
ABSTRACT
Coxiella burnetii is an intracellular bacterial pathogen which causes Q fever, a human infection with the ability to cause chronic disease with potentially life-threatening outcomes. In humans, Coxiella infects alveolar macrophages where it replicates to high numbers in a unique, pathogen-directed lysosome-derived vacuole. This compartment, termed the Coxiella-containing vacuole (CCV), has a low internal pH and contains markers both of lysosomes and autophagosomes. The CCV membrane is also enriched with CLTC (clathrin heavy chain) and this contributes to the success of the CCV. Here, we describe a role for CLTC, a scaffolding protein of clathrin-coated vesicles, in facilitating the fusion of autophagosomes with the CCV. During gene silencing of CLTC, CCVs are unable to fuse with each other, a phenotype also seen when silencing genes involved in macroautophagy/autophagy. MAP1LC3B/LC3B, which is normally observed inside the CCV, is excluded from CCVs in the absence of CLTC. Additionally, this study demonstrates that autophagosome fusion contributes to CCV size as cell starvation and subsequent autophagy induction leads to further CCV expansion. This is CLTC dependent, as the absence of CLTC renders autophagosomes no longer able to contribute to the expansion of the CCV. This investigation provides a functional link between CLTC and autophagy in the context of Coxiella infection and highlights the CCV as an important tool to explore the interactions between these vesicular trafficking pathways.
Introduction
Intracellular bacterial pathogens possess diverse strategies that allow them to evade the bactericidal characteristics of the host cell and form a replicative niche. Most intracellular bacterial pathogens, such as Shigella flexneri and Legionella pneumophila, actively evade cellular autophagic surveillance to avoid clearance [Citation1]. However, some intracellular bacteria take advantage of the host autophagy machinery in order to thrive. Examining the interactions between bacterial pathogens and the autophagy pathway provides a physiologically relevant tool to elucidate both the intricacies of autophagic processes and how they contribute to host-pathogen interactions.
Coxiella burnetii is an intracellular bacterial pathogen and the causative agent of the human disease Q fever, which commonly presents as a mild febrile illness. However, Coxiella can cause serious chronic infections, such as endocarditis, that can be life threatening [Citation2]. Humans can contract Q fever through inhalation of contaminated aerosols. The environmental source for Coxiella is commonly livestock, mainly ruminants, that can contaminate the local environment through bodily excretions particularly during parturition. Such was the case during a recent outbreak in the Netherlands from 2007–2010, where over 4000 cases of human Q fever were reported and linked to heavily contaminated goat farms [Citation3].
When individuals inhale aerosols containing Coxiella, the bacteria are taken up into the lung, where Coxiella primarily infects alveolar macrophages [Citation4]. Once Coxiella is engulfed by a human cell, the phagosome matures through the endocytic pathway, and Coxiella becomes resident in a lysosome [Citation5,Citation6]. This compartment is remodeled by the pathogen to support intracellular replication and is termed the Coxiella-containing vacuole (CCV). Features of the CCV, such as a low pH of approximately 4.8 and the presence of proteolytic and degradative enzymes, are closely aligned to those of lysosomes [Citation7]. This environment is essential for the metabolic activity of Coxiella and the biogenesis of the replicative CCV [Citation8,Citation9]. Over a period of several days the pathogen directs the expansion of the CCV such that it can occupy the majority of the host cytoplasm. The ability of Coxiella to develop this unique intracellular niche is entirely dependent on a type IV Dot/Icm secretion system [Citation10,Citation11]. This is a multi-protein apparatus which spans both the bacterial and vacuolar membranes, to enable the translocation of bacterial proteins, termed effectors, into the host cell [Citation12]. The secretion system itself, and a subset of the effectors translocated by this system are essential for intracellular replication [Citation10,Citation11,Citation13]. The cohort of approximately 150 effector proteins are collectively responsible for manipulating host cellular functions to facilitate the intracellular replication of Coxiella [Citation10].
Not only does the CCV have lysosomal characteristics, but it is also known to promiscuously fuse with endosomes, autophagosomes, clathrin-coated vesicles and other CCVs to facilitate expansion. Hence, the CCV acquires markers of these various cellular vesicles. Lysosomal markers, including LAMP1 (lysosomal associated membrane protein 1), the vacuolar-type H+-translocating ATPase, and CTSD (cathepsin D), surround the CCV [Citation14]. Additionally, the autophagy marker MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) is located inside the CCV, and is recruited there during autophagosome fusion with the CCV [Citation5,Citation13,Citation15]. These fusion events benefit the establishment of the CCV, since induction of autophagy by starvation allows for more infected cells and larger CCVs [Citation16]. LC3B recruitment to the CCV has been shown to be dependent on the Coxiella Dot/Icm effector protein, Cig2 [Citation15]. Indeed, LC3B is no longer recruited to the lumen of CCVs of cig2 mutant Coxiella. Under normal conditions, the fusion of CCVs with each other ensures only one replicative CCV exists inside cells, even if multiple bacteria infect the same cell. However, the cig2 mutant displays a distinct multi-vacuole phenotype, where multiple replicative CCVs form within individual host cells due to a defect in CCV homotypic fusion [Citation15]. Interestingly, this phenocopies what is observed during silencing of genes involved in autophagy, indicating that homotypic fusion of CCVs likely requires them to be in an autolysosomal state.
CLTC (clathrin heavy chain) is also enriched on the CCV membrane [Citation17,Citation18]. Clathrin-mediated endocytosis is the process by which host cells internalize extracellular material, termed cargo, from the plasma membrane. Additionally, CLTC is found at endosomes and the trans-Golgi network to facilitate cargo movement from these structures. Cargo molecules are recognized by adaptor protein complexes, such as AP2 (adaptor related protein complex 2), which form a link to CLTC itself, which acts as a scaffold forming a cage-like structure around clathrin-coated vesicles [Citation19]. The Coxiella Dot/Icm effector CvpA has been shown to bind AP2, and is required for both intracellular replication of Coxiella and CLTC recruitment to the CCV [Citation17]. Gene silencing studies demonstrate that CLTC and AP2 are important for CCV biogenesis and intracellular replication of Coxiella. In the absence of CLTC, CCVs remain small and support minimal Coxiella replication [Citation17,Citation18]. More recently, the Coxiella effector Cig57 was shown to interact with FCHO2 (FCH domain only 2), an accessory protein in clathrin-mediated endocytosis, and contribute to Coxiella virulence, as Cig57 is essential for intracellular replication of Coxiella and CCV biogenesis [Citation13,Citation18]. Both Cig57 and CvpA play vital, non-redundant roles in facilitating intracellular replication of Coxiella through their interactions with clathrin-dependent processes, however the biochemical functions of these important effectors remain to be determined.
A link between CLTC and autophagy has recently been discovered with the observation that CLTC is required both for autophagosome formation and in the later stages for regulation of autophagic lysosomal reformation [Citation20,Citation21]. In addition, it has been reported that CLTC colocalizes with LC3B upon induction of autophagy [Citation22]. CLTC has also been shown to bind ATG16L1 (autophagy related 16 like 1), an essential protein in the autophagy process, and OCRL (inositol polyphosphate-5-phosphatase), a protein that colocalizes with lysosomes upon induction of autophagy [Citation20,Citation23]. Hence, there is a growing body of research that indicates a role for CLTC in autophagy, although the functional role of CLTC at autophagosomes remains unknown.
Here, we present data, in the context of Coxiella infection, to support the hypothesis that CLTC is intimately linked to autophagy. Interaction between autophagosomes and the CCV, a modified lysosomal compartment, is dependent upon CLTC.
Results
Autophagy and CLTC are required for normal CCV formation
To confirm the reports that autophagy is needed for development of a normal CCV, the phosphatidylinositol 3-kinase inhibitor 3-methyladenine (3-MA) was used to block autophagy during infection. Bafilomycin A1 and chloroquine, other common autophagy inhibitors, cannot be used at the onset of a Coxiella infection as they alter lysosomal homeostasis and disrupt biogenesis of the CCV. Treatment with 10 mM 3-MA at the time of infection, and for 3 days post-infection, resulted in a multi-vacuole phenotype ()). When quantified, 87.1 ± 2.9% of infected HeLa cells contained more than 1 CCV during 3-MA treatment compared to 7.0 ± 1.6% in the untreated cells ()).
Figure 1. Coxiella infection in the absence of autophagy or CLTC leads to a multi-vacuolar phenotype. (a) HeLa cells were treated with 10 mM 3-MA, or left untreated, and infected with Coxiella, with a multiplicity of infection (MOI) of 50, for 3 days. At this time, cells were fixed and stained with anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue). Representative images show CCVs with a white asterisk. Scale bar: 10 µm. (b) At least 100 infected cells, from each of 3 independent experiments, were scored for having either 1 or more than 1 CCV. Error bars represent SD. (c) HeLa cells were reverse transfected (+) with siRNA targeting CLTC (clathrin heavy chain), STX17 (syntaxin 17), or a non-targeting siRNA (si-OTP) (–) and immunoblots of whole cell lysates demonstrated knockdown efficiency on day 0 (D0, day of infection, 2 days post-transfection) and day 3 (D3, 5 days post-knockdown). ACTB (actin beta) was used as a loading control. (d) Representative confocal microscopy images of HeLa cells treated with si-OTP, si-STX17, or si-CLTC and infected with Coxiella for 3 days before being fixed and stained. Anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue) demonstrate the formation of replicative CCVs, highlighted with white asterisks. Scale bar: 10µm. (e) Quantification of the number of CCVs observed in infected HeLa cells, where greater than 100 cells were scored for having either 1 CCV or more than 1 CCV for each of 3 independent experiments. Error bars represent SD.
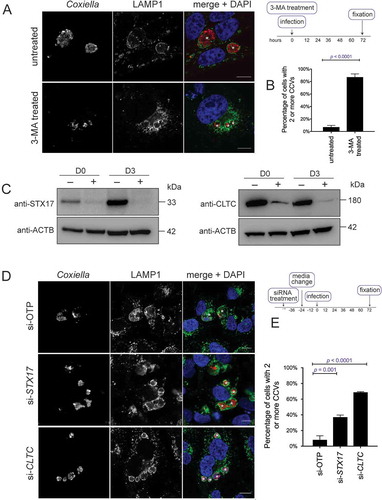
Consistent with previous studies, this multi-vacuole phenotype was also observed when expression of STX17 (syntaxin 17) was silenced to block autophagy progression [Citation13,Citation24] ()). STX17 is responsible for facilitating fusion of autophagosomes with lysosomes [Citation25]. Two days post-siRNA treatment, cells were infected with wild-type (WT) Coxiella for 3 days. As displayed in ), and as per previously published data [Citation24], a robust multi-vacuolar phenotype was observed in the absence of STX17. The same experiment was performed with cells treated with CLTC siRNA to silence expression of the clathrin heavy chain ()). Under these conditions, as with STX17 silencing, a significantly increased proportion of cells contained more than 1 CCV ()). Across 3 independent experiments, the percentage of cells displaying more than 1 CCV was quantified ()). Only 8.1 ± 3.0% of cells treated with ON-TARGET plus non-targeting control (OTP) siRNA contained more than 1 CCV compared to 68.7 ± 0.4% of infected cells possessing 2 or more CCVs during CLTC silencing, and 37.1 ± 1.6% of cells during STX17 silencing. Silencing expression of OCRL (Fig. S1A) in HeLa cells also led to a high percentage of cells harboring more than 1 CCV per cell (Fig. S1B and C). These data demonstrate that, in the absence of CLTC, CCV biogenesis phenocopies the multi-vacuole phenotype seen when autophagy is inhibited. This observation led to the hypothesis that there is a functional link between CLTC and autophagy during Coxiella infection.
CLTC recruitment is required for LC3B localization in the CCV
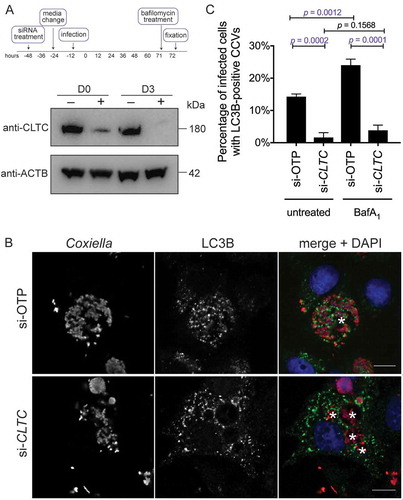
In addition to having only one large CCV per cell, a normal Coxiella infection of HeLa cells results in recruitment of LC3B to the lumen of the CCV, representing significant fusion of the CCV with autophagosomes [Citation13,Citation15]. The multi-vacuole phenotype observed above is indicative of a CCV fusion defect in the absence of CLTC. Therefore, LC3B recruitment to the CCV was examined in the absence of CLTC. CLTC was silenced with siRNA (), and 3 days post-infection HeLa cells were examined for LC3B localization. As shown in ), LC3B is recruited to the WT CCV, but is excluded from CCVs formed in the absence of CLTC. At 3 days post-infection, 14.3 ± 0.5% of CCVs were positive for LC3B, but this dropped to 1.6 ± 1.0% in the absence of CLTC ()). Furthermore, when cells were treated with bafilomycin A1 for 1 h before fixation, blocking the vacuolar-type H+-translocating ATPase and subsequently the degradative capacity of the CCV, the percentage of LC3B-positive CCVs increased to 24.0 ± 1.1%. However, bafilomycin A1 did not significantly alter LC3B recruitment during CLTC silencing, with only 3.8 ± 0.9% of CCVs observed as LC3B positive. These results rule out the idea that the loss of LC3B localization is due to an increased degradative potential of the CCV in the absence of CLTC. Rather, they indicate that CLTC is required for creation of 1 large CCV and the delivery of LC3B to the CCV.
Figure 2. CLTC is required for LC3B localization to the CCV. (a) HeLa cells were treated with non-targeting (–) siRNA or targeting siRNA against CLTC (+) for 2 days and infected with Coxiella at day 0 (D0) for 3 days, at which time whole cell lysate samples were collected for immunoblot analysis (D3) using antibodies against CLTC and ACTB. (b) Representative confocal images of HeLa cells treated with non-targeting siRNA (si-OTP) or CLTC siRNA (si-CLTC) and infected for 3 days with WT Coxiella. Fixed samples were stained with anti-Coxiella (red), anti-LC3B (green), and DAPI (blue). CCVs are highlighted with white asterisks. Scale bar: 10 µm. (c) Quantification of the percentage of infected HeLa cells that were observed to have LC3B-positive CCVs. Cells were either left untreated or treated with bafilomycin A1 (BafA1; 100 ng/ml) for 1 h before fixation. Here, at least 100 cells were scored for each of 3 independent experiments, and error bars represent SD.
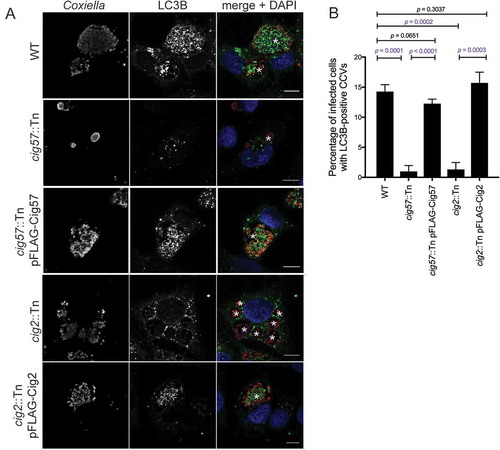
It was previously reported that the Coxiella effector Cig2 directs the delivery of LC3B to the CCV, as CCVs formed during infection with cig2::Tn Coxiella no longer recruit LC3B [Citation13]. Another Dot/Icm effector, Cig57, is required for recruitment of CLTC to the CCV [Citation18]. During infection with cig57::Tn Coxiella, CLTC is no longer enriched on the CCV membrane. To examine the relationship between CCV localization of LC3B, CLTC, and these important effectors, LC3B localization was examined during infection with the effector mutants. HeLa cells were infected for 3 days with the relevant strains of Coxiella, then fixed and stained for LC3B. As previously observed, LC3B was located within the CCV during WT infection. In contrast, LC3B was excluded from CCVs during infection with both cig2::Tn and cig57::Tn Coxiella, and this phenotype could be complemented, as infection with cig2::Tn pFLAG-Cig2 or cig57::Tn pFLAG-Cig57 Coxiella restored LC3B location to the CCV ()). In concert with previous studies, 14.3 ± 0.7% of WT CCVs were positive for LC3B, similar to the 15.7 ± 1.0% observed for cig2::Tn pFLAG-Cig2 CCVs, compared to only 1.3 ± 0.7% of CCVs formed in the absence of Cig2. Interestingly, in the absence of Cig57, 1.0 ± 0.6% CCVs were positive for LC3B, which was restored to 12.3 ± 0.4% for the Cig57-complemented Coxiella strain ()). This finding recapitulates the importance of Cig2 in CCV-autophagosome fusion and indicates that Cig57 function is also linked to this aspect of CCV maturation.
Figure 3. Dot/Icm effectors control the localization of LC3B to the CCV. (a) HeLa cells were infected with the indicated Coxiella strains (red) for 3 days, then stained for LC3B (green), and nuclei (DAPI, blue). The CCVs are highlighted with white asterisks. Scale bars: 10 µm. (b) Quantification of the percentage of HeLa cells that have LC3B-positive CCVs. At least 100 infected cells for each of 3 independent experiments were scored for recruitment of LC3B. Error bars represent SD.
Our previous investigations have demonstrated that Cig57 interacts with the human protein FCHO2, and this interaction was shown to facilitate efficient Coxiella intracellular replication and CCV biogenesis [Citation18]. Hence, a functional link between FCHO2 and autophagy was investigated by observing LC3B recruitment to the CCV in the absence of FCHO2. Figure S2 shows that FCHO2 is not involved in LC3B recruitment to the CCV (Fig. S2A), as the percentage of cells with LC3B-positive CCVs was unaffected during infection of FCHO2 knockout (KO) cells (Fig. S2B). FCHO2 is also not required for autophagosomes to fuse with the CCV (Fig. S2C). In FCHO2 KO HeLa cells, CCV expansion also occurred in response to starvation (Fig. S2D and E). LC3B is also still lipidated in the absence of FCHO2 (Fig. S2F). This indicates that Cig57 may influence autophagy in an FCHO2-independent manner.
CLTC recruitment to the CCV is linked to autophagy
To investigate a link between the multi-vacuole phenotype and CLTC, the localization of CLTC was examined in the absence of Cig2. HeLa cells were infected with WT, cig2::Tn, or cig2::Tn pFLAG-Cig2 Coxiella for 3 days and stained for CLTC. CCV localization of CLTC was observed with infection of both WT and cig2::Tn pFLAG-Cig2 CCVs, however CLTC was absent from CCVs formed by the cig2::Tn Coxiella mutant ()). When quantified, WT CCVs had an intensity ratio of CLTC on the CCV compared to the cytoplasm of 2.4 ± 0.1. During cig2::Tn Coxiella infection the ratio was 1.1 ± 0.01, and during cig2::Tn pFLAG-Cig2 infection the ratio was 2.2 ± 0.03 ()). To further validate this observation, HeLa cells infected with our cohort of Coxiella strains were stained for CLTC and LAMP1 and observed by confocal microscopy. Figure S3 shows that WT and complemented Coxiella mutants form CCVs with LAMP1 and CLTC enriched at the CCV membrane, however this is not the case for cig57::Tn or cig2::Tn Coxiella. Independent confirmation that CLTC is indeed located on the CCV was achieved by mechanically lysing infected HeLa cells and using a histodenz gradient to isolate intact CCVs. The CCV enriched fraction was collected and stained for CLTC and LAMP1. As anticipated, CCVs isolated from WT Coxiella infected cells were positive for CLTC, as were the complemented cig57::Tn and cig2::Tn mutants, but both cig57::Tn and cig2::Tn CCVs did not stain positive for CLTC ()). The observation that cig2::Tn mutant CCVs, in which autophagosomes are not able to fuse, were negative for CLTC suggested that functional autophagy is required for CLTC deposition onto the CCV. To further investigate this, cells were treated with 3-MA at the point of infection, and for 3 days post-infection, and then stained for CLTC. In these conditions, there was no enrichment of CLTC with untreated cells showing a CCV:cytoplasm CLTC ratio of 2.3 ± 0.1 and 3-MA treated cells having a ratio of 1.0 ± 0.04 ()).
Figure 4. CLTC enrichment on the CCV membrane is linked to autophagy. (a) HeLa cells were infected with WT, cig2::Tn, or cig2::Tn pFLAG-Cig2 Coxiella for 3 days and then stained with anti-Coxiella (red), anti-CLTC (green), and DAPI (blue). Scale bar: 10µm. White asterisks denote CCVs and images are representative of 3 independent experiments. (b) Quantification of CLTC intensity on the CCV as a ratio compared to the intensity in the cytoplasm. Five independent measurements of CLTC intensity were taken for both the cytoplasm and at the CCV membrane, and a ratio of CCV:cytoplasm CLTC intensity was calculated. This intensity ratio was quantified for more than 20 cells in each of 3 independent experiments. Error bars represent SD and dotted line represents a ratio of 1, where there is equal CLTC on the CCV and in the cytoplasm. (c) HeLa cells were infected with the indicated strains of Coxiella, and intact CCVs were purified from cells. CCVs were spun onto coverslips and stained with anti-LAMP1 (red), anti-CLTC (green), and DAPI (blue). Scale bar: 10 µm. Images are representative of 2 independent experiments. (d) Confocal images of HeLa cells treated with 3-MA or left untreated and infected with WT Coxiella at the time of treatment. Cells were stained with anti-Coxiella (red), anti-CLTC (green), and DAPI (blue). Scale bar: 10 µm. Asterisks denote the CCVs and images are representative of 3 independent experiments. (e) Quantification of CLTC intensity on the CCV during 3-MA treatment as described in A. Error bars represent SD and results are from 3 independent experiments. (f) Confocal images of HeLa cells treated with non-targeting siRNA (si-OTP) or siRNA against STX17 (si-STX17) and infected with WT Coxiella for 3 days. Following fixation, samples were stained with anti-Coxiella (red), anti-CLTC (green), and DAPI (blue). Scale bar: 10 µm. White asterisks highlight CCVs and images are representative of 3 independent experiments. (g) The average CLTC intensity ratio calculated for at least 20 cells for each of 3 experiments represented in F. Error bars represent the SD across 3 independent experiments. (h) Confocal images of HeLa cells that were infected with Coxiella for 60 h before being starved in HBSS or left in DMEM for 12 h prior to fixation. Samples were stained with anti-CLTC (green), anti-Coxiella (red), and DAPI (blue). Scale bar: 10µm. CCVs are highlighted with white asterisks and images are representative of 3 experiments. (i) Quantification of CLTC intensity relating to experiments performed in H, the CLTC intensity ration was performed for a minimum of 20 cells within each of 3 experiments. Error bars represent SD.
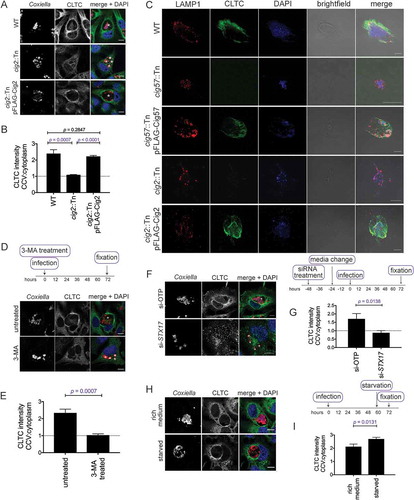
This experiment was also performed in the context of STX17 knockdown. Consistent with the Cig2 and 3-MA finding, in the absence of STX17, and therefore the absence of functional autophagy, CLTC was not recruited to the CCV membrane ()). Across 3 independent experiments, the CCV:cytoplasm ratio of CLTC intensity was 1.7 ± 0.2 in si-OTP conditions, compared to 0.9 ± 0.1 in si-STX17 conditions ()). Together these data indicate that CLTC recruitment to the CCV is linked to autophagosome fusion with the CCV. Thus, we hypothesized that increased autophagy would lead to increased CLTC on the CCV. In order to examine this, infected cells were starved for 12 h before immunofluorescent detection of CLTC ()). Indeed, quantification of CLTC intensity demonstrated further enrichment of CLTC on the CCV during starvation conditions. The CLTC intensity ratio was 2.1 ± 0.1 during incubation in rich medium compared to 2.7 ± 0.07 during starvation conditions ()).
CCV size can be modulated by autophagy
Given that autophagy is required for a normal CCV phenotype, it is reasonable to assume that normal infection induces autophagy in order to promote CCV biogenesis. A time course experiment was established, in which HeLa cells were infected with WT Coxiella, and samples were collected for western blot at 12, 24, 48 and 72 h post-infection. SDS-PAGE separated samples were then probed with an antibody against LC3B. Increasing amounts of lipidated LC3B were observed at 48 and 72 h post-infection. Thus, in agreement with previous literature [Citation26], it was observed that LC3B-I to LC3B-II conversion occurs specifically in response to Coxiella infection ()).
To determine whether autophagy induction is effector driven, HeLa cells were either left uninfected, or infected with WT Coxiella or the Dot/Icm-deficient icmL::Tn strain, for 72 h. LC3B conversion was then determined by western blot. A large amount of LC3B-II was formed during WT infection but was absent during infection when no Coxiella effector proteins are translocated into the host ()). The specific effector proteins Cig2 and Cig57 were then tested for their ability to induce autophagy. HeLa cells were infected with WT, cig57::Tn, cig57::Tn pFLAG-Cig57, cig2::Tn, or cig2::Tn pFLAG-Cig2 Coxiella and LC3B conversion was examined at 72 h post-infection. LC3B-II was present in all samples except for uninfected and cig57::Tn infection ()). This observation raised the possibilities that Cig57 may act to induce LC3B lipidation during infection, or that autophagy induction is a host response to CCV expansion, which is significantly perturbed during cig57::Tn Coxiella infection. To test a direct induction of autophagy by Cig57, HeLa cells engineered to express FLAG-Cig57 in a doxycycline-dependent manner were employed. LC3B-II formation was monitored in response to FLAG-Cig57 expression and no autophagy induction was observed ()). This data indicates that Cig57 alone is not capable of inducing LC3B lipidation.
Figure 5. Coxiella infection leads to increased LC3B-II (a) HeLa cells were infected with WT Coxiella (+), or left uninfected (–), for 12, 24, 48 or 72 h. Lysates were collected and probed with LC3B antibodies and anti-ACTB. Shown is the conversion of LC3B-I (18 kDa) to LC3B-II (16 kDa). (b) HeLa cells were infected with WT or icmL::Tn Coxiella, or left uninfected for 3 days. Lysates were collected and separated by SDS-PAGE before being probed with anti-LC3B and anti-ACTB. (c) HeLa cells were infected with the indicated strains of Coxiella, or left uninfected, for 3 days. Samples were then collected for SDS-PAGE analysis and immunoblotted for LC3B or ACTB. Shown is LC3B-I (18 kDa) conversion to LC3B-II (16 kDa) (d) A HeLa parent cell line, or stable HeLa cell line expressing FLAG-Cig57 under the control of a doxycycline inducible promotor, were left untreated (–), or treated with 20 ng/ml of doxycycline (+) for 2 days, after which samples were taken for SDS-PAGE and probed with anti-LC3B, anti-FLAG, and anti-ACTB. (E) HeLa cells were infected with the indicated strain of Coxiella, or left uninfected, for 60 h, at which point cells were either left in growth medium (–) or incubated in HBSS (+) for 12 h. Cell lysates were collected and separated by SDS-PAGE for immunoblotting with antibodies against SQSTM1 and ACTB and image shown is representative of 3 independent experiments. Quantification represents the SQSTM1 signal relative to ACTB from the representative immunoblot.
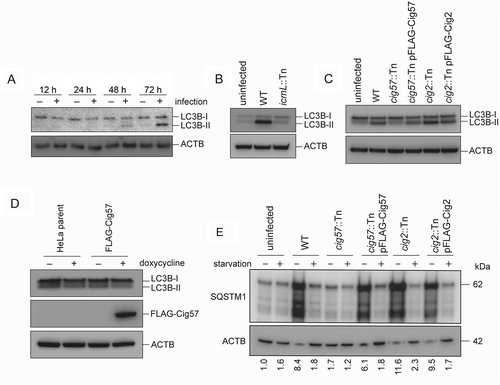
The induction of autophagy through lipidation of LC3B leads to an increased presence of autophagosomes in the cell, however it remains to be tested whether these autophagosomes are degraded efficiently through fusion with lysosomes. Thus, autophagic flux was measured by probing the levels of SQSTM1 (sequestosome 1) during infection with Coxiella, with or without starvation. Cells were infected with the relevant strain of Coxiella for 60 h, before cells were either starved in HBSS or maintained in growth media. Cell lysates were collected for SDS-PAGE analysis at 72 h post-infection. In agreement with previous studies [Citation26], there was a lack of SQSTM1 degradation during infection with WT Coxiella. In fact, SQSTM1 levels were increased under these conditions, but could be rescued with starvation ()). In accordance with the diminished LC3B lipidation during infection with cig57::Tn Coxiella, SQSTM1 levels were not increased during infection with this mutant, but were increased when cells were infected with cig2::Tn strains.
In order to clarify the importance of autophagy induction on CCV expansion, experiments were performed to further increase the amount of autophagy in infected cells. CCV size was compared in HeLa cells infected for 48 h, and then either starved for 12 h or maintained in nutrient-rich medium. It was observed that CCVs were notably larger upon starvation-induced autophagy ()). When quantified, CCVs were approximately 250% larger in starved cells, 240.9 ± 14.1 µm2, compared to 96.0 ± 17.8 µm2 in rich media ()). To visualize the variability in CCV area, individual data points from 1 experiment are shown in ), demonstrating that starvation leads to a wider range of CCV sizes.
Figure 6. CCV size depends on the autophagic state of the host cell. (a) HeLa cells infected with WT Coxiella for 60 h were starved in HBSS for 12 h before being fixed and stained with anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue). Images are representative of CCV formation. Scale bar: 10 µm. CCV areas were (b) measured over 3 experiments, or (c) displayed as individual data points (n = 50) from 1 experiment. Error bars represent SD. (d) HeLa cells were infected with WT Coxiella for 48 h and treated with 10 mM of 3-MA for 24 h, or left untreated. Samples were then fixed and stained with anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue). Scale bar: 10 µm. CCV areas were measured (e) over 3 experiments, or (f) displayed as individual data points (n = 50) from 1 experiment. Error bars represent SD.
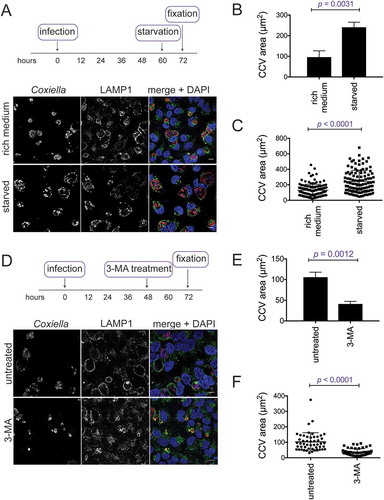
The converse experiments, where autophagy was blocked, were performed to examine the impact that this had on CCV size. After 48 h of infection, HeLa cells were treated with 3-MA for 24 h, before being fixed and stained for Coxiella and LAMP1. CCV size was significantly reduced by 3-MA treatment, likely representing autophagy inhibition stunting CCV expansion ()). CCV sizes were quantified over 3 experiments. Untreated cells averaged a CCV area of 105.8 ± 7.0 µm2 and CCVs treated with 3-MA for the final 24 h were significantly smaller 41.1 ± 3.6 µm2 ()). ) illustrates that the distribution of the CCV sizes was greater in untreated cells than treated cells. Together, these results highlight that CCV size is dependent upon the amount of autophagy within the host cell. Where more autophagy is induced, more autophagosomes can fuse with the CCV to contribute to its size, and, when little autophagy occurs, CCVs do not expand.
CLTC recruitment to the CCV is required for starvation dependent CCV expansion
Given that autophagy contributes to CCV size, and CLTC contributes to LC3B accumulation within the CCV, a relationship between CLTC and CCV expansion due to starvation was examined. After silencing of CLTC or STX17 ()), HeLa cells were infected for 48 h and then starved in HBSS for 12 h, after which cells were fixed and stained ()). As expected, in si-OTP-treated cells, CCVs formed during starvation were larger, 185.3 ± 5.3 µm2, than those left in nutrient rich conditions, 94.6 ± 5.5 µm2. Similarly, in the absence of CLTC but without starvation, CCVs were smaller than si-OTP-treated cells, 28.0 ± 5.2 µm2. However, starvation after silencing of CLTC did not alter the size of the CCVs, 25.5 ± 2.3 µm2 ()). Similar findings were observed during STX17 silencing, where starvation did not alter the CCV size. In nutrient rich conditions, CCVs formed in the absence of STX17, were 55.7 ± 5.1 µm2 compared to 54.4 ± 2.1 µm2 during starvation. Individual CCV areas plotted from 1 representative experiment confirm that there is a greater spread of CCV sizes when si-OTP treated cells were starved, whereas when either CLTC or STX17 silenced cells were starved, the CCV areas remained restricted ()). This indicates that CLTC is required for autophagosome fusion to contribute to CCV size.
Figure 7. Autophagy-dependent CCV expansion requires CLTC. (a) Immunoblots showing CLTC or STX17 and ACTB during respective siRNA treatments (+) or non-targeting treatment (–), where samples were taken at the day of infection (D0, 2 days post siRNA treatment) and 3 days post infection (D3). (b) Confocal images of HeLa cells, treated with non-targeting (si-OTP) siRNA, siRNA targeting CLTC (si-CLTC), or STX17 (si-STX17) before being infected with Coxiella for 60 h and then incubated in either normal DMEM + 10% FBS (rich medium) or in HBSS starvation medium (starved) for 12 h before fixation. Samples were stained with anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue) to highlight CCV formation. Scale bar: 10 µm. (c) Quantification of CCV sizes during the above conditions. Data represents the average CCV area and SD over 3 independent experiments. (d) Individual data points from 1 experiment quantifying CCV size (n = 50) to show the spread of the data. (e) Representative confocal images of HeLa cells infected with WT, cig57::Tn, or cig57::Tn pFLAG-Cig57 Coxiella for 60 h before starvation treatment for 12 h. As above, anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue) highlight the formation of CCVs. Scale bar: 10 µm. (f) Quantification of CCV area over 3 independent experiments shown in E. Error bars represent SD. (g) Individual data points (n = 50) from 1 experiment showing the observed variation in CCV size. Error bars represent SD.
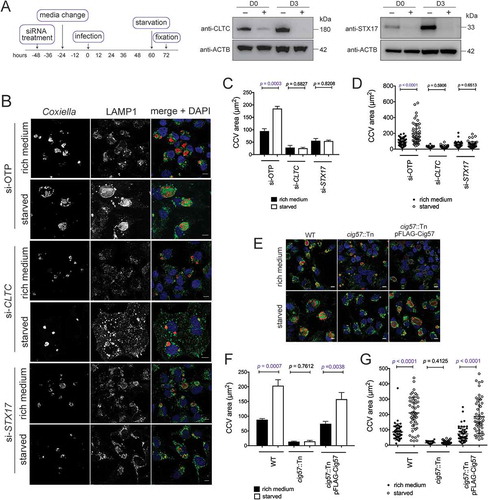
Analogous findings were observed when the cig57::Tn Coxiella strain was used to examine the link between CLTC recruitment to the CCV and CCV expansion. This effector mutant does not show enriched CLTC localization to the CCV membrane [Citation18]. Comparison of cig57::Tn Coxiella infections in nutrient rich and starvation conditions demonstrated that increased autophagy did not influence the size of this CCV ()). There was no difference in CCV size during infection with cig57::Tn Coxiella under starvation conditions, 13.8 ± 1.1 µm2, compared to nutrient rich conditions, 14.6 ± 2.1 µm2. This indicates that CLTC recruitment to the CCV, in a Cig57-dependent manner, is required for autophagosomes to contribute to CCV expansion ()).
CLTC is located at sites of autophagosome fusion with the CCV
The locations of LC3B and CLTC were next examined during infection with WT or mutant Coxiella during starvation conditions. HeLa cells were infected for 48 h with the relevant strain of Coxiella and starved in HBSS for 12 h before being fixed and stained with antibodies to LC3B and CLTC. ) provides a representative image of the large LC3B-positive autophagosomes that were observed around the CCV of WT Coxiella. Interestingly, CLTC was concentrated in the areas in which these autophagosomes meet the CCV, and often lay in between the 2 structures. However, during infection with the cig57::Tn Coxiella mutant, large LC3B-positive autophagosomes were not observed, and CLTC was rarely seen to associate with LC3B, a phenotype which was able to be complemented. When cells were infected with cig2::Tn Coxiella, large CLTC-covered autophagosomes were observed inside the cell, yet these structures were not located in proximity of the CCV membrane to the same extent as in WT infection. Again, this observation could be complemented, likely owing to the function of Cig2 in allowing autophagosomes to fuse with the CCV. The occurrence of these CLTC-positive LC3B autophagosomes was quantified, and the results presented in ).
Figure 8. Colocalization of CLTC and LC3B during starvation conditions. (a) HeLa cells were left uninfected or infected with the various strains of Coxiella for 60 h and starved in HBSS for 12 h before fixation. Samples were stained with anti-LC3B (red), anti-CLTC (green), and DAPI (blue). White boxes highlight regions of interest amplified to the right. Scale bar: 10 µm. Images are representative of at least 3 independent experiments. (b) The frequency of LC3B-CLTC vesicles at the CCV membrane as shown in A. At least 50 cells were observed for the number of LC3B-CLTC-positive vesicles in proximity of the CCV. Results are representative of 3 independent experiments and error bars represent SD. (c) HeLa cells were treated with control si-RNA (si-OTP) or with siRNA against CLTC, infected with WT Coxiella for 60 h, and starved in HBSS for 12 h. After fixation, cells were stained with antibodies against Coxiella (red), LC3B (green), and DAPI (blue). Images are representative of 3 independent experiments. (d) HeLa cells were reverse transfected with siRNA to target CLTC (si-CLTC) or left untreated (mock), then infected with WT Coxiella before being transfected to express GFP-LC3B (green) and starved in HBSS. DRAQ5 (red) was added to cells 5 min before imaging. Cells were imaged every minute for 1 h and selected images over the time course are shown. Arrows indicate LC3B puncta of interest. Scale bar: 10 µm.
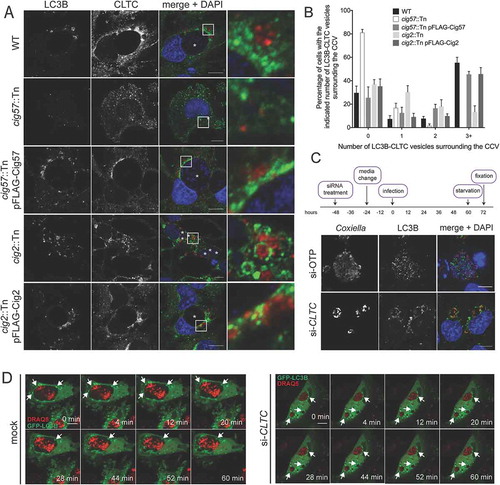
We next questioned the contribution of CLTC in the formation of the large autophagosomes, so CLTC was silenced with siRNA, and cells were starved before being stained for LC3B and Coxiella. As expected, LC3B was present inside CCVs of control knockdown cells, but in the absence of CLTC, LC3B-positive autophagosomes were still formed, and localized to the membrane of the CCV, but not inside the CCV ()).
Images presented in ) cannot distinguish whether the LC3B-positive vesicles are about to fuse with the CCV or are actually budding from the CCV. To discern the direction of movement of the vesicles, live confocal microscopy was performed on cells that were either subjected to CLTC siRNA or not, transfected with pGFP-LC3B, and starved. GFP-LC3B structures were observed fusing with the CCV in control cells over the 1 h time period, however, in the absence of CLTC, autophagosomes remain stagnant at the CCV membrane ()).
These results indicate that CLTC is located at sites of autophagosomal fusion with the CCV and supports the hypothesis that CLTC is involved in the fusion of autophagosomes with the CCV, and is delivered onto the CCV by the autophagosomal structures.
Discussion
Originally discovered as a process for bulk degradation of cellular components, autophagy has since been recognized to be important for host defense and immunity [Citation27,Citation28]. Formation of a double-membrane structure inside host cells, and the recruitment of ATG proteins and LC3B facilitates the establishment of mature autophagosomes which ultimately fuse with lysosomes to degrade the internal components, such as invading bacteria. However, some intracellular bacterial pathogens have evolved techniques to interfere with or take advantage of the autophagy machinery for intracellular survival [Citation29]. Thus, investigating the interactions between pathogens and the autophagy pathway can provide novel insights into the molecular mechanisms required for successful autophagy.
Coxiella is an intracellular bacterial pathogen which requires a late stage of autophagy, fusion of mature autophagosomes with the lysosome, to develop the normal CCV. Failure of autophagosomes to fuse with lysosomes results in a multi-vacuolar phenotype, as silencing of STX17 resulted in this unusual CCV formation [Citation13,Citation24]. Whether Coxiella infection induces the initiation of autophagy remains under debate. It has been reported that LC3B lipidation is induced during Coxiella infection of macrophages, although this was not dependent upon the Dot/Icm secretion system [Citation26]. Another report found no difference in the LC3B lipidation profile between uninfected and infected HeLa cells [Citation13]. Within this study we observed autophagy induction in a Dot/Icm-dependent manner during infection of HeLa cells, which has previously been reported in THP1 cells [Citation26]. This induction likely assists the CCV in expanding to its normal size, since further autophagy, induced through starvation, increased the CCV size significantly. Thus, the membrane capacity of the CCV is expanded as autophagosomes deliver extra membrane to the growing CCV. In agreement with previously published work [Citation26], the data presented here indicates there is a block in the autophagic flux during Coxiella infection due to the accumulation of SQSTM1 in cells. This has previously led to the conclusion that autophagy is not induced by the bacteria, rather that LC3B-II is not being degraded, however we can show that increasing autophagy through starvation leads to further CCV expansion. Therefore, in support of previously published research [Citation16], we confirm that increased autophagy during infection aids the intracellular success of Coxiella, whether it is actively triggered by the pathogen or not. This is not incompatible with a block in autophagic flux, seen by increased SQSTM1, as the observation that LC3B accumulates inside the CCV gives evidence to believe that the cargo of autophagosomes that fuse with the CCV are not being degraded as readily.
Recent studies have identified CLTC as playing a role in autophagy, showing that it participates in the regulation of autophagic lysosomal reformation [Citation21]. CLTC is also present on mature autophagosomes, colocalizing with LC3B during initiation of autophagy [Citation22]. Additionally, given that CLTC has been shown to interact with ATG16L1, which mediates earlier stages of autophagosome development, it is possible that CLTC may also contribute to other stages of autophagy [Citation20]. In this study, Coxiella is used as a tool to demonstrate that fusion of autophagosomes with lysosomes involves CLTC. In the absence of CLTC, LC3B is not delivered to the CCV, indicating that LC3B-positive autophagosomes are no longer fusing and delivering the LC3B. CCVs also fail to coalesce into one without CLTC. This study is the first to link CLTC with the fusion of vesicles, indeed CLTC is well characterized as being involved in the opposite procedure, budding of vesicles from the plasma membrane and other organelles. Hence, this data suggests the possibility of a new, additional function of CLTC in the context of autophagy. Recent studies have demonstrated that CLTC exists in 2 isoforms, CHC17 and CHC22, and that these isoforms have different functions [Citation30,Citation31]. CHC17 binds to clathrin light chains (CLC) and AP2 at the plasma membrane for endocytosis, however CHC22 binds neither CLC or AP2 and has been described to aid in trafficking of SLC2A4/GLUT4 (solute carrier family 2 member 4) [Citation32]. This provides an intriguing example of how different CLTC isoforms are functionally distinct, and future work may elucidate whether a specific isoform is involved in the autophagosome-CCV interaction. Both CLTC isoforms are capable of forming lattices, and this may provide a clue as to the function of CLTC at autophagosomes, as it is potentially the stabilization and membrane structure properties of CLTC which are being taken advantage of in the fusion of autophagosomes with lysosomes. There is no precedence for CLTC to be involved in a membrane fusion event, so it is more plausible that CLTC is not mediating this process directly. As examined in a recent review [Citation33], CLTC recruits downstream effectors on the lysosomal membrane during autophagic lysosome reformation to alter the phosphoinositide signature of the membrane [Citation21]. It is intriguing to consider a similar role for CLTC at the autophagosomal membrane to facilitate interaction with lysosomes.
The data presented here indicate that fusion of autophagosomes with lysosomes not only contributes to the capacity of CCVs to homotypically fuse, but also to the massive expansion of CCVs that is observed following activation of the Dot/Icm system. It is possible that the fusion of autophagosomes with the CCV encourages nutrient deposition into the CCV in the form of autophagosomal cargo to cater for growth of the bacteria. However, when the CCV is not in an autolysosomal state, such as in the absence of Cig2, or when autophagy is blocked, Coxiella is still able to replicate to the same extent as when autophagy is functional, albeit in a modified vacuole [Citation13,Citation24]. Despite no obvious replication defect, the altered, multi-vacuole CCV state appears to significantly affect the virulence of Coxiella. In Galleria mellonella, injection of WT Coxiella results in efficient killing of larvae and this is attenuated in the absence of cig2 [Citation15,Citation34]. Similarly, cig2::Tn bacteria were recovered from the spleen and lung of severe combined immunodeficiency (SCID) mice at significantly lower levels than WT Coxiella [Citation35]. Hence, the CCV being maintained in an autolysosomal state, through Cig2 activity, is required for full virulence of Coxiella.
Recent research into the pathogenesis of Coxiella has identified several important Dot/Icm effectors including Cig2, which binds phosphatidylinositol-3-phosphate and phosphatidylserine [Citation34] and is necessary for LC3B delivery to the CCV, and Cig57, which interacts with FCHO2 and is necessary for recruitment of CLTC to the CCV [Citation18]. Given that we did not see LC3B-I conversion to LC3B-II during infection with cig57::Tn, there may be a role for Cig57 in linking CLTC to autophagosomes. Ectopic expression of FLAG-Cig57 did not induce autophagy, however it cannot be ruled out that Cig57 induces autophagy through actions that require infection conditions. Importantly, in the absence of Cig57 a multi-vacuole phenotype is not observed. Another possible reason for the lack of autophagy induction in the absence of Cig57 is the size of the CCVs [Citation13]. Perhaps maturation events including commencement of CCV expansion, which is perturbed in the absence of Cig57, need to occur before autophagy is triggered. Whether this is directly effector mediated or a host response to detection of the Dot/Icm-dependent formation of a large CCV remains to be determined.
There is a precedence for effectors of other pathogens to manipulate components of the autophagy pathway and block autophagy to prevent delivery to the degradative lysosomal compartment. A well-characterized example is Legionella pneumophila, which possesses the effector RavZ to catalyze the irreversible uncoupling of Atg8-family proteins from phosphatidylethnolamine on autophagosonal membranes, allowing L. pneumophila to inhibit autophagy [Citation36,Citation37]. Additionally, the effector Lpg1137 has recently been reported to proteolytically cleave STX17, a protein involved in the fusion of autophagosomes with the lysosome, and a further effector LpSpI, inhibits autophagy through its ability to target host sphingosine biosynthesis [Citation38,Citation39]. In contrast, rather than being bactericidal, the lysosome provides the appropriate environment to induce metabolic activity of Coxiella and promote intracellular replication [Citation8]. Hence, autophagy induction and autophagosomal fusion is in this pathogen’s favor. Our research indicates that Coxiella may possess factors that not only induce autophagy but also facilitate clathrin-dependent autophagosome fusion with the CCV.
The observations reported support the hypothesis that CLTC forms a link between autophagosomes and the CCV, promoting fusion of these vesicles. CLTC is delivered to the CCV through autophagosomal fusion, as is it not localized to the CCV membrane without autophagy functionality. This fits with the observation that CLTC localizes to lysosomes upon autophagy induction [Citation21,Citation22]. We propose a model in which autophagy is induced, directly by the bacteria or as a host response to the CCV, and the resulting autophagosomes acquire CLTC, with the aid of Coxiella effectors such as Cig57. These clathrin-coated autophagosomes then fuse with the CCV facilitated by Cig2. Fusion of these vesicles with the CCV leads to deposition of CLTC on the CCV.
This research has demonstrated that CLTC is an important protein for both functional autophagy and the Coxiella life cycle. Through bacterial-driven induction of autophagy, autophagosomes function to expand the CCV, allow it to undergo homotypic fusion, and deliver CLTC, an important protein for CCV biogenesis and bacterial growth, to the CCV membrane. Studies that employ Coxiella as a tool provide an opportunity to study a large, stable autolysosome, the CCV, in eukaryotic cells. Future research using Coxiella will allow for further understanding of how CLTC and autophagy may be exploited by intracellular bacteria.
Materials and methods
Bacterial strains and tissue culture cells
Coxiella burnetii Nine Mile Phase II, strain RSA 439, was cultured axenically in acidified citrate cysteine media 2 (ACCM-2; made in-house) as previously described [Citation40]. When required, chloramphenicol (Boehringer Mannheim, 634,433) and/or kanamycin (Amresco, 0408) were added to ACCM-2 at 3 µg/ml and 300 µg/ml respectively. HeLa human cervical epithelial cells (ATCC, CCL-2) were cultured in Dulbecco’s Modified Eagle’s Medium GlutaMAX (DMEM; Gibco, 10,566,016) supplemented with 10% fetal bovine serum (FBS; Gibco, 16,000,044) at 37°C with 5% CO2. HeLa cells stably expressing an inducible FLAG-Cig57 were engineered using lentiviral transduction; pFTRE-3G-FLAG-Cig57 (A gift from the laboratory of John Silke, WEHI) was transfected into HEK293T cells (a gift from the laboratory of Elizabeth Hartland, Hudson Institute) alongside pCMV-VSV-G (A gift from the laboratory of John Silke, WEHI) and pCMV-dR8.2dvpr (A gift from the laboratory of John Silke, WEHI), and viral particles were collected and infected onto HeLa cells. Transduced cells were selected for with puromycin (5 µg/ml; ThermoFisher Scientific, A1113803), and effector expression was induced with doxycycline (20 ng/ml; Sigma Aldrich, D3447).
Coxiella infection of HeLa cells
HeLa cells were seeded at 2.5 × 104 cells per well into 24 well tissue culture trays (Corning Costar, LS3524-100EA) with or without coverslips (12mm, #1; ThermoFisher Scientific, 1014355112NR1). The following day, Coxiella cultures were measured for the number of bacteria per ml using the Quant-iT PicoGreen dsDNA Assay Kit (ThermoFisher Scientific, P7589) [Citation41]. Cells were infected at a multiplicity of infection (MOI) of 50 for 4 h, at which point the wells were washed once with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4), and media replaced with DMEM containing 5% FBS. This time point was allocated as day 0. Coverslips were fixed for fluorescence microscopy 3 days post infection, or samples for western blot were taken at desired time points throughout infection.
Western blot analysis
Samples were resuspended in 2× SDS-PAGE sample buffer (125 mM Tris, 4% SDS [Amresco, 0227], 20% glycerol [The British Drug Houses, 158,920,010], 0.2% bromophenol blue [Bio-Rad, 1,610,404], 5% 2-mercaptoethenol [Sigma, M6250], pH 6.8) and boiled for 10 min before being loaded on 4–12% NuPAGE Bis-Tris gels (Life Technologies, NP0321BOX) with either 1× MES (Life Technologies, NP000202) or 1× MOPS (Life Technologies, NP0001) running buffer. Proteins were transferred to PVDF membranes using the iBlot2 system (Life Technologies, IB24001). Membranes were blocked in 5% skim milk in TBST (TBS [20 mM Tris, 150 mM NaCl, pH 8.0] containing 0.1% Tween [ThermoFisher Scientific, 28,320]) for at least 1 h at room temperature. Primary antibodies were diluted in 5% BSA (Roche, 10,735,094,001) in TBST and incubated with the membranes overnight. Secondary antibodies were diluted in TBST containing 5% skim milk and incubated with the membranes for approximately 1 h. Membranes were washed extensively before the use of Clarity Western ECL Reagents (BioRad, 1,705,061) and MF-ChemiBIS, version 3.2 (DNR Bio-Imaging Systems, Ltd.) to develop the blots. Blots were exposed for between 10 sec and 1 min and were assessed with GelCapture, version 7.0.18 software (DNR Bio-Imaging Systems, Ltd.) to ensure band detection was within the linear range. Antibodies used in this study for immunoblotting were: anti-CLTC (1:2000; Abcam, ab21679), anti-STX17 (1:1000; ThermoFisher Scientific, PA5-40,127), anti-LC3B (1:3000; Novus, NB100-2220), anti-OCRL (1:500; ThermoFisher Scientific, PA5-27,844), anti-SQSTM1 (1:2000; ThermoFisher Scientific, PA5-20,839), anti-ACTB (1:5000; Sigma Aldrich, A5316), HRP-conjugated goat anti-mouse (1:3000; BioRad, 1,721,011), HRP-conjugated goat anti-rabbit (1:3000; Perkin Elmer, NEF812001EA).
Confocal fluorescence microscopy
For LAMP1 and CLTC staining, cells were fixed at room temperature for 20 min with 4% paraformaldehyde (Sigma Aldrich, 158,127), and permeabilized and blocked with 0.05% saponin (Sigma Aldrich, 47,036) and 2% BSA in PBS (blocking solution) for 1 h. Primary antibodies were diluted in blocking solution and incubated with sample at room temperature for 1 h. Cells were washed 3 times in PBS before secondary antibodies were applied in blocking solution for 1 h at room temperature. Cells were washed in 4ʹ,6-diamidino-2-phenylindole (DAPI; Life Technologies, D1306) made up in PBS (1:10,000) for 5 min before 2 more washes in PBS and mounting on glass slides with ProLong Gold reagent (Life Technologies, P36934). For LC3B staining, cells were fixed and permeabilized in ice-cold methanol for 5 min on ice and blocked in 2% BSA in PBS for 1 h. Primary and secondary antibodies were diluted in 2% BSA in PBS and incubated at room temperature for 1 h each, with 3 PBS washes in between. Coverslips were then treated with DAPI and mounted as above. Samples were imaged using a Zeiss LSM710 instrument with Zen software. Antibodies for immunofluorescence microscopy used in this study were as follows: polyclonal anti-Coxiella (1:10,000; a gift from the laboratory of Craig Roy, Yale) anti-LAMP1 (1:250; Developmental Hybridoma Studies Bank, H4A3-C), anti-LC3B (1:200; Nanotools, 0260–100/LC3-2G6), anti-CLTC (1:1000; Abcam, ab21679), anti-mouse and anti-rabbit Alexa Fluor 488 (1:2000; Life Technologies, A11001 and A11008), anti-mouse and anti-rabbit Alexa Fluor 568 (1:2000; Life Technologies, A11004 and A11011).
CLTC on the CCV was measured using Fiji analysis software (version 2.0.0), as previously described [Citation18]. Briefly, 5 measurements of CLTC fluorescence intensity in a small square area were taken on both the CCV membrane and in the cytoplasm. Fluorescence intensity averages were calculated and expressed as a ratio of CCV intensity:cytoplasm intensity. A minimum of 20 cells for each condition in each of 3 independent experiments were quantified in this way. CCV areas were measured in Fiji using the freehand selection tool to outline the CCV with the aid of the Coxiella and LAMP1 stains. A minimum of 50 CCVs were measured for each experimental condition.
Live-cell imaging was performed on a Zeiss LSM700 with an incubated chamber. 2 × 105 cells were seeded into a 35 mm µ-dish (Ibidi, 81,166), and infected with Coxiella at an MOI of 100 for 24 h, then transfected with pGFP-LC3B using Lipofectamine 3000 (Invitrogen, L3000001) for 12 h, then starved in Hanks’ balanced salt solution (HBSS; 1.26 mM CaCl2, 5.33 mM KCl, 0.44 mM KH2PO4, 0.5 mM MgCl2-6H2O, 0.41 mM MgSO4-7H2O, 138 mM NaCl, 4 mM NaHCO3, 0.3 mM Na2HPO4, 5.6 mM glucose, pH 7.2) for a further 12 h before imaging. DRAQ5 (ThermoFisher Scientific, 62,251) was added to cells 5 min before imaging at a final concentration of 1 nM. Images were acquired every minute over a 1 h time period.
RNA interference
HeLa cells were reverse transfected with small-interfering RNA (si-RNA) using siGenome SMART-pools (Dharmacon, GE Life Sciences) against human CLTC (M-004001–00), STX17 (M-020965–01), OCRL (M-010026–01) or with ON-TARGETplus (OTP) non-targeting pool (D-001810–10) using DharmaFECT 1 transfection reagent (Dharmacon, GE Life Sciences, T-2001). Cells were seeded into 24 well plates at a density of 5 × 104 cells per well with a final concentration of 50 nM siRNA. After a 2-day incubation, with a media change after 1 day, cells were infected with Coxiella at an MOI of 50 for 4 h before being washed once with PBS and being incubated with fresh media. Day 0 western blot samples were collected at this time. The infection was allowed to proceed for 3 days, at which point cells were collected for western blot analysis and/or fixed for confocal fluorescence microscopy as described above.
Starvation or treatment with 3-methyladenine (3-MA)
For induction of autophagy, HeLa cells were infected as above, and the infection allowed to proceed for 60 h, at which time cells were washed twice with HBSS and the media was replaced with HBSS. Cells were then incubated for a further 12 h before fixation for immunofluorescence microscopy or collection for western blot as above. For inhibition of autophagy post-infection, HeLa cells were infected for 48 h at an MOI of 50, then the media was replaced with DMEM containing 10% FBS and 10 mM 3-MA (Sigma Aldrich, 5142–23-4), and cells were incubated for a further 24 h before fixation or resuspension in SDS-PAGE sample buffer. For inhibition of autophagy at the time of infection, cells were infected with Coxiella at an MOI of 50, and at the 4-h wash time point 3-MA was added to the media at a concentration of 10 mM and left on for the duration of the infection.
Isolation of CCVs
HeLa cells were grown to 30% confluence in 2 T75 flasks per condition, and subsequently infected with the required strain of Coxiella at an MOI of 300. The infection was allowed to progress for 5 days, at which time cells were trypsinized (Gibco, 25,300,062) and resuspended in ice-cold HS buffer (20 mM HEPES, 0.25 M sucrose, 1 mM EDTA, pH 7.2) supplemented with protease inhibitors (Complete, EDTA-free; Roche, 11,873,580,001). Cells were homogenized with a ball-bearing homogenizer (Isobiotech) using a ball with an 18-µm clearance and 11 strokes. Lysates were precleared by centrifugation at 600 × g for 15 min at 4°C, and the resuspended pellet was added to a PBS-Histodenz gradient consisting of 5.75 mL of 10% Histodenz (Sigma Aldrich, 66,108–95-0) placed on top of 5.75 mL 35% Histodenz. Gradients were centrifuged for 1 h at 3,250 × g at 4°C, and 1.5-mL fractions were collected and spun onto poly-L-lysine (Sigma Aldrich, P9155) coated coverslips. Isolated CCVs were stained as described above.
Statistical analysis
Statistical analyses were performed with Prism (GraphPad Software Inc.) by use of the unpaired Student’s t test. p values are displayed on graphs and if less than 0.05, the value is highlighted purple.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ACTB | = | actin beta |
| AP2 | = | adaptor related protein complex 2 |
| ATG16L1 | = | autophagy related 16 like 1 |
| CCV | = | Coxiella-containing vacuole |
| CLTC | = | clathrin heavy chain |
| Cig2 | = | co-regulated with icm gene 2 |
| Cig57 | = | co-regulated with icm gene 57 |
| DAPI | = | 4ʹ,6-diamidino-2-phenylindole |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FCHO2 | = | FCH domain only 2 |
| FBS | = | fetal bovine serum |
| GFP | = | green fluorescent protein |
| HBSS | = | Hanks’ balanced salt solution |
| KO | = | knockout |
| LAMP1 | = | lysosomal associated membrane protein 1 |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3 beta |
| MOI | = | multiplicity of infection |
| OCRL | = | inositol polyphosphate-5-phosphatase |
| OTP | = | on-target plus control siRNA |
| PBS | = | phosphate-buffered saline |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| siRNA | = | small interfering ribonucleic acid |
| SQSTM1 | = | sequestosome 1 |
| STX17 | = | syntaxin 17 |
| WT | = | wild type |
Supplemental Material
Download Zip (62.5 MB)Acknowledgments
Confocal imaging was performed at the Biological Optical Microscopy Platform, The University of Melbourne (www.microscopy.unimelb.edu.au). We thank Professor Jason Mackenzie for the gift of the GFP-LC3B expression construct, Professor John Silke for lentiviral plasmids and Professor Elizabeth Hartland for HEK293T cells.
Disclosure statement
No potential conflict of interest was reported by the authors.
SUPPLEMENTARY MATERIAL
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Mostowy S. Autophagy and bacterial clearance: a not so clear picture. Cell Microbiol. 2013 Mar;15(3):395–402. PubMed PMID: 23121192; PubMed Central PMCID: PMCPMC3592990.
- Madariaga MG, Rezai K, Trenholme GM, et al. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003 Nov;3(11):709–721. PubMed PMID: 14592601.
- Schneeberger PM, Wintenberger C, van der Hoek W, et al. Q fever in the Netherlands – 2007–2010: what we learned from the largest outbreak ever. Med Mal Infect. 2014 Aug;44(8):339–353. PubMed PMID: 25108615.
- Fernandes TD, Cunha LD, Ribeiro JM, et al. Murine alveolar macrophages are highly susceptible to replication of coxiella burnetii phase II in vitro. Infect Immun. 2016 Sep;84(9):2439–2448. PubMed PMID: 27297388; PubMed Central PMCID: PMCPMC4995897.
- Beron W, Gutierrez MG, Rabinovitch M, et al. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002 Oct;70(10):5816–5821. PubMed PMID: 12228312; PubMed Central PMCID: PMCPMC128334.
- Newton HJ, Roy CR. The Coxiella burnetii Dot/Icm system creates a comfortable home through lysosomal renovation. MBio. 2011;2(5). PubMed PMID: 22010216; PubMed Central PMCID: PMCPMC3195215. DOI:10.1128/mBio.00226-11.
- Howe D, Shannon JG, Winfree S, et al. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun. 2010 Aug;78(8):3465–3474. PubMed PMID: 20515926; PubMed Central PMCID: PMCPMC2916283.
- Hackstadt T, Williams JC. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A. 1981 May;78(5):3240–3244. PubMed PMID: 6942430; PubMed Central PMCID: PMCPMC319537.
- Maurin M, Benoliel AM, Bongrand P, et al. Phagolysosomes of Coxiella burnetii-infected cell lines maintain an acidic pH during persistent infection. Infect Immun. 1992 Dec;60(12):5013–5016. PubMed PMID: 1452331; PubMed Central PMCID: PMCPMC258270.
- Carey KL, Newton HJ, Luhrmann A, et al. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 2011 May;7(5):e1002056. PubMed PMID: 21637816; PubMed Central PMCID: PMCPMC3102713.
- Beare PA, Gilk SD, Larson CL, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio. 2011;2(4):e00175–11. PubMed PMID: 21862628; PubMed Central PMCID: PMCPMC3163939.
- Seshadri R, Paulsen IT, Eisen JA, et al. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5455–5460. PubMed PMID: 12704232; PubMed Central PMCID: PMCPMC154366.
- Newton HJ, Kohler LJ, McDonough JA, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014 Jul;10(7):e1004286. PubMed PMID: 25080348; PubMed Central PMCID: PMCPMC4117601.
- Heinzen RA, Scidmore MA, Rockey DD, et al. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996 Mar;64(3):796–809. PubMed PMID: 8641784; PubMed Central PMCID: PMCPMC173840.
- Kohler LJ, Reed SR, Sarraf SA, et al. Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. MBio. 2016;7(4). PubMed PMID: 27435465. DOI:10.1128/mBio.01127-16
- Gutierrez MG, Vazquez CL, Munafo DB, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005 Jul;7(7):981–993. PubMed PMID: 15953030.
- Larson CL, Beare PA, Howe D, et al. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci U S A. 2013 Dec 3;110(49):E4770–9. PubMed PMID: 24248335; PubMed Central PMCID: PMCPMC3856779.
- Latomanski EA, Newton P, Khoo CA, et al. The effector Cig57 hijacks FCHO-mediated vesicular trafficking to facilitate intracellular replication of Coxiella burnetii. PLoS Pathog. 2016 Dec;12(12):e1006101. PubMed PMID: 28002452; PubMed Central PMCID: PMCPMC5176192.
- Robinson MS. Forty years of clathrin-coated vesicles. Traffic. 2015 Dec;16(12):1210–1238. PubMed PMID: 26403691.
- Ravikumar B, Moreau K, Jahreiss L, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010 Aug;12(8):747–757. PubMed PMID: 20639872; PubMed Central PMCID: PMCPMC2923063.
- Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012 Sep;14(9):924–934. PubMed PMID: 22885770.
- Guo Y, Chang C, Huang R, et al. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci. 2012 Apr 01;125(Pt 7):1706–1715. PubMed PMID: 22328508.
- De Leo MG, Staiano L, Vicinanza M, et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol. 2016 Aug;18(8):839–850. PubMed PMID: 27398910; PubMed Central PMCID: PMCPMC5040511.
- McDonough JA, Newton HJ, Klum S, et al. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. MBio. 2013;4(1):e00606–12. PubMed PMID: 23362322; PubMed Central PMCID: PMCPMC3560531.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 07;151(6):1256–1269. PubMed PMID: 23217709.
- Winchell CG, Graham JG, Kurten RC, et al. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun. 2014 Jun;82(6):2229–2238. PubMed PMID: 24643534; PubMed Central PMCID: PMCPMC4019161.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010 Sep;12(9):814–822. PubMed PMID: 20811353; PubMed Central PMCID: PMCPMC3616322.
- Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015 Oct;16(10):1014–1024. PubMed PMID: 26382870.
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014 Feb;12(2):101–114. PubMed PMID: 24384599.
- Dannhauser PN, Camus SM, Sakamoto K, et al. CHC22 and CHC17 clathrins have distinct biochemical properties and display differential regulation and function. J Biol Chem. 2017 Nov 2. PubMed PMID: 29097553. DOI:10.1074/jbc.M117.816256.
- Liu SH, Towler MC, Chen E, et al. A novel clathrin homolog that co-distributes with cytoskeletal components functions in the trans-Golgi network. EMBO J. 2001 Jan 15;20(1–2):272–284. PubMed PMID: 11226177; PubMed Central PMCID: PMCPMC140205.
- Vassilopoulos S, Esk C, Hoshino S, et al. A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science. 2009 May 29;324(5931):1192–1196. PubMed PMID: 19478182; PubMed Central PMCID: PMCPMC2975026.
- Liu X, Klionsky DJ. Regulation of autophagic lysosome reformation by kinesin 1, clathrin and phosphatidylinositol-4,5-bisphosphate. Autophagy. 2017 Oct 5:1–2. PubMed PMID: 28980869. DOI:10.1080/15548627.2017.1386821.
- Martinez E, Allombert J, Cantet F, et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A. 2016 Jun 7;113(23):E3260–9. PubMed PMID: 27226300.
- van Schaik EJ, Case ED, Martinez E, et al. The SCID mouse model for identifying virulence determinants in coxiella burnetii. Front Cell Infect Microbiol. 2017;7:25. PubMed PMID: 28217558; PubMed Central PMCID: PMCPMC5289997.
- Choy A, Dancourt J, Mugo B, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012 Nov 23;338(6110):1072–1076. PubMed PMID: 23112293; PubMed Central PMCID: PMCPMC3682818.
- Kirisako T, Ichimura Y, Okada H, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000 Oct 16;151(2):263–276. PubMed PMID: 11038174; PubMed Central PMCID: PMCPMC2192639.
- Arasaki K, Mikami Y, Shames SR, et al. Legionella effector Lpg1137 shuts down ER-mitochondria communication through cleavage of syntaxin 17. Nat Commun. 2017 May 15;8:15406. PubMed PMID: 28504273; PubMed Central PMCID: PMCPMC5440676.
- Rolando M, Escoll P, Nora T, et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc Natl Acad Sci U S A. 2016 Feb 16;113(7):1901–1906. PubMed PMID: 26831115; PubMed Central PMCID: PMCPMC4763766.
- Omsland A, Cockrell DC, Howe D, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009 Mar 17;106(11):4430–4434. PubMed PMID: 19246385; PubMed Central PMCID: PMCPMC2657411.
- Martinez E, Cantet F, Bonazzi M. Generation and multi-phenotypic high-content screening of Coxiella burnetii transposon mutants. J Vis Exp. 201599:e52851. PubMed PMID: 25992686; PubMed Central PMCID: PMCPMC4542693. DOI:10.3791/52851