 phosphorylation at Y685 and Y731 was obviously increased in the rab26−/- mice lung microvascular ECs, consistent with the increased abundance of phosphorylated SRC protein (). Immunohistochemical staining also showed that CDH5 phosphorylation at Y685 and Y731 was increased in rab26−/- mice, compared with wild-type mice (). Moreover, histological examination of lungs (HE staining) demonstrated more obvious congestion, alveolar collapse and thickening of alveolar walls in lung tissue in the rab26−/- mice than in wild-type mice in response to LPS stimulation, whereas rab26−/- mice did not exhibit significant pathological changes under physiological conditions ()). We also observed a dramatic increase in bronchoalveolar lavage fluid (BALF) protein content, lung edema and pulmonary microvascular permeability in the lungs of rab26–/ – mice compared with those of controls (), which is in accordance with the severe lung injury in the model. We next gave mice a sublethal dose of LPS, and, as expected, rab26−/- mice exhibited considerably higher mortality than wild-type controls ()). Together, these results show that Rab26 deletion might be a prerequisite for the full opening of EC junctions and the induction of lung injury in the presence of inflammatory agents, but may not increase vascular permeability under physiological conditions.
Figure 2. Deletion of Rab26 in mice enhances CDH5 phosphorylation. (a) Dissected lungs from mice; WB was performed to detect the expression of the indicated proteins. GAPDH was used as the standard for verifying equivalent loading. (b) Quantitative data for expression of the indicated protein expression normalized to GAPDH expression. * p < 0.01, ^ p < 0.01, & p < 0.01 versus the indicated wild-type mice (n = 3). (c) Representative immunohistochemical images demonstrating the expression of p-Tyr685 and p-Tyr731 CDH5 in rab26−/- and wild-type mice. (d) Semiquantitative analysis of p-Tyr685 and p-Tyr731 CDH5 expression as measured by the integrated optical density (IOD). * p < 0.01, ^ p < 0.01compared with the wild-type mice (n = 5).
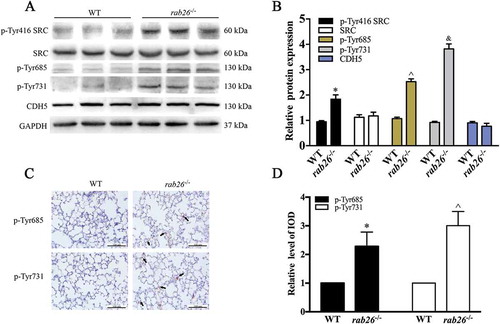
Figure 3. Deletion of Rab26 in mice aggravates the LPS-induced ALI. (a) Lung tissue samples were dissected and subjected to H&E staining. (b) rab26−/- and wild-type mice were challenged with PBS or 15 mg/kg LPS. Pulmonary transvascular permeability to EBA was measured; n = 5/group. * p < 0.01 versus the 6 h group. (c, d) rab26−/- and wild-type mice were challenged with PBS or 15 mg/kg LPS. Lung edema and BALF protein content were measured; n = 5/group. * p < 0.01 versus the 6 h group. (e) rab26−/- and wild-type mice were challenged with 40 mg/kg LPS i.p., and survival was monitored; n = 8/group. Differences in mortality were analyzed by the log-rank test. * p < 0.01 by the log-rank test.
Downregulation of Rab26 can induce CDH5 internalization and disrupt AJ integrity
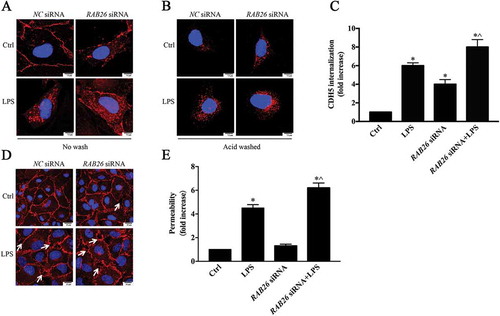
To explore the mechanism of RAB26 in the regulation of vascular endothelial permeability, we examined its direct impact on AJs in ECs. Immunofluorescence studies were conducted to visualize CDH5 internalization. The ECs were treated with chloroquine (CQ), after which an acid wash was or was not performed to reveal the internalized CDH5. The results showed that RAB26 depletion obviously increased CDH5 internalization, and CDH5 endocytosis was exacerbated under LPS stimulation (). Because CDH5 internalization was clearly correlated with the disruption of AJ integrity, we further assessed the morphological changes of CDH5. Visualization of CDH5 in AJs via immunostaining revealed that siRNA-mediated knockdown of RAB26 also enhanced the formation of intercellular gap areas, which could be further aggravated by LPS stimulation ()). Transient expression of RAB26 siRNA also significantly damaged the EC barrier function ()). These results demonstrate that knockdown of RAB26 resulted in concomitant degradation of EC barrier function and that RAB26 can promote EC integrity through the regulation of CDH5 internalization.
Figure 4. Downregulation of RAB26 induced internalization of CDH5 and disrupted EC barrier integrity. HPMECs were transfected with RAB26 siRNA and incubated with LPS for an additional 6 h. (a, b) HPMECs were transfected with RAB26 siRNA, exposed to 1 µg/ml LPS for 6 h, and labeled with anti-CDH5 antibody and treated with 100 μM chloroquine for 6 h at 37°C. Then the cells were washed or not washed with mild acid buffer to visualize the CDH5 internalization. Scale bars: 7.5 μm. The images show 3 independent experiments. (c) Quantification of CDH5 internalization. * p < 0.01 versus the control group, ^ p < 0.01 versus the LPS group (n = 3). (d) AJ integrity of HPMECs was determined by CDH5 immunofluorescence staining. Arrows demonstrate intercellular gap formation. Scale bars: 25 μm. (e) The effect of RAB26 siRNA on the monolayer permeability of HPMECs. * p < 0.01 versus the control group, ^ p < 0.01 versus the LPS group (n = 3).
Knockdown of RAB26 enhances CDH5 phosphorylation in ECs
To investigate the association between RAB26 expression and CDH5 phosphorylation, we incubated HPMECs in the presence of LPS (1 µg/mL) at several time points. Using western blot analysis, we observed that the RAB26 level was significantly inhibited (). As shown by immunofluorescence staining, RAB26 was largely localized in the perinuclear region, and RAB26 expression was decreased at 6 h after LPS treatment ()), which is consistent with the western blotting results. We then examined the abundance of phosphorylated CDH5 protein. The results indicated that CDH5 phosphorylation at Y685 and Y731 were significantly higher in the LPS-treated cells than in the control cells at all time points (). We transfected the cells with RAB26 siRNA to explore the influence of RAB26 on CDH5 phosphorylation (Fig. S3A). The siRNA-induced depletion of RAB26 obviously increased CDH5 phosphorylation at Y685 and Y731 in response to LPS treatment but had no influence on total CDH5 expression (). In addition, we used a co-immunoprecipitation assay to further determine the phosphorylation of CDH5. The amount of phosphotyrosine in the immunoprecipitates, as measured by immunoblotting, was significantly higher in the RAB26 depletion cells than the controls (-h)). These results clearly indicated that RAB26 can promote EC integrity through inhibition of CDH5 phosphorylation.
Figure 5. Knockdown of RAB26 increases the CDH5 phosphorylation in ECs. (a, b) HPMECs were treated with LPS (1 μg/mL) for various periods of time (0, 3, 6, 9 and 12 h). Lysates from cells were incubated with antibodies targeting RAB26, and phosphorylation of the indicated tyrosine of CDH5. (c) Western blot results indicating that CDH5 phosphorylation at Y685 and Y731 was increased in the RAB26-depleted HPMECs. GAPDH was used as the standard for verifying equivalent loading. (g and h) HPMECs were transfected with RAB26 siRNA, and lysates from cells were immunoprecipitated (IP) with CDH5 antibodies and then immunoblotted with anti-phosphotyrosine antibodies. (i) Immunofluorescence staining showing the RAB26 protein expression profile in LPS-treated HPMECs at 0 h and 6 h. Nuclei are indicated with DAPI. Scale bars: 25 μm. (d and e) Quantitative data for the indicated proteins were normalized to GAPDH expression. * p < 0.01 and ^ p < 0.01versus the indicated 0 h group (n = 3). (f) Quantitative data of the indicated proteins were normalized to GAPDH expression. * p < 0.01 versus the control group and ^ p < 0.01versus the LPS group (n = 3).
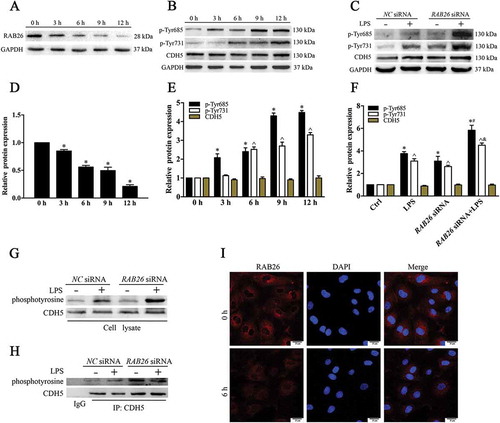
RAB26 can promote autophagy-mediated degradation of phosphorylated SRC
Because SRC kinase-dependent CDH5 phosphorylation is an important regulator of the remodeling and plasticity of AJs, we examined the expression of SRC protein. LPS exposure increased the level of phosphorylated SRC, and there was no significant difference in total SRC expression ()). In addition, because SRC plays a vital role in CDH5 phosphorylation, we measured changes in SRC expression in response to the RAB26 siRNA ()). The data showed that RAB26 siRNA partially increased the expression of phosphorylated SRC but had no effect on the total SRC level. Furthermore, phosphorylated SRC was also downregulated in cells transfected with wild-type RAB26 ()), whereas the expression of unphosphorylated SRC expression was not affected (Fig. S4). These data demonstrated that increased phosphorylated SRC expression in ECs was secondary to compromised RAB26 availability.
Figure 6. RAB26 promoted the autophagic degradation of phosphorylated SRC. (a) HPMECs were treated with LPS (1 µg/mL) for various periods of time (0, 3, 6, 9 and 12 h). Lysates from cells were incubated with anti-SRC and anti-p-Tyr416 SRC. (b) Western blot results showing increased phosphorylated SRC expression in the RAB26-knockdown HPMECs. GAPDH was used as the standard for verifying equivalent loading. (c and d) HPMECs were transfected with the pEGFP-C1 vector (Ctrl) or HA-tagged wild-type RAB26 (RAB26WT); lysates from cells were immunoprecipitated (IP) with LC3 antibodies and then immunoblotted with anti-p-Tyr416 SRC antibody. (e) The effect of phosphorylated SRC dominant-negative mutants pcSRC416 and SRC siRNA on the monolayer permeability of RAB26-depleted HPMECs. * p < 0.01 versus the control group and ^ p < 0.01versus the LPS group (n = 3). (f) HPMECs were transfected with the HA-tagged wild-type RAB26 (RAB26WT). Immunofluorescence staining showing the location of phosphorylated SRC and LC3B. Nuclei are indicated with DAPI. Scale bars: 25 μm. (g) The effect of wild-type RAB26 on monolayer permeability. HPMECs were transfected with the HA-tagged wild-type RAB26 (RAB26WT) and then treated with LPS. * p < 0.01 versus the control group and ^ p < 0.01versus the LPS group (n = 3). (h) Western blot results showing decreased CBL expression in the CBL siRNA-transfected HPMECs. (i) The pEGFP-C1 vector (Ctrl) or HA-tagged wild-type RAB26 (RAB26WT) was co-transfected with CBL siRNA in HPMECs. Western blot results showing the expression of the phosphorylated SRC.
Previous reports show that autophagy can promote degradation of phosphorylated SRC [Citation25]. Because LC3 participates in mediating autophagy substrate degradation, we next examined whether phosphorylated SRC interacts with LC3. We observed a pronounced colocalization of phosphorylated SRC and LC3 in cells transfected with wild-type RAB26 ()). In addition, the data showed reduced expression of phosphorylated SRC in ECs that overexpressed RAB26 ()), and the expression of phosphorylated SRC in the immunoprecipitates was increased in cells transfected with wild-type RAB26 ()), indicating that a regulatory complex existed between phosphorylated SRC and LC3 during the autophagic process. To investigate whether tyrosine phosphorylation of CDH5 was secondary to the high expression of phosphorylated SRC in EC integrity, we transfected RAB26-depletion lung microvascular ECs with phosphorylated SRC dominant-negative mutant pcSRC416 and SRC siRNA. We found that disruption of SRC function or expression partially rescued EC hyperpermeability ()). In addition, RAB26 overexpression attenuated cell permeability under LPS treatment ()). We next knocked down C-CBL expression in RAB26-overexpressing ECs (-i)), and found that CBL siRNA promoted the restoration of phosphorylated SRC, implying that CBL may mediate the degradation of phosphorylated SRC in ECs. These findings indicated that RAB26 participates in regulating EC permeability via modulation of CDH5 phosphorylation by targeting phosphorylated SRC for autophagic degradation.
Effect of autophagy on EC barrier intergrity
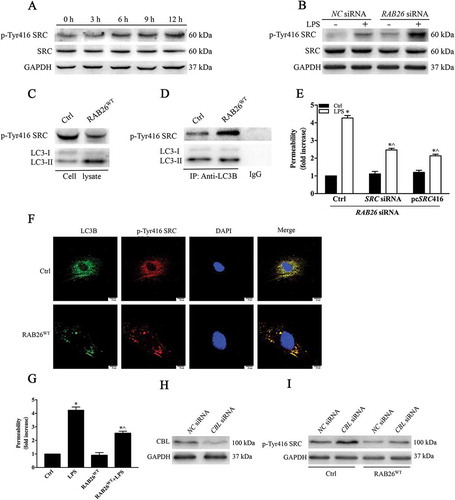
Next, we addressed whether autophagy is involved in LPS-induced EC hyperpermeability. LPS exposure reduced the LC3-II protein levels in a time-dependent manner (). The receptor protein SQSTM1/p62 is required for the degradation of ubiquitinated substrates [Citation27]. The results showed that LPS treatment increased the level of SQSTM1 (), confirming that LPS attenuates autophagy activity. Autophagic flux refers to the dynamic process of autophagy. To measure autophagic flux, we examined the levels of LC3-II and GFP-LC3-positive autophagosomes in the absence or presence of chloroquine (CQ), a specific lysosome inhibitor that prevents autophagy by blocking the fusion of autophagosomes with lysosomes. We found that the CQ challenge elevated the protein expression of LC3-II and the formation of GFP-LC3-positive autophagosomes in ECs (), (c) and (e)), indicating that LPS stimulation actually reduced the baseline levels of autophagy instead of increasing degradation capacity in HPMECs.
Figure 7. Autophagy alleviates LPS-induced destruction of EC barrier integrity. (a) HPMECs were treated with LPS (1 µg/mL) for different periods of time (0, 3, 6, 9 and 12 h). Lysates from cells were incubated with anti-LC3 and anti-SQSTM1 antibodies. (b) Western blot results showing the levels of LC3 after LPS treatment with or without CQ (20 μM). (c) Immunofluorescence showing the formation of GFP-LC3-positive green puncta. Scale bars: 7.5 μm. (d) Quantitative data for the indicated proteins, normalized to GAPDH expression. * p < 0.01 and ^ p < 0.01compared with the indicated 0 h group (n = 3). (e) Quantitative data for the indicated proteins, normalized to GAPDH expression. * P < 0.01, compared with the control group (n = 3) and ^ p < 0.01versus the LPS group (n = 3). (f, g) Western blot analysis confirming the siRNA-mediated knockdown of ATG5 and ATG7. The LC3-II and SQSTM1 expression indicated that depletion of ATG5 and ATG7 markedly inhibited the autophagy process. (h) The monolayer permeability of HPMECs were examined after ATG5 siRNA, ATG7 siRNA, and CQ treatment, followed by LPS stimulation. * p < 0.01, compared with the control group (n = 3) and ^ p < 0.01versus the LPS group (n = 3). (i) The HPMECs were treated with ATG5 siRNA or ATG7 siRNA. The AJ integrity of the HPMECs was determined by CDH5 immunofluorescence staining. Scale bars: 25 μm. Arrows indicate intercellular gap formation.
To further ascertain the impact of autophagy on the LPS-induced deterioration of HPMEC barrier function, we employed the autophagy inhibitor CQ and siRNA-mediated knockdown of the ATG5 gene. Transfection with ATG5 siRNA or ATG7 siRNA markedly decreased the LC3 level and increased SQSTM1 expression (). These results clearly indicated that the autophagy level was inhibited. We then examined the localization of CDH5. CDH5 immunostaining revealed that autophagy inhibition aggravated the formation of intercellular gaps after LPS stimulation. Knockdown of ATG5 or ATG7 or treatment with CQ ultimately enhanced both basal and LPS-induced AJ disassembly (indicated with arrows, )). We then determined the effect of autophagy on EC barrier function. We found that the transendothelial permeability of HPMECs to the tracer FITC-dextran was increased following autophagy inhibition ()). Taken together, these data demonstrate that autophagy activity is required for the maintenance of EC barrier integrity.
RAB26-induced autophagosomes depend on its GTP-bound form
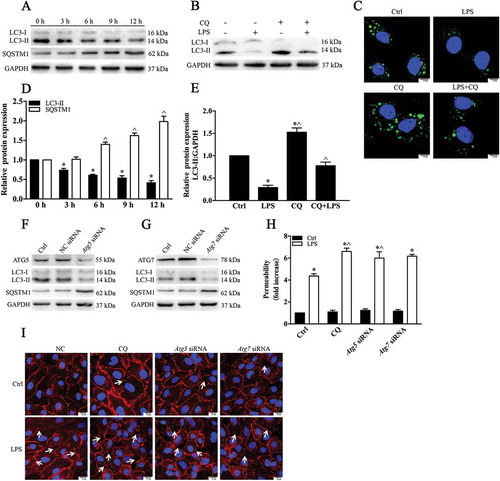
To elucidate the effects of RAB26 and its mutants in the autophagic process, we examined the formation of puncta in ECs. HA-tagged RAB26, its constitutively active mutant RAB26Q123L (RAB26QL), or its dominant-negative mutant RAB26N177I (RAB26NI), was expressed together with GFP-LC3. Co-transfection of cultured ECs with a plasmid expressing HA-RAB26WT and GFP-LC3 resulted in significantly increased autophagosome formation (). Treatment with CQ actually enhanced the amounts of green puncta, demonstrating that RAB26 overexpression induced autophagic flux. The effect of RAB GTPases is mediated through their GTP-GDP exchange cycles. Overexpression of the GTP-bound form, HA-RAB26QL, increased autophagosome formation, whereas the dominant-negative mutant HA-RAB26NI had nearly no effect on the formation of green puncta (). In this instance, the results revealed that RAB26 promotes the autophagic process and that the correlation between RAB26 and autophagic activity depends mainly on the GTP-bound form.
Figure 8. RAB26 promotion of autophagy in ECs depends on the GTP-bound form (a, b) After co-transfection of GFP-LC3-expressing cells with the plasmid expressing HA-tagged wild-type RAB26 (RAB26WT), or its mutants, HA-tagged RAB26Q123L (RAB26QL) and HA-tagged RAB26N177I (RAB26NI), HPMECs were incubated with or without CQ (20 μM) for 12 h. The formation of GFP-LC3 autophagosomes was detected using confocal microscopy, and at least 50 cells were counted in each experiment. *p < 0.01, compared with the control group. ^p < 0.01, compared with the RAB26WT group. &p < 0.01 compared with the RAB26QL group. #p < 0.01, compared with the RAB26NI group. (c) HPMECs were transfected with RAB26 siRNA and incubated with LPS for an additional 6 h. The levels of LC3 and SQSTM1 were detected by western blot. (d) Immunofluorescence images demonstrating the formation of autophagosomes. After co-transfection with RAB26 siRNA and a plasmid encoding GFP-LC3, HPMECs were incubated with or without LPS for 6 h. Scale bars: 7.5 μm. (e) Quantitative data of the indicated proteins normalized to GAPDH expression. * p < 0.01, compared with the indicated group. ^ p < 0.01versus the LPS group (n = 3). (f) The formation of GFP-LC3 autophagosomes was detected using confocal microscopy, and at least 50 cells were counted in each experiment. *p < 0.01, compared with the control group. ^p < 0.01, compared with the LPS group.
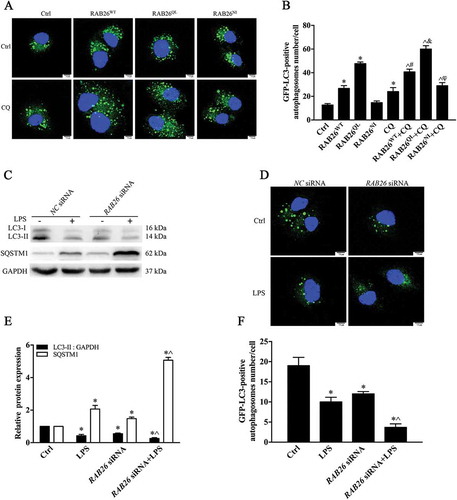
We then determined the effect of siRNA-mediated knockdown of RAB26 on autophagy. We found that siRNA-mediated RAB26 depletion clearly inhibited LC3-II expression. Meanwhile, the SQSTM1 protein level was increased (). These experiments demonstrated that the proper function of RAB26 is necessary for the autophagic process. To further confirm these results, we observed autophagosome formation directly using a GFP-LC3 plasmid to monitor the formation of puncta. A plasmid encoding GFP-LC3 was co-transfected with RAB26 siRNA in ECs, and the cells were continuously incubated with LPS. The results showed that the formation of green puncta decreased significantly in the RAB26 siRNA group () and was aggravated by LPS stimulation, further indicating that RAB26 depletion inhibited autophagy activity.
RAB26 directly interacts with ATG16L1 to promote autophagy
To identify the intracellular component by which RAB26 regulates the autophagy pathway, we next utilized an antibody against the autophagy protein ATG16L1. The autophagy pathway is initiated by conjugation of the ubiquitin-like protein ATG12 to ATG5, followed by the recruitment of ATG16L1. As displayed via confocal microscopy, endogenous RAB26 was localized chiefly within the perinuclear zone. The subcellular localization of GFP-fused RAB26, including both wild-type RAB26 and the GTP-bound form RAB26QL, was very similar to that of endogenous RAB26, whereas the dominant-negative mutant RAB26NI was predominantly diffusely distributed in the cytoplasm ()). Upon staining for endogenous ATG16L1, we observed some strong colocalization with endogenous RAB26, GFP-RAB26WT, and active mutant GFP-RAB26QL in HPMECs ()) but not with GFP-RAB26NI. These results indicate that the relevance of RAB26 in the autophagy process depends mainly on the GTP-bound form.
Figure 9. RAB26 interacts with ATG16L1 via its GTP-bound form. (a) HPMECs were transfected with the plasmids expressing GFP-tagged wild-type RAB26 (RAB26WT), or its mutants, GFP-tagged RAB26Q123L (RAB26QL) and GFP-tagged RAB26N177I (RAB26NI), and co-stained with anti-ATG16L1 (red). Scale bars, 7.5 µm. (b and c) HPMECs were transfected with the HA-tagged wild-type RAB26 (RAB26WT) or its mutants HA-tagged RAB26Q123L (RAB26QL) and HA-tagged RAB26N177I (RAB26NI), and lysates from cells were immunoprecipitated (IP) with HA antibodies and then immunoblotted with anti-ATG16L1 antibodies. endo, endogenous.
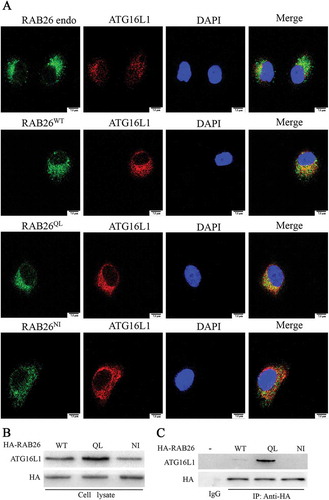
The colocalization of RAB26 and ATG16L1 prompted us to investigate whether those 2 proteins were directly associated in a biochemical regulatory complex. To explore this possibility, we performed a co-immunoprecipitation experiment. HPMECs were transfected with all 3 HA-tagged RAB26 variants and were immunoprecipitated with anti-HA antibody. The results showed that immunoprecipitated ATG16L1 was highly abundant in ECs expressing HA-RAB26QL, whereas little ATG16L1 was detected in ECs expressing HA-RAB26WT or HA-RAB26NI (). These findings provided evidence that RAB26 and ATG16L1 were present in a biochemical complex, and that the interaction preferentially depends on the GTP-bound form.
Discussion
Here, we have demonstrated the important role of the small GTPase RAB26 in strengthening the stability of endothelial junctions. These results were in agreement with our previous findings that RAB26 siRNA-loaded DNA nanovectors destroy EC barrier function [Citation28]. Together, these data provide evidence that RAB26 activates autophagy via ATG16L1 and causes the auophagic degradation of SRC. This degradation of phosphorylated SRC limits the ability of this kinase to phosphorylate CDH5, thereby promoting adherens junction integrity. Endotoxin markedly compromises RAB26 activity and promotes active SRC-mediated CDH5 phosphorylation and internalization, which is a constitutive mechanism that reduces AJ formation and exacerbates EC permeability. This study is the first time that RAB26-induced autophagy has been confirmed to contribute to EC barrier function. The results provide evidence for us to clarify the underlying signaling pathway by which RAB26 acts as a regulator of EC barrier function.
Autophagy plays different roles in ALI: both protective and deleterious aspects have been detected in different lung injury models in response to various stimuli, indicating that the role of autophagy in ALI is complex. Mice lacking Atg5 in macrophage cells produce Hc/complement C5a, which aggravates ALI resulting from intestinal ischemia-reperfusion through autophagy-induced alveolar macrophage apoptosis [Citation29]. In the epithelium, autophagy appears to be beneficial in LPS-induced ALI. Mice lacking Mtor in bronchial or alveolar epithelial cells show attenuated airway inflammation and barrier disruption [Citation30]. In addition, after bleomycin treatment, Atg4b-deficient mice display significantly heightened inflammatory responses and bronchial epithelial cell apoptosis [Citation31]. However, the role of autophagy in lung ECs remains complex. Domigan et al. observed depletion of VEGF in the endothelium, leading to increased autophagy that results in cell death [Citation32]. Another study showed that autophagy activation plays a special role in the pathogenesis of hepatopulmonary syndrome and that inhibition of autophagy ameliorates pulmonary microvascular dilation [Citation33]. Other studies in ischemia-reperfusion-induced lung injury models have revealed that autophagy has a vital role in protecting endothelial function mediated by ITGAV-ITGB5/integrin αvβ5 [Citation34], and that the ability of mesenchymal stem cells to alleviate the severity of LPS-induced lung injury is dependent on autophagy via a phosphatidylinositol 3-kinase-dependent signaling pathway [Citation35,Citation36]. Similar to these latter studies, our results demonstrate that inhibition of autophagy via inhibitor or siRNA-mediated depletion of ATG5 or ATG7 can aggravate the destruction of EC integrity, suggesting that autophagy plays a positive role in the regulation of EC function under endotoxin insult. The underlying mechanisms by which autophagy exerts both protective and injurious effects in endothelial injury remain unclear, and further investigation of the double-edged sword of autophagy in pulmonary diseases is needed.
RAB proteins belong to the RAS GTPase superfamily and are key regulators in the vesicle trafficking process. The function of RAB GTPases is regulated in exchange cycles between GTP-bound active forms and GDP-bound inactive forms [Citation37–Citation40]. Numerous studies have revealed that RAB proteins participate in regulating different stages of the autophagy process. RAB1, RAB4, RAB5, RAB7, RAB9, RAB11, RAB23 and RAB32 can contribute to autophagosome formation, and RAB7, RAB8, RAB9, RAB24 and RAB33 promote autophagosome maturation [Citation41–Citation44]. Consistent with previous studies [Citation14], we found that interaction between ATG16L1 and RAB26 might be involved in the autophagic process and mainly involves the GTP-bound active form. The main reason for this finding is that ATG16L1 regulates not only autophagy but also vesicle secretion [Citation41,Citation42]. By contrast, other RAB GTPases in the same functional group, including RAB3A, RAB3B, RAB3C, RAB3D, RAB27A, RAB27B, and RAB37 [Citation6], do not exhibit a similar role in autophagy. Autophagy was obviously unregulated in secretory-deficient RAB3−/- β-cells [Citation45,Citation46]. The siRNA-mediated knockdown of RAB27A blocks autophagy induced by hepatitis C virus [Citation47]. In addition, RAB37 is relatively less well investigated. Our results demonstrated that RAB26 did not perform similar autophagy functions to other proteins in their functional group. Our data are also the first to reveal that RAB26 may play a significant role in the regulation of autophagy in ECs in addition to its secretory function and may have a special role in the regulation of EC function.
Cell surface CDH5 phosphorylation is directly linked to EC barrier integrity. The dynamic balance action between the SRC kinase-mediated CDH5 phosphorylation and PTPRB-dependent CDH5 dephosphorylation controls the integrity of blood vessels [Citation48–Citation50]. Phosphorylation of CDH5 at tyrosine residues induces opening of endothelial AJs during inflammation [Citation51]. Post-translational modifications of CDH5 at tyrosine residues are considered to participate in vascular permeability and leukocyte transmigration. Phosphorylation of Tyr731 is necessary for leukocyte transmigration mediated by ICAM1, and phosphorylation of Tyr685 can control vascular permeability [Citation52–Citation55].
The ubiquitin-proteasome pathway and the autophagy-lysosome pathway are the major mechanisms of SRC protein degradation [Citation56–Citation58]. Previous studies demonstrated that the E3 ubiquitin ligase CBL targets phosphorylated SRC for autophagy and converts proteasome-dependent degradation of the protein to autophagic-lysosomal-mediated turnover [Citation26]. Thus, it appears that ubiquitin for selective autophagy is also involved in phosphorylated SRC degradation. Our research indicates that CDH5 phosphorylation in response to LPS stimulation is time-dependent. CDH5 phosphorylation is increased in RAB26-deficient cells, depending on phosphorylated SRC expression, whereas our results showed that CDH5 phosphorylation did not lead to destruction of barrier function in rab26−/- mice under normal conditions. These results are in line with previous studies [Citation54,Citation59], and one possible explanation is that phosphorylation of CDH5 is necessary but not sufficient for the opening of EC junctions to permeability-inducing agents. The fact that the phosphorylated SRC dominant-negative mutant or SRC siRNA was not sufficient to rescue vascular barrier function in RAB26-depleted ECs indicates that other RAB26 targets, such as ADRA2A [Citation6] or some types of exocrine granules [Citation9,Citation13], might also participate in the protective effects of RAB26.
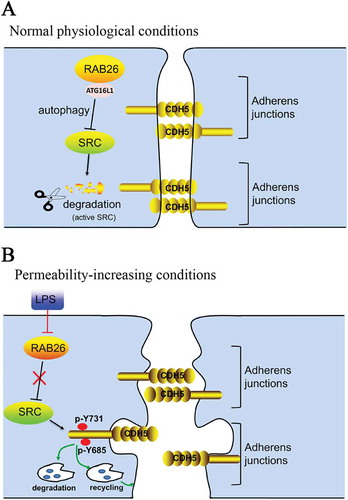
In conclusion, our current study suggests that inhibition of RAB26 expression and subsequent activation of CDH5 phosphorylation in the pulmonary endothelium are essential for LPS-induced endothelial hyperpermeability in vitro and ALI pathogenesis in vivo. RAB26 targets phosphorylated SRC for autophagy-dependent degradation, forming the critical mechanism in mediating CDH5 phosphorylation. Taken together, these findings reveal new perspectives on the correlation between RAB26 and autophagy signaling, which may provide novel targets for preventing ALI secondary to bacterial infection ().
Figure 10. The signaling pathway regulating RAB26-induced SRC degradation (a) Under normal physiological conditions, RAB26 interacts with ATG16L1, resulting in the induction of autophagy, which targets phosphorylated SRC for degradation. Basal levels of CDH5 are expressed in HPMECs to retain the endothelial barrier. (b) Under endotoxin stimulation, the inhibition of RAB26 activity results in autophagy inhibition, and phosphorylated SRC accumulates in the cytoplasm. The interaction between phosphorylated SRC and CDH5 phosphorylated at p-Tyr685 and p-Tyr731 promotes CDH5 internalization, thus reducing AJ formation and endothelial barrier integrity.
Material and methods
Reagents and antibodies
Reagents were as follows: ATG5 siRNA (Santa Cruz Biotechnology, sc-41,445), lipopolysaccharides from Escherichia coli O111:B4 (Sigma, L4391), lipopolysaccharides from Escherichia coli 055:B5 (Sigma, L2880), ViaFectTMTransfection Reagent (Promega Corporation, E4982), X-tremeGENE siRNA Transfection Reagent (Roche, 04476093001), Protein A/G PLUS-Agarose Immunoprecipitation Reagent (Santa Cruz Biotechnology, sc-2003), FITC-dextran (Santa Cruz Biotechnology, sc-263,323), chloroquine (Sigma, C6628), Tris-buffered saline (TBS; Beijing Zhongshan Jinqiao Biological Technology, ZLI-9073), Tween 20 (Amresco, 9005–64-5), phosphate-buffered saline (PBS; HyClone, SH30256.01), M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, 78,501), Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, 23,225), T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific, 78,510).
Antibodies were as follows: Non-phospho-SRC (Tyr416; Cell Signaling Technology, 2102), phospho-SRC family (Tyr416; Cell Signaling Technology, 6943), SRC (Cell Signaling Technology, 8077), phospho-tyrosine (p-Tyr-100; Cell Signaling Technology, 9411), ATG16L1 (Cell Signaling Technology, D6D5) for western blot (WB), ATG16L1 (Santa Cruz Biotechnology, sc-393,274) for immunofluorescence (IF), SQSTM1/p62 (Cell Signaling Technology, 5114), HA (Santa Cruz Biotechnology, sc-7392), LC3B (Cell Signaling Technology, 3868) for WB, LC3B (Sigma, SAB4200361) for IF and immunoprecipitation (IP), ATG7 (Abcam, ab133528), RAB26 (Abcam, ab126996) for WB, RAB26 (abcam, ab187151) for IF, GAPDH (Beijing Biosynthesis Biotechnology, bs10900R), CDH5/VE-cadherin (phosphoTyr731; Sigma, SAB4504676), CDH5/VE-cadherin (phosphoTyr685; Abcam, ab119785), CDH5/VE-cadherin (Cell Signaling Technology, 2500), CDH5/VE-cadherin (Santa Cruz Biotechnology, sc-9989), CDH5/VE-cadherin (BV9; Santa Cruz Biotechnology, sc-52,751), ATG5 (Beijing Biosynthesis Biotechnology, bs-4005R), CBL/C-CBL (Proteintech Group, 66,576–1-Ig), Alexa Flour 488-conjugated secondary antibody (Beijing Zhongshan Jinqiao Biological Technology,ZF-0512), Alexa Flour 594-conjugated secondary antibody (Beijing Zhongshan Jinqiao Biological Technology,ZF-0516), Alexa Fluor 647-conjugated secondary antibody (Beyotime Biotechnology, A0468).
Cell culture and transfection with small interfering RNA (siRNA)
HPMECs were obtained from ScienCell Research Laboratories (Sciencell, 3000) and cultured according to the instructions. HPMECs were transfected with RNAi using X-tremeGENE. The RAB26 siRNA sequence was UAGUAGGCAUGGGUAACACTT, the siRNA sequence target to SRC was TGTTCGGAGGCTTCAACTCCT, the siRNA sequence target to CBL was GCCUGAUUGGGCUCAUGAATT, the siRNA sequence target to ATG7 was GGAUCCUGGACUCUCUAAATT, the ATG5 siRNA was purchased from Santa Cruz Biotechnology and the negative control was UUCUCCGAACGUGUCACGUTT.
Plasmid constructs and transfection
pcSRC416 was purchased from Addgene (17,676; deposited by David Shalloway) [Citation60], RAB26WT, RAB26QL, RAB26NI and pEGFP-C1 were gifts from Guangyu Wu (Department of Pharmacology and Toxicology, Georgia Regents University). GFP-LC3 plasmid was purchased from Genomeditech (GM-1314P101H). HPMECs were transfected with the plasmids using ViaFectTM Transfection reagent according to the manufacturer’s instructions. Briefly, ViaFectTM Transfection reagent (1.5 μL) and the plasmid (0.5 μg) were diluted in 50 μL of OPTI-MEM (Life Technologies Corporation, 31,985–070) without antibiotics or fungicides. The mixture was incubated for 10–15 min. Then, the transfection mixture was added to the culture dishes. The experiments were performed after 60 h.
Co-immunoprecipitation measurement
The co-immunoprecipitation assay was performed as previously reported [Citation61]. After stimulation, cells were washed with ice-cold PBS and lysed in M-PER reagent. The protein concentrations were detecting using the BCA protein assay kit. Cell lysates from HPMECs were incubated with either an anti-p-SRC (TYR416) antibody, an anti-CDH5 antibody, or an equal amount of IgG and then further incubated with protein A- and protein G-conjugated Sepharose beads (Santa Cruz Biotechnology, sc-2003), and proteins were detected by western blot (WB) analysis.
Generation of RAB26-null mice
The MRab26 gene (GenBank accession number: NM_177375.1; Ensembl: ENSMUSG00000079657) is located on mouse chromosome 17. Nine exons have been identified, with the ATG start codon in exon 1 and TGA stop codon in exon 9. Exon 1 was selected as the TALEN target site. TALENs were constructed using the Golden Gate Assembly method and confirmed by sequencing. TALEN mRNA was generated by in vitro transcription and injected into fertilized eggs for knockout mouse production; Rab26 heterozygous mice were provided by CyagenCompany of China. Tail DNA was isolated using a Mouse Direct PCR Kit (Biomake, B40013). The following PCR primers were used: (F) CCAGGAAGAAGACCCCCAAGAGC and (R) AAGGTGCTGTGCCAATTCAGCG. The mice were genotyped via sequence analysis to ensure that homozygous mice were obtained.
Immunofluorescence and immunohistochemistry
HPMVECs were grown on coverslips in a 24-well plate, fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 (Sigma, T9284). Slides were stained with primary antibodies and appropriate fluorescence-conjugated secondary antibodies. The cells were viewed with a Leica confocal microscope (Leica microsystems, Germany) and analyzed using ImageJ software (NIH, USA) or Leica LAS AF 2.3.0 software (Leica microsystems, Germany). Lung tissues were fixed and then subjected to immunohistochemical staining with the appropriate antibodies. Images were obtained with the Leica microscope (Leica microsystems, Germany). The positively-stained areas were quantified using the Image Pro Plus software.
CDH5 endocytosis assay
To perform the internalized CDH5 assay, ECs were labeled with an anti-CDH5 extracellular domain-targeting antibody at 4°C for 1 h as previously reported [Citation62]. Then, cells were incubated at 37°C for 6 h in the presence of 100 μM chloroquine and washed in mild acid buffer (in PBS, pH 2.7, containing 25 mM glycine, 3% BSA [Amresco, 0332]) for 15 min to visualize the internalized CDH5. After that, the cells were processed for immunofluorescence staining. The number of internalized CDH5 vesicles was determined by counting CDH5 vesicles/cell.
Monitoring endothelial cell permeability
HPMECs were cultured on 0.4-μm transwell inserts. After stimulation, the upper wells were incubated with FITC-dextran (1 mg/ml), and then, after 30 min, 50 μl of medium from the bottom chamber was aspirated. The absorbance of the medium at the excitation wavelength of 488 nm and the emission wavelength of 520 nm was read with a microplate reader.
Sepsis mouse model
All animal experiments were performed in accordance with the guidelines of the Animal Care and Use Committee of the Third Military Medical University. Mice were injected with 15 mg/kg LPS into the peritoneal cavity. Mice were sacrificed 6 h after LPS administration. Lung tissues were immediately removed, fixed with 4% paraformaldehyde, and paraffin embedded. Paraffin-embedded tissue sections (4 μm) were observed after H&E staining for histological evaluation.
Pulmonary transvascular flux assay using evans blue-albumin
After LPS (15 mg/kg) was administered via peritoneal cavity injection, the mice were injected with Evans blue-albumin (40 mg/kg; Sigma, E-2129) via the tail vein, and it was allowed to circulate in the blood vessels for 30 min. Intravascular Evans blue was washed through the right ventricle with saline for 5 min. Mice lungs were removed, weighed, homogenized in 1 ml of PBS, and extracted in 2 ml of formamide for 24 h at 60°C. The Evans blue content was measured at OD620 and OD740.
Lung wet:dry ratio measurement
After stimulation, the mice lungs were excised and the excess blood was removed. The lung wet weight was measured, and then the lungs were dried in an oven at 60°C for 72 h to determine the dry weight.
Survival rate assay
C57BL/6 and rab26−/- mice were challenged with 40 mg/kg LPS i.p., and then, the survival percentage in each group was monitored for 5 days.
Western blotting
Protein was extracted from cells that were prepared using M-PER reagent. Tissue protein extracts were prepared using T-PER reagent. Western blotting was performed as reported [Citation37]. In brief, Protein samples were electrophoresed in 8–15% SDS-PAGE polyacrylamide gels, incubated with the appropriate antibodies and measured by enhanced chemiluminescence detection system (Thermo Fisher Scientific, 34,079). The densitometric analysis were performed using ImageJ software.
Statistical analysis
Quantification of replicate experiments is expressed as the mean ± SD, and the experiments were conducted more than 3 times. One-way analysis of variance (ANOVA) and log-rank tests were applied to determine statistical significance, with P values set to less than 0.01.
Supplemental Material
Download MS Word (1.8 MB)Acknowledgments
We thank Prof. Guangyu Wu and David Shalloway for providing plasmids.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740.
- Dejana E. Endothelial cell-cell junctions: happy together. Nature Reviews Molecular Cell Biology. 2004;5(4):261–270.
- Yadav H, Thompson BT, Gajic O. Fifty years of research in ARDS. is acute respiratory distress syndrome a preventable disease? Am J Respir Crit Care Med. 2017;195(6):725–736.
- Maca J, Jor O, Holub M, et al. Past and present ARDS mortality rates: a systematic review. Respir Care. 2017;62(1):113–122.
- Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med. 2017;5(6):524–534.
- Li C, Fan Y, Lan TH, et al. Rab26 modulates the cell surface transport of alpha2-adrenergic receptors from the Golgi. J Biol Chem. 2012;287(51):42784–42794.
- Yoshie S, Imai A, Nashida T, et al. Expression, characterization, and localization of Rab26, a low molecular weight GTP-binding protein, in the rat parotid gland. Histochem Cell Biol. 2000;113(4):259–263.
- Wagner AC, Strowski MZ, Goke B, et al. Molecular cloning of a new member of the Rab protein family, Rab 26, from rat pancreas. Biochem Biophys Res Commun. 1995;207(3):950–956.
- Nashida T, Imai A, Shimomura H. Relation of Rab26 to the amylase release from rat parotid acinar cells. Arch Oral Biol. 2006;51(2):89–95.
- Lo HG, Jin RU, Sibbel G, et al. A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev. 2017;31(2):154–171.
- Jin RU, Mills JC. RAB26 coordinates lysosome traffic and mitochondrial localization. J Cell Sci. 2014;127(Pt 5):1018–1032.
- Mills JC, Taghert PH. Scaling factors: transcription factors regulating subcellular domains. Bioessays. 2012;34(1):10–16.
- Tian X, Jin RU, Bredemeyer AJ, et al. RAB26 and RAB3D are direct transcriptional targets of MIST1 that regulate exocrine granule maturation. Mol Cell Biol. 2010;30(5):1269–1284.
- Binotti B, Pavlos NJ, Riedel D, et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife. 2015;4:e05597.
- Lo S, Yuan SS, Hsu C, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg. 2013;257(2):352–363.
- Herrera-Garcia AM, Dominguez-Luis MJ, Arce-Franco M, et al. Prevention of neutrophil extravasation by alpha2-adrenoceptor-mediated endothelial stabilization. J Immunology. 2014;193(6):3023–3035.
- Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121(Pt 13):2115–2122.
- Taddei A, Giampietro C, Conti A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10(8):923–934.
- Flemming S, Burkard N, Renschler M, et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res. 2015;107(1):32–44.
- Cai J, Wei J, Li S, Suber T, Zhao J. AM966, an Antagonist of Lysophosphatidic Acid Receptor 1, Increases Lung Microvascular Endothelial Permeability through Activation of Rho Signaling Pathway and Phosphorylation of VE-Cadherin. Mediators Inflamm. 2017; 2017: 6893560.
- Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nature Reviews Molecular Cell Biology. 2016;17(10):611–625.
- Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front Physiol. 2015;6:365.
- Mikelis CM, Simaan M, Ando K, et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun. 2015;6:6725.
- Ha CH, Xu XB, Wong C, et al. Novel role of VE-cadherin as a scaffold protein to modulate VEGF-induced Src, Akt and eNOS activation in endothelial cells. Circulation. 2006;114(18):229.
- Sandilands E, Schoenherr C, Frame MC. p70S6K is regulated by focal adhesion kinase and is required for Src-selective autophagy. Cell Signal. 2015;27(9):1816–1823.
- Sandilands E, Serrels B, McEwan DG, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14(1):51–60.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544.
- Li H, He B, Liu X, et al. Regulation on toll-like receptor 4 and cell barrier function by Rab26 siRNA-loaded DNA nanovector in pulmonary microvascular endothelial cells. Theranostics. 2017;7(9):2537–2554.
- Hu R, Chen ZF, Yan J, et al. Complement C5a exacerbates acute lung injury induced through autophagy-mediated alveolar macrophage apoptosis. Cell Death Dis. 2014;5:e1330.
- Hu Y, Lou J, Mao YY, et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12(12):2286–2299.
- Cabrera S, Maciel M, Herrera I, et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11(4):670–684.
- Domigan CK, Warren CM, Antanesian V, et al. Autocrine VEGF maintains endothelial survival through regulation of metabolism and autophagy. J Cell Sci. 2015;128(12):2236–2248.
- Xu D, Chen B, Gu J, et al. Inhibition of autophagy ameliorates pulmonary microvascular dilation and PMVECs excessive proliferation in rat experimental hepatopulmonary syndrome. Sci Rep. 2016;6:30833.
- Zhang D, Li C, Song Y, et al. Integrin alphavbeta5 inhibition protects against ischemia-reperfusion-induced lung injury in an autophagy-dependent manner. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L384–L394.
- Li J, Zhou J, Zhang D, et al. Bone marrow-derived mesenchymal stem cells enhance autophagy via PI3K/AKT signalling to reduce the severity of ischaemia/reperfusion-induced lung injury. J Cell Mol Med. 2015;19(10):2341–2351.
- Zhou Z, You Z. Mesenchymal stem cells alleviate LPS-induced acute lung injury in mice by MiR-142a-5p-controlled pulmonary endothelial cell autophagy. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2016;38(1):258–266.
- Yang J, Yao W, Qian G, et al. Rab5-mediated VE-cadherin internalization regulates the barrier function of the lung microvascular endothelium. Cellular and Molecular Life Sciences: CMLS. 2015;72(24):4849–4866.
- Yang J, Sun H, Zhang J, et al. Regulation of beta-adrenergic receptor trafficking and lung microvascular endothelial cell permeability by Rab5 GTPase. Int J Biol Sci. 2015;11(8):868–878.
- Wang G, Wu G. Small GTPase regulation of GPCR anterograde trafficking. Trends Pharmacol Sci. 2012;33(1):28–34.
- Dong C, Yang L, Zhang X, et al. Rab8 interacts with distinct motifs in alpha2B- and beta2-adrenergic receptors and differentially modulates their transport. J Biol Chem. 2010;285(26):20369–20380.
- Itoh T, Fujita N, Kanno E, et al. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008;19(7):2916–2925.
- Ishibashi K, Uemura T, Waguri S, et al. Atg16L1, an essential factor for canonical autophagy, participates in hormone secretion from PC12 cells independently of autophagic activity. Mol Biol Cell. 2012;23(16):3193–3202.
- Jain N, Ganesh S. Emerging nexus between RAB GTPases, autophagy and neurodegeneration. Autophagy. 2016;12(5):900–904.
- Szatmari Z, Sass M. The autophagic roles of Rab small GTPases and their upstream regulators: a review. Autophagy. 2014;10(7):1154–1166.
- Gonzalez CD, Lee MS, Marchetti P, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7(1):2–11.
- Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy. 2009;5(2):280–282.
- Shrivastava S, Devhare P, Sujijantarat N, et al. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J Virol. 2015;90(3):1387–1396.
- Chichger H, Duong H, Braza J, et al. p18, a novel adaptor protein, regulates pulmonary endothelial barrier function via enhanced endocytic recycling of VE-cadherin. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2015;29(3):868–881.
- Nawroth R, Poell G, Ranft A, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. The EMBO Journal. 2002;21(18):4885–4895.
- Gong H, Rehman J, Tang H, et al. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest. 2015;125(2):652–664.
- Wessel F, Winderlich M, Holm M, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15(3):223–230.
- Turowski P, Martinelli R, Crawford R, et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121(Pt 1):29–37.
- Vestweber D, Wessel F, Nottebaum AF. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin Immunopathol. 2014;36(2):177–192.
- Orsenigo F, Giampietro C, Ferrari A, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208.
- Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454.
- Hakak Y, Martin GS. Ubiquitin-dependent degradation of active Src. Current Biology: CB. 1999;9(18):1039–1042.
- Li X, Lonard DM, Jung SY, et al. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124(2):381–392.
- Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nature Reviews Molecular Cell Biology. 2014;15(2):81–94.
- Adam AP, Sharenko AL, Pumiglia K, et al. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285(10):7045–7055.
- Kmiecik TE, Shalloway D. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 1987;49(1):65–73.
- Gong H, Shen B, Flevaris P, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327(5963):340–343.
- Xiao K, Allison DF, Buckley KM, et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163(3):535–545.