
ABSTRACT
Picornaviruses, one of the major causes of human diseases ranging from the common cold to acute flaccid paralysis, have a short cytosolic lifecycle that, in cultured cells, ends in cell lysis. For years, the prevailing model was that these viruses exit from cells exclusively through cell lysis. However, over the last several years it has become apparent that for some picornaviruses, a macroautophagy/autophagy-related pathway can result in release of virus particles wrapped in a membrane containing autophagic markers. It has been proposed that this enveloped release predominates within hosts, allowing cell-to-cell movement of virus while minimizing exposure to the immune system. One reason that picornaviruses induce the autophagy pathway is to provide membrane scaffolds for RNA replication complexes. Perhaps more importantly, acidified autophagosomes (known as amphisomes) provide havens for maturation of new viral particles into infectious viruses. In back-to-back papers recently published in Cell Reports, our labs investigated a basic question: if picornavirus particles are maturing inside amphisomes, then how are they avoiding the typical degradative fate of autophagic cargo and exiting the cell intact?
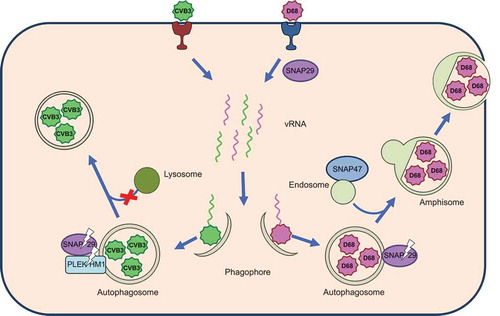
Working in parallel, our labs investigated the role of the autophagic soluble N-ethylmaleimide-sensitive factor activating protein receptor proteins (SNAREs) in coxsackievirus B3 (CVB3) and enterovirus D-68 (EV-D68), 2 medically important picornaviruses [Citation1,Citation2]. We discovered that SNAP29, a SNARE protein required for autophagosome-lysosome fusion, is cleaved by the 3C protease, encoded by all picornaviruses. This cleavage separates 2 coiled-coil domains in the protein: one interacting with STX17 (syntaxin 17) on autophagosomes or amphisomes and the other binding VAMP8 (vesicle-associated membrane protein 8) on lysosomes, thereby inhibiting the fusion of these vesicles. This would suggest that the build-up of double-membrane vesicles that we show are likely either autophagosomes or amphisomes, but not autolysosomes.
A recent study examining enterovirus A71 (EV71) found that viral non-structural protein 2BC interacts with both STX17 and SNAP29. Furthermore, the STX17-SNAP29 interaction, required for autophagosome-lysosome fusion, was no longer present during viral infection. Bona fide autophagy results from EV71 2BC transfection, so during infection 2BC interaction with autophagic SNAREs could promote degradation, or it might be related to a larger viral strategy to disrupt autophagosome-lysosome fusion. There are differences in how these 2 viruses deal with the pathway: while there is a buildup of STX17 protein during EV-D68 infection, CVB3 infection instead causes a decrease in STX17 protein levels late in infection. The CVB3 result is supported by recent work demonstrating downregulation of STX17 at the transcriptional level following CVB3 infection.
Our data provide some insight into the functions of SNAP29 during viral lifecycles. Full-length SNAP29 appears to suppress intracellular replication of CVB3 while also facilitating viral exit and spread. For EV-D68, the roles of SNAP29 are slightly different; it is required for a very early step in the virus lifecycle, but has a separate, inhibitory role late in the virus lifecycle.
These viruses interact with other members of the autophagy fusion machinery, such as PLEKHM1 (pleckstrin homology domain containing, family M [with RUN domain] member 1), an adaptor/tethering protein that facilitates formation of SNARE complexes. PLEKHM1 is also cleaved during CVB3 infection, contributing to impaired formation of SNARE complexes. For EV-D68, the data indicate that the virus requires the SNARE protein SNAP47, with a significant drop in viral titer if SNAP47 is knocked down. SNAP47 does not appear to be cleaved, so its function is likely important for virus replication. We also describe a previously unknown role for SNAP47 in autophagy, which appears more important to autophagic flux than SNAP29.
These data indicate that picornaviruses are crippling the fusion machinery required for normal autophagic flux. Measurement of flux by standard methods is difficult during infection. This is because, as previously shown by the Luo lab and confirmed in the recent Cell Reports work, the SQSTM1 cargo receptor normally used as a marker for autophagic degradation is cleaved by a viral protease. The cleavage is functionally interesting because it not only impairs the function of SQSTM1 in cargo recognition and selective degradation, but also generates C-terminal polypeptides that act as dominant-negatives, preventing normal cargo loading. As a practical matter, cleavage of SQSTM1 can be misinterpreted as active autophagy, as levels of full-length SQSTM1 are reduced due to cleavage during infection.
In summary, during infection, autophagic cargo loading is inhibited and fusion with lysosomes is reduced (). The first consequence of these changes is obvious: viruses within autophagic vesicles will not be degraded, avoiding the innate immune function of autophagy. So, then, what is the fate of viruses captured within autophagosomes?
Figure 1. EV-D68 and CVB3 harness autophagic membranes for RNA replication complexes and require its acidified chambers, called amphisomes, for viral maturation. During early infection, EV-D68 requires the SNARE proteins SNAP29 and SNAP47 for viral replication. Both EV-D68 and CVB3 disrupt autophagosome-lysosome fusion by cleaving SNAP29 through the activity of viral protease 3C. Additionally, CVB3 cleaves the tethering adaptor protein PLEKHM1. Virus-associated amphisomes or autophagosomes are redirected to the cell periphery to mediate non-lytic release.
For the past several years, Picornavirologists have provided evidence for a previously unknown stage in the virus lifecycle: an enveloped stage, proposed to occur primarily for trafficking from cell-to-cell within the host. First identified by Stanley Lemon’s lab as a non-autophagic event for hepatitis A virus, it is now known that several picornaviruses have an enveloped stage. CVB3 was the first shown to have an envelope with an autophagic origin. The enveloped-stage viruses presumably use host-derived membrane to avoid immune detection more efficiently than naked viruses.
The presumption has been that the enveloped virus vesicles are redirected autophagosomes or early, double-membraned, amphisomes. For at least 2 picornaviruses, CVB3 and PV, extracellular vesicles contain LC3 on the surrounding membrane. It has been known for years that picornavirus virions themselves localize within autophagic vesicles inside the cell. These 2 findings link the extracellular vesicles to autophagic secretion. Our work demonstrates a mechanism by which these picornaviruses switch the focus of the pathway from cargo degradation to secretion ().
While the link between autophagy and secretion is well established, how the 2 modes of autophagy interact with one another during infection is an open question. Does STX17 find another fusion partner in the absence of SNAP29? There are only 4 known SNAP proteins, also called Qbc SNARES: SNAP29, SNAP47, neuron-specific SNAP25, and SNAP23, the latter of which is found at the plasma membrane. For neurotropic picornaviruses, SNAP25 may play a role in infection. However, SNAP23 is the most interesting candidate for a SNAP role in release in non-neuronal cells.
Several other questions remain. For example, we do not know if picornaviruses are targeted to phagophores (autophagosome precursors) through a selective uptake mechanism, if they are taken up because viral translation, replication, and assembly take place in proximity to autophagosome formation, or if uptake is incidental, as with most cytoplasmic contents.
Studies of picornaviruses continue to teach us how autophagy is normally wired by showing us how the virus subverts and short-circuits the pathway. Whether SNAP29 is also regulated by cells as a switch between degradative and secretory autophagy is an interesting question for future research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mohamud Y, Shi J, Qu J, et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018;22(12):3292–3303. PMID:29562184.
- Corona AK, Saulsbery HM, Corona Velazquez AF, et al. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018;22(12):3304–3314. PMID:29562185.