
ABSTRACT
Macroautophagy/autophagy is a multistep cellular process that sequesters cytoplasmic components for lysosomal degradation. BECN1/Beclin1 is a central protein that assembles cofactors for the formation of a BECN1-PIK3C3-PIK3R4 complex to trigger the autophagy protein cascade. Discovering the regulators of BECN1 is important for understanding the mechanism of autophagy induction. Here, we demonstrate that TRIM59, a tripartite motif protein, plays an important role in autophagy regulation in non-small cell lung cancer (NSCLC). On the one hand, TRIM59 regulates the transcription of BECN1 through negatively modulating the NFKB pathway. On the other hand, TRIM59 regulates TRAF6 induced K63-linked ubiquitination of BECN1, thus affecting the formation of the BECN1-PIK3C3 complex. We further demonstrate that TRIM59 can mediate K48-linked ubiquitination of TRAF6 and promote the proteasomal degradation of TRAF6. Taken together, our findings reveal novel dual roles for TRIM59 in autophagy regulation by affecting both the transcription and the ubiquitination of BECN1.
Abbreviations: ACTB: actin beta; BECN1: beclin 1; CHX: cycloheximide; CQ: chloroquine; GFP: green fluorescent protein; HA: haemagglutinin tag; His: polyhistidine tag; LC3B: microtubule associated protein 1 light chain 3 beta; NFKB: nuclear factor kappa B; NFKBIA: NFKB inhibitor alpha; NSCLC: non-small cell lung cancer; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; RELA: RELA proto-oncogene, NF-kB subunit; SQSTM1: sequestosome 1; tGFP: Turbo green fluorescent protein; TRAF6: TNF receptor associated factor 6; TRIM59: tripartite motif containing 59; B: ubiquitin
Introduction
Autophagy is an evolutionarily conserved cellular process that is involved in various aspects of cell functions [1,2]. In normal cells, autophagy is used to remove superfluous and damaged organelles and cytosolic proteins for degradation, serving as a surveillance mechanism and a survival strategy to maintain cell viability under stress conditions [Citation3,Citation4]. Disruption of autophagy is involved in diverse human diseases [Citation5,Citation6]. In cancer progression, autophagy plays dual roles, either as a tumor suppressor or promoting tumor progression [Citation7].
Various proteins participate in autophagy. BECN1 is a coiled-coil protein that interacts with BCL2 and is also a central component of the BECN1-PIK3C3-PIK3R4 complex which is indispensable for autophagy induction [Citation1]. BECN1 consists of 3 domains: BH3, coiled-coil domain (CCD) and BARA (the latter includes the ECD), and the roles of BECN1 in autophagy initiation depend on the proteins that bind to these domains [Citation1,Citation8]. For example, the class Ⅲ phosphatidylinositol 3-kinase (PtdIns3K) interacts with the CCD and ECD to form a complex with BECN1 [Citation9]. This complex increases the production of phosphatidylinositol-3-phosphate (PtdIns3P) that favors phagophore elongation and allows the recruitment of ATG proteins to the phagophore [Citation1]. Thus, exploring the regulation mechanisms of BECN1 is important for us to reveal the mysteries of autophagy and its functions in human diseases.
The RBCC/TRIM (tripartite motif) proteins is a large family that contains an N-terminal ring domain, 1 or 2 B-box domains and a CCD [Citation10]. TRIM proteins participate in multiple cellular processes including cell proliferation, transcriptional regulation, immunity and cancer progression [Citation11–Citation14]. A series of studies indicated that many TRIM proteins are linked to autophagy [Citation15–Citation27]. Recently, several studies demonstrated that TRIM proteins can act as both autophagy cargo receptors and platforms assembling autophagosome-formation machinery. These TRIMs recruit ULK1 and BECN1 complexes and bind to mammalian Atg8-family paralogs via an LC3-interacting region, while recognizing cargo through the SPRY and potentially other domains [Citation26,Citation27]. In our previous studies, we explored the potential roles of TRIM proteins in non-small cell lung cancer (NSCLC) by the mRNA expression profiling of all TRIM proteins in NSCLC cells [Citation28]. We identified TRIM59 as a potential tumor-promoting gene, but the precise molecular mechanism was obscure. TRIM59 has been reported to have oncogenic activity in mouse models and can be used as a multiple tumor marker for detecting early tumorigenesis [Citation29,Citation30]. In gastric tumors, TRIM59 promotes gastric carcinogenesis through ubiquitinating and degrading TP53 [Citation11]. However, the role of TRIM59 in regulating autophagy has not been studied yet. In this study, we found an unexpected role of TRIM59 in autophagy regulation. The expression of TRIM59 was reciprocally correlated with BECN1 expression in NSCLC, and TRIM59 knockdown significantly increased the basal levels of autophagy in NSCLC cells. We discovered that TRIM59 could affect the transcription of the BECN1 gene by negatively regulating the NFKB pathway. What is more intriguing was that TRIM59 could also regulate autophagy initiation through modulating TRAF6-mediated K63-linked ubiquitination of BECN1. TRIM59 overexpression significantly reduced the TRAF6-induced ubiquitination of BECN1 and disturbed the formation of the BECN1-PIK3C3-PIK3R4 complex, whereas TRIM59 knockdown had an opposite effect. Further studies demonstrated that TRIM59 could directly ubiquitinate TRAF6 in a K48-linkage for proteasomal degradation. Thus, our studies point out a new role of TRIM59 in NSCLC cells and elucidate a novel mechanism of autophagy regulation.
Results
TRIM59 knockdown induces autophagy
In our previous study, we demonstrated that TRIM59 promoted the growth of non-small cell lung cancer [Citation28]. Here, we found an interesting phenomenon that the protein expression level of TRIM59 was inversely related to that of BECN1, an autophagy-related protein, in NSCLC tissues and cancer cells (). This prompted us to explore if TRIM59 could affect autophagy. To detect the formation of autophagosomes, we transfected a plasmid encoding MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) fused with the green fluorescent protein (GFP) into H1299 cells. As can be seen from left panel, the GFP-LC3B puncta were significantly increased when TRIM59 was knocked down. The percentage of the cells with GFP-LC3B puncta increased significantly in TRIM59 knockdown cells compared with control H1299 cells (), right panel).
Figure 1. Knocking down TRIM59 induces autophagy. (a) Western blot analysis of the expression levels of BECN1 and TRIM59 in NSCLC tissues (top panel). BECN1 and TRIM59 expression relative to ACTB were quantified. Data represent the average of 3 independent experiments (mean± SD) (bottom panel). (b) Western blot analysis of the expression levels of BECN1 and TRIM59 in NSCLC cells (top panel). BECN1 and TRIM59 expression relative to ACTB were quantified. Data represents the average of 3 independent experiments (mean± SD) (bottom panel). (c) H1299 cells stably expressing pEGFP-C2-LC3B (GFP-LC3B) were transiently transfected with TRIM59 siRNAs. After 42 h, the cells were treated with or without 20 μM chloroquine (CQ) for 6 h and then analyzed by fluorescence microscopy (Olympus IX83). Scale bar: 10 μm (left panel). The cell numbers with GFP-LC3B puncta were counted under 200× magnification. **P ≤ 0.01 (right panel). (d) TRIM59 was knocked down in H1299 cells stably expressing GFP-LC3B and treated with or without 20 μM chloroquine (CQ) for 6 h. The autophagy-related proteins were analyzed using the indicated antibodies.
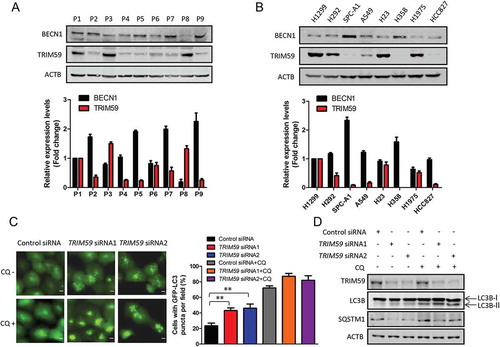
We next assessed the expression levels of LC3-II and SQSTM1/p62 in TRIM59-silenced cells. As expected, the expression of the autophagosome-associated lipidated form, LC3-II, increased and the expression of SQSTM1 decreased when TRIM59 was knocked down ()). As we know, autophagy is a highly dynamic process, and the accumulation of autophagosomes and LC3-II levels can be caused by either the induction of autophagy or the inhibition of autophagic flux [Citation31]. In order to discriminate between the 2 distinct mechanisms in our study, we used the autophagy inhibitor chloroquine (CQ) to treat cells. As can be seen in ), the percentage of the cells with LC3B puncta significantly increased when treated with CQ in both control and TRIM59-silenced cells. The expression of LC3-II also increased remarkably following CQ treatment compared with the CQ untreated group ()). These data proved that TRIM59 knockdown promoted autophagy induction, not autophagy inhibition.
TRIM59 affects the transcription of BECN1 through regulating the NFKB pathway
As the expression of BECN1 was reciprocally correlated with TRIM59 (), we presumed that TRIM59 affected autophagy by regulating the expression of BECN1. With the increasing expression of exogenous TRIM59 in H1299 cells, the expression of BECN1 decreased in a concentration dependent manner ()). We next examined the effects of TRIM59 on the mRNA level of BECN1. Overexpression of TRIM59 decreased the BECN1 mRNA level, and knocking down TRIM59 significantly increased the mRNA level of BECN1 (). These results indicated that TRIM59 might regulate the transcription of BECN1. We generated constructs with different fragments of the BECN1 promoter region in the pGL3-enhancer vector and then performed a luciferase activity assay. From , we found that the promoter fragments + 1000 to −934 and + 500 to −934 showed higher activity than the others. We then detected the effects of TRIM59 on the activity of these promoter fragments. Overexpression of TRIM59 reduced the promoter activity of the + 1000 to −934 and + 500 to −934 fragments, whereas the + 1 to −934 and −500 to −934 fragments remained unchanged ()). These results proved that the promoter region containing + 500 to + 1 might be important for TRIM59 to regulate the transcription of BECN1.
Figure 2. TRIM59 affects the transcription of BECN1. (a) H1299 cells were transfected with pCMV-HA-TRIM59 (HA-TRIM59) with a gradual increase in the amounts of the plasmids. The expressions of BECN1 and exogenous TRIM59 were detected by western blot using anti-BECN1 and anti-HA antibodies. The BECN1 intensity was calculated by normalizing against ACTB. (b) The mRNA expression of BECN1 and TRIM59 in control and TRIM59-overexpressing conditions were determined by Q-PCR. Data represent the average of 3 independent experiments (mean± SD). (c) The mRNA expression of BECN1 and TRIM59 in control (CTL) and TRIM59-silenced conditions were determined by Q-PCR. Data represent the average of 3 independent experiments (mean± SD). (d) pGL3-enhancer vectors containing different fragments of the BECN1 promoter and Renilla control plasmid were co-transfected into H1299 cells. The relative levels of luciferase activity were normalized to the levels of the + 1000 to −934 fragment and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (e) pGL3-enhancer vectors containing different fragments of the BECN1 promoter were transfected into H1299 cells, co-transfected with Renilla control plasmid and HA-TRIM59 or empty vector. The relative levels of luciferase activity were normalized to the levels of the vector control and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (f) pGL3-enhancer vector containing the + 500 to −130 region of the BECN1 promoter were transfected into H1299 cells, co-transfected with Renilla control plasmid and pCMV-HA-TRIM59 plasmid. The relative levels of luciferase activity were normalized to the levels of vector control and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (g) pGL3-enhancer vector containing the + 500 to −130 region of the BECN1 promoter were transfected into H1299 cells, co-transfected with Renilla control plasmid and control siRNA or TRIM59 siRNAs. The relative levels of luciferase activity were normalized to the levels of control siRNA and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (h) pGL3-enhancer vector containing the + 500 to −130 region of the BECN1 promoter was transfected into H1299 cells, co-transfected with Renilla control plasmid and pcDNA3.0-RELA plasmid. The relative levels of luciferase activity were normalized to the levels of vector control and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (i) pGL3-enhancer vector containing the + 500 to −130 region of the BECN1 promoter was transfected into H1299 cells, co-transfected with Renilla control plasmid and control siRNA or RELA siRNAs. The relative levels of luciferase activity were normalized to the levels of control siRNA and to the levels of luciferase activity of the Renilla control plasmid. Data represent the average of 3 independent experiments (mean± SD). (j) The pCMV-HA-TRIM59 plasmid was transfected into H1299 cells. After 48 h, the expression of the indicated proteins was determined by western blot. The BECN1 intensity was calculated by normalizing against ACTB. (k) The control siRNA or TRIM59 siRNAs were transfected into H1299 cells. After 48 h, the expression of the indicated proteins was determined by western blot. The BECN1 intensity was calculated by normalizing against ACTB.
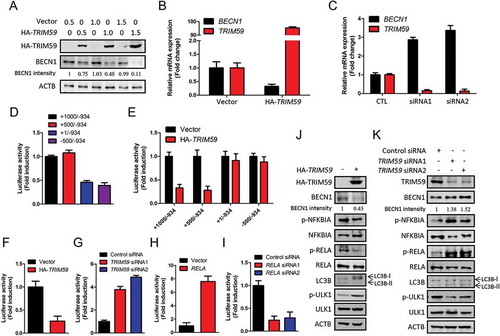
We next inserted the promoter region containing + 500 to −130 into the pGL3-enhancer vector and performed a luciferase activity assay in TRIM59-overexpressing and TRIM59-silenced conditions. and g show that the activity of this promoter region was sensitive to the expression level of TRIM59. The activity of this promoter fragment decreased when TRIM59 was overexpressed and increased when TRIM59 was knocked down. These results confirmed that TRIM59 regulated the transcription of BECN1 through the + 500 to + 1 region of the BECN1 promoter.
Because TRIM59 is not a transcription factor, we presumed that TRIM59 regulated the transcription of BECN1 through modulating the activity of a certain transcription factor. By analysis of the promoter region in + 500 to + 1, we identified a RELA/p65 (a key subunit of NFKB) binding site: GGGGATTTCC, located in + 317 to + 308. We next detected the promoter activity of the + 500 to −130 fragment by modulating the expression of RELA. The promoter activity increased remarkably when RELA was overexpressed and decreased when RELA was knocked down (, i)). Overexpressing RELA promoted the expression of BECN1, and knocking down RELA reduced the expression of BECN1 (Fig. S1A-S1D). These results demonstrated that RELA could regulate the transcription of BECN1. A similar result was also reported by Copetti et al. in 2009 [Citation32].
We next detected the effects of TRIM59 on the NFKB pathway and autophagy. Overexpression of TRIM59 blocked the activation of the NFKB pathway, as can be seen from the reduced expression of phosphorylated RELA and phosphorylated NFKBIA. ULK1 (unc-51 like autophagy activating kinase 1) is an important regulator in the autophagy pathway, and the phosphorylation of ULK1 at Ser757 is closely related to the repression of autophagy induction [Citation33]. TRIM59 overexpression increased the expression of p-ULK1 (Ser757) and decreased the expression of LC3-II, indicating the repression of autophagy ()), whereas TRIM59 knockdown activated the NFKB pathway and induced autophagy ()). According to these results, we demonstrated that TRIM59 could affect the transcription of BECN1 through regulating the NFKB pathway, and thus affected autophagy.
TRIM59 affects the ubiquitination level of BECN1
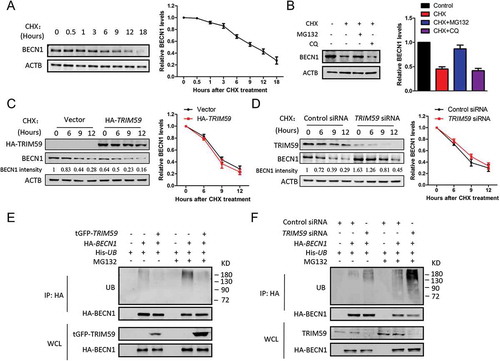
As TRIM59 possessed E3 ligase function [Citation11], we examined if TRIM59 could affect the stability of the BECN1 protein. We first tested the stability of BECN1 in physiological conditions by adding cycloheximide (CHX) to block protein translation in H1299 cells. The protein level of BECN1 decreased significantly when cells were treated with CHX for 6 h and this effect could be recovered by adding MG132, a proteasomal inhibitor, but not the lysosomal inhibitor chloroquine (CQ) (). These results indicated that BECN1 was degraded via a proteasomal degradation pathway. To determine whether TRIM59 regulates the stability of BECN1, we examined the protein levels of BECN1 in the presence of CHX under TRIM59-overexpressing or -silenced conditions. and d showed that TRIM59 did not affect the stability of BECN1. We next detected whether the ubiquitination of BECN1 was affected by TRIM59. Unexpectedly, the ubiquitination level of BECN1 was inversely related to TRIM59 expression levels. Knocking down TRIM59 remarkably increased the ubiquitination level of BECN1 and overexpressing TRIM59 had an opposite effect () and Fig. S2A, S2B). From these results, we concluded that TRIM59 did not affect the stability of the BECN1 protein through a proteasomal degradation pathway. However, TRIM59 strongly affected the ubiquitination level of BECN1 and these changes in ubiqutination did not influence the stability of the BECN1 protein.
Figure 3. TRIM59 affects the ubiquitination of BECN1. (a) H1299 cells were treated with 25 μg/ml cycloheximide (CHX) for different times and the expression of BECN1 was detected by western blot (left panel). BECN1 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (b) H1299 cells were treated with 25 μg/ml CHX alone or 25 μg/ml CHX plus 20 μM MG132 or 25 μg/ml CHX plus 20 μM chloroquine (CQ) for 9 h. The BECN1 expression was detected by western blot (left panel). BECN1 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (c) H1299 cells were transfected with HA-TRIM59, treated with 25 μg/ml CHX for different time points and then immunoblotted with antibodies against HA, BECN1 and ACTB. The BECN1 intensity was calculated by normalizing against ACTB (left panel). BECN1 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (d) H1299 cells were transfected with control or TRIM59 siRNA, treated with 25 μg/ml CHX for different time points and then immunoblotted with antibodies against TRIM59, BECN1 and ACTB. The BECN1 intensity was calculated by normalizing against ACTB (left panel). BECN1 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (e) H1299 cells were co-transfected with plasmids encoding HA-BECN1, His-UB and with or without tGFP-TRIM59. After 42 h, the cells were treated with or without MG132 for 6 h. Proteins were immunoprecipitated with HA antibody. The ubiquitination was detected using anti-UB antibody. Exogenous BECN1 and TRIM59 were detected using anti-HA and anti-tGFP antibodies. (f) H1299 cells were co-transfected with plasmids encoding HA-BECN1, His-UB and with or without TRIM59 siRNA. After 42 h, the cells were treated with or without MG132 for 6 h. Then, proteins were immunoprecipitated with HA antibody, and the ubiquitination was detected using anti-UB antibody. The exogenous BECN1 and endogenous TRIM59 were detected using anti-HA and anti-TRIM59 antibodies.
TRIM59 affects traf6-mediated ubiquitination of BECN1
Previous studies have demonstrated that upregulating K63-linked ubiquitination of BECN1 promotes autophagy [Citation34]. According to our previous results in , we supposed that TRIM59 might also regulate autophagy through modifying K63-linked ubiquitination of BECN1. To test this hypothesis, we used a K63R mutant of ubiquitin to test its effects on the ubiquitination of BECN1 when TRIM59 was knocked down. showed that the increased ubiquitination of BECN1 induced by TRIM59 knockdown was not observed when the wild-type ubiquitin was replaced with the K63R mutant. We used an antibody specific for K63-linked ubiquitin for western blot. The results revealed that TRIM59 knockdown significantly increased the K63-linked ubiquitination of BECN1 ()). TRAF6 was reported to directly ubiquitinate BECN1 through a K63 linkage [Citation34]. In H1299 cells, we found that TRAF6 could bind to BECN1 ()). Thus, we examined if TRIM59 affected TRAF6-mediated ubiquitination. We found that overexpression of TRAF6 increased the ubiquitination of BECN1, and overexpression of TRIM59 remarkably reduced the TRAF6-induced ubiquitination ()). These results indicated that, except for the influence on the transcription of BECN1, TRIM59 might also affect autophagy through interfering with TRAF6 mediated K63-linked ubiquitination of BECN1.
Figure 4. TRIM59 inhibits TRAF6-mediated ubiquitination of BECN1. (a) H1299 cells were co-transfected with plasmids encoding His-UB or His-UB-K63R, HA-BECN1 and either TRIM59 siRNA or control siRNA. Proteins were immunoprecipitated with HA antibody, and the ubiquitination was detected using an antibody specifically targeting ubiquitin. Exogenous BECN1 was detected using anti-HA antibody. Endogenous TRIM59 was detected using anti-TRIM59 antibody. (b) H1299 cells were co-transfected with His-UB, HA-BECN1 and either TRIM59 siRNA or control siRNA. Proteins were immunoprecipitated with HA antibody and the K63 linked ubiquitination was detected using an antibody specifically targeting ubiquitin that was K63 linked. (c) H1299 cells were co-transfected with plasmids encoding GFP-TRAF6 and either HA-BECN1 or empty vector. The cell lysates were immunoprecipitated with HA antibody and blotted with anti-HA or anti-GFP antibodies. (d) 293T cells were co-transfected with the indicated plasmids. Proteins were immunoprecipitated with HA antibody, and the ubiquitination was detected using an antibody specifically targeting ubiquitin. Exogenous BECN1, TRIM59 and TRAF6 were detected using anti-HA, anti-tGFP and anti-GFP antibodies. (e) H1299 cells stably expressing GFP-LC3B were co-transfected with pCMV-HA-TRAF6 (HA-TRAF6) and either pCMV-HA-TRIM59 (HA-TRIM59) or empty vector. After 48 h, the cells were analyzed by fluorescence microscopy (Olympus IX83). Scale bar: 10 μm (left panel). The cell numbers with GFP-LC3B puncta were counted under 200× magnification. *P ≤ 0.05, **P ≤ 0.01 (right panel). (f) H1299 cells stably expressing GFP-LC3B were co-transfected with a plasmid encoding HA-TRAF6 and either HA-TRIM59 or empty vector. After 46 h, the cells were treated with or without 20 μM chloroquine (CQ) for 2 h. The autophagy-related proteins were analyzed by western blot. (g) H1299 cells were co-transfected with the indicated exogenous genes and siRNAs. After 48 h, the cells were lysed and immunoprecipitated with HA antibody. FLAG-PIK3C3 and HA-BECN1 were detected using anti-FLAG and anti-HA antibodies. TRIM59 was detected using anti-TRIM59 antibody. (h) 293T cells were co-transfected with HA-BECN1 and the indicated exogenous genes. Two days later, the cells were lysed and immunoprecipitated with HA antibody. FLAG-PIK3C3 and HA-BECN1 were detected using anti-FLAG and anti-HA antibodies. GFP-TRAF6 and tGFP-TRIM59 were detected with anti-GFP and anti-tGFP antibodies.
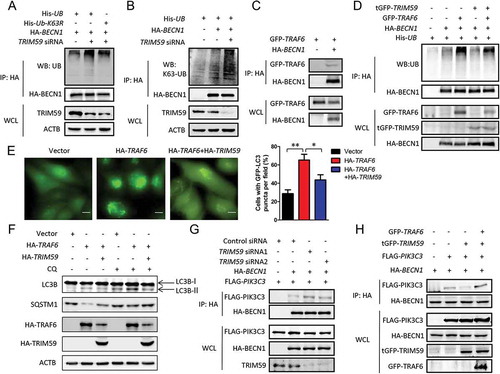
We wondered whether TRIM59-regulated TRAF6 induced autophagy. H1299 cells stably expressing GFP-LC3B were transfected with a plasmid encoding HA-TRAF6 alone or co-transfected with HA-TRAF6- and HA-TRIM59-encoding plasmids. The autophagy induced by TRAF6 overexpression was greatly prohibited by TRIM59 overexpression. The cell numbers with GFP-LC3B puncta were increased when transfecting with a TRAF6 plasmid alone, whereas a significant reduction was observed when the cells were co-transfected with plasmids encoding TRAF6 and TRIM59 ()). Overexpression of TRIM59 also decreased the expression of LC3B-II, whereas it increased SQSTM1 expression compared with overexpression of TRAF6 alone ()).
BECN1 plays a central role in autophagy through participating in the formation of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex [Citation1]. PIK3C3 is the catalytic subunit of PtdIns3K and interacts with BECN1 to form a complex when autophagy is induced. As TRIM59 could affect autophagy by modifying the ubiquitination of BECN1, we tested the effects of TRIM59 on the formation of the BECN1-PIK3C3 complex. From , we could see that TRIM59 knockdown enhanced the interaction between BECN1 and PIK3C3. However, overexpression of TRIM59 significantly reduced the PIK3C3 protein levels precipitated by HA-BECN1 and this effect was recovered by TRAF6 overexpression (). These results demonstrated that TRIM59 could also affect autophagy through regulating the TRAF6-induced K63-linked ubiquitination of BECN1, thus modulating the formation of the PtdIns3K complex.
TRIM59 regulates the degradation of TRAF6 by ubiquitination
We already proved that TRIM59 could interfere with the E3 ubiquitin ligase function of TRAF6. An important question that needed to be answered was how TRIM59 regulated TRAF6. As shown in , knocking down TRIM59 apparently increased the expression of TRAF6. When the expression of exogenous TRIM59 increased, the expression of TRAF6 decreased in a concentration-dependent manner ()). To investigate if TRIM59 affected the stability of TRAF6, we detected the stability of TRAF6 protein in physiological conditions by adding CHX for different times in H1299 cells. The expression of TRAF6 decreased significantly when treated with CHX for 3 h ()) and the decreased expression of TRAF6 could be recovered by adding MG132, but not CQ (Fig. S3A). This observation indicated that TRAF6 protein was degraded in a proteasomal degradation pathway.
Figure 5. TRIM59 regulates the degradation of TRAF6 by ubiquitination. (a) TRIM59 was knocked down in H1299 cells and protein expression was detected using the indicated antibodies. (b) H1299 cells were transfected with a plasmid encoding HA-TRIM59 with gradually increasing the amount of the plasmid. The expression of TRAF6 and TRIM59 was detected by western blot using anti-TRAF6 and anti-HA antibodies. (c) H1299 cells were treated with 25 μg/ml cycloheximide (CHX) for different times and the TRAF6 expression was detected by western blot (left panel). TRAF6 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (d) H1299 cells were transfected with a plasmid encoding HA-TRIM59, treated with 25 μg/ml CHX for different time points and then immunoblotted with antibodies against HA, TRAF6 and ACTB (left panel). TRAF6 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (e) H1299 cells were transfected with control or TRIM59 siRNA, treated with 25 μg/ml CHX for different time points and then immunoblotted with antibodies against TRIM59, TRAF6 and ACTB (left panel). TRAF6 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (f) H1299 cells were co-transfected with a plasmid encoding HA-TRAF6 and either tGFP-TRIM59 or empty vector. The cell lysates were immunoprecipitated with HA antibody and blotted with HA and tGFP antibodies. (g) The lysate of H1299 cells was immunoprecipitated using TRAF6 antibody or normal rabbit IgG, and blotted with TRAF6 and TRIM59 antibodies. (h) GST-TRIM59 purified with glutathione Sepharose beads was incubated with extracts from HA-TRAF6-transfected 293T cells. Western blot was performed to detect the indicated proteins using anti-HA and anti-GST antibodies. (i) H1299 cells were transfected with TRIM59 siRNAs. After 42 h, the cells were treated with MG132 for 6 h. The lysates were immunoprecipitated using TRAF6 antibody and blotted with the indicated antibodies. (j) H1299 cells were co-transfected with a plasmid encoding His-UB, HA-TRAF6 and either tGFP-TRIM59 or empty vector. After 42 h, the cells were treated with or without MG132 for 6 h. Proteins were immunoprecipitated with HA antibody. The ubiquitination was detected using an antibody specific for ubiquitin, and TRAF6 and TRIM59 were detected using anti-HA and anti-tGFP antibodies. (k) H1299 cells were co-transfected with a plasmid encoding His-UB, HA-TRAF6 and either tGFP-TRIM59 or empty vector. Proteins were immunoprecipitated with HA antibody. The K48-linked ubiquitination was detected using an antibody specifically targeting ubiquitin that was K48 linked. TRAF6 and TRIM59 were detected using anti-HA and anti-tGFP antibodies. (l) The purified GST-TRIM59 was incubated with the indicated reaction component. The reaction mixture was subjected to western blot using antibodies against ubiquitin, Flag and GST.
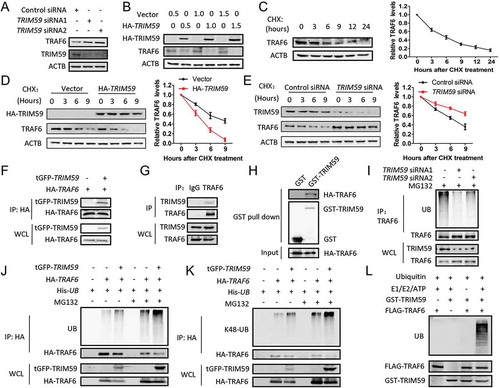
To determine whether TRIM59 regulates the stability of TRAF6, we examined the protein levels of TRAF6 in the presence of CHX under TRIM59-overexpressing or -silenced conditions. and e show that overexpression of TRIM59 accelerated the degradation rate of TRAF6, whereas knocking down TRIM59 stabilized the protein level of TRAF6. In order to figure out if TRIM59 could interacted with TRAF6, we performed co-immunoprecipitation and GST affinity isolation experiments and found that TRIM59 could bind to TRAF6 (-h) and Fig. S3B). As TRIM59 has an E3 ubiquitin ligase function, we detected the effect of TRIM59 on the ubiquitination of TRAF6. showed that TRIM59 knockdown reduced the ubiquitination level of endogenous TRAF6. Overexpression of TRIM59 increased the ubiquitination level of TRAF6, whereas it decreased the protein expression of TRAF6, indicating the ubiquitin-mediated degradation of TRAF6 ().
TRAF6 was reported to possess both K48-linked and K63-linked ubiquitination [Citation35–Citation39], we next used ubiquitin mutants specific for K48 (K48R) or K63 (K63R) to check the changes in the ubiquitination level of TRAF6 induced by TRIM59. The increased ubiquitination levels of TRAF6 induced by TRIM59 overexpression was not observed when transfecting with a plasmid encoding the K48R mutant; however, the increased ubiquitination levels of TRAF6 could still be observed when transfecting with a plasmid encoding the K63R mutant, indicating that TRIM59 induced a K48-linked ubiquitination of TRAF6 (Fig. S4A, S4B). We further examined these results using an antibody specific for K48-linked ubiquitination. showed that the enhanced ubiquitination of TRAF6 induced by TRIM59 was indeed K48-linked. To provide evidence that TRIM59 directly ubiquitinated TRAF6, we performed an in vitro ubiquitination assay. As can be seen in , the polyubiquitin bands were observed when purified TRAF6 was added to the reaction system containing ubiquitin, UBA1/UBE1, UBE2D1, ATP and TRIM59. All these results pointed out that TRIM59 could directly regulate the degradation of TRAF6 in a ubiquitin-mediated proteasomal degradation process.
As TRAF6 was originally described as an important regulator for the NFKB pathway [Citation40,Citation41], we wanted to explore if the inhibition of BECN1 expression was also dependent on TRAF6 degradation induced by TRIM59. Figure S5A and S5B show that TRAF6 overexpression could not rescue the reduction of both the mRNA and protein level of BECN1/BECN1 triggered by TRIM59. These results demonstrated that TRIM59 affected the expression of BECN1 though a TRAF6-independent NFKB pathway.
The ring domain of TRIM59 is important for regulating traf6-induced autophagy
The ring domain of TRIM family proteins has been shown to be essential for their protein degradation function [Citation10,Citation11]. To get further insight into the mechanism for TRIM59 to ubiquitinate TRAF6, we constructed a TRIM59 mutant lacking the ring domain (ΔR). We found that this mutant could still interact with TRAF6 ()). However, the mutant lost the ability to destabilize the TRAF6 protein ()). Besides, the ability to ubiquitinate TRAF6 was also abolished ()). These findings demonstrated that the ring domain of TRIM59 was important for the degradation of TRAF6.
Figure 6. The ring domain of TRIM59 is important for regulating TRAF6-induced autophagy. (a) H1299 cells were co-transfected with a plasmid encoding GFP-TRAF6 and HA-ΔR. The cell lysates were immunoprecipitated with HA antibody or normal mouse IgG, and blotted with HA and GFP antibodies. (b) H1299 cells were transfected with HA-ΔR, treated with 25 μg/ml CHX for different time points and then immunoblotted with antibodies against HA, TRAF6 and ACTB (left panel). TRAF6 expression relative to ACTB was quantified. Data represent the average of 3 independent experiments (mean± SD) (right panel). (c) H1299 cells were co-transfected with a plasmid encoding His-UB, HA-TRAF6 and either GFP-ΔR or empty vector. After 42 h, the cells were treated with or without MG132 for 6 h. Proteins were immunoprecipitated with HA antibody. The ubiquitination was detected using an antibody specific for ubiquitin, and TRAF6 and ΔR were detected using anti-HA and anti-GFP antibodies. (d) H1299 cells stably expressing GFP-LC3B were co-transfected with a plasmid encoding HA-TRAF6 and either HA-ΔR or empty vector. After 42 h, the cells were treated with or without 20 μM chloroquine (CQ) for 6 h. The autophagy-related proteins were analyzed by western blot.
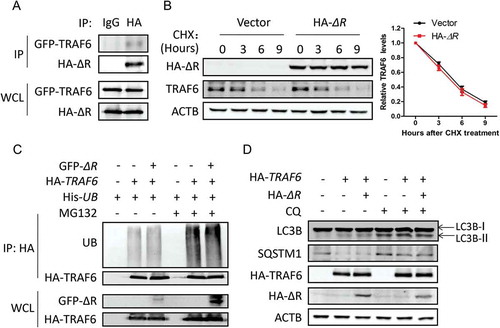
We next detected the inhibitory effect of ΔR on the TRAF6-induced autophagy. In cells co-transfected with plasmids encoding TRAF6 and ΔR, the expression of LC3-II was not changed when compared with the cells transfected with the TRAF6 plasmid alone. Also, the expression levels of SQSTM1 were similar between these 2 groups ()). In the group co-transfected with plasmids for HA-TRAF6 and HA-ΔR, the percentage of the cells with GFP-LC3B puncta was not changed significantly compared with the group transfected with the HA-TRAF6 plasmid alone (Fig. S6A). These data demonstrated that overexpressing ΔR did not block the autophagy process induced by TRAF6. Then, we tested the ubiquitination function of this mutant on BECN1. Overexpressing ΔR did not change the ubiquitination of BECN1, and the inhibitory effect of TRIM59 on autophagy was not observed for this mutant (Fig. S6B, S6C). Taken together, the ring domain of TRIM59 is important for regulating the ubiquitination of TRAF6 and the TRAF6-induced autophagy.
Both the transcription and the ubiquitination of BECN1 are necessary for the autophagy induced by TRIM59 knockdown
As TRIM59 affects both the expression and the K63-linked ubiquitination of BECN1, we next wanted to examine whether both effects were necessary for autophagy induced by TRIM59 knockdown. RELA siRNAs were co-transfected with TRIM59 siRNA into H1299 cells stably expressing GFP-LC3B. showed that the GFP-LC3B puncta were significantly increased when TRIM59 was knocked down. In addition, the percentage of the cells with GFP-LC3B puncta increased significantly; however, knocking down RELA decreased the total GFP-LC3B puncta and the cell numbers with GFP-LC3B puncta. The expression of LC3-Ⅱ was increased when knocking down TRIM59, whereas knocking down RELA reversed these effects. In , TRAF6 siRNAs were transfected into H1299 cells with TRIM59 knockdown. We discovered increased GFP-LC3B puncta and enhanced expression of LC3-II when TRIM59 was knocked down, whereas TRAF6 knockdown remarkably reduced the LC3B puncta and LC3-II expression induced by TRIM59 knockdown.
Figure 7. Both the transcription of BECN1 and the ubiquitination of BECN1 are necessary for the autophagy induced by TRIM59 knockdown. (a) TRIM59 siRNA was co-transfected with or without RELA siRNAs into H1299 cells stably expressing GFP-LC3B. After 48 h, the cells were analyzed by fluorescence microscopy (Olympus IX83). Scale bar: 10 μm (upper panel). The cell numbers with GFP-LC3B puncta were counted under 200× magnification. **P ≤ 0.01 (middle panel). The expression of related proteins was examined by western blot (bottom panel). (b) TRIM59 siRNA was co-transfected with or without TRAF6 siRNAs into H1299 cells stably expressing GFP-LC3B. After 48 h, the cells were analyzed by fluorescence microscopy (Olympus IX83). Scale bar: 10 μm (upper panel). The cell numbers with GFP-LC3B puncta were counted under 200× magnification. *P ≤ 0.05, **P ≤ 0.01 (middle panel). The expression of related proteins was examined by western blot (bottom panel). (c) HA-BECN1 was co-transfected with or without TRAF6 siRNAs into H1299 cells stably expressing GFP-LC3B. After 48 h, the cells were analyzed by fluorescence microscopy (Olympus IX83). Scale bar: 10 μm (upper panel). The cell numbers with GFP-LC3B puncta were counted under 200× magnification. **P ≤ 0.01, ***P ≤ 0.001 (middle panel). The expression of related proteins was examined by western blot (bottom panel).
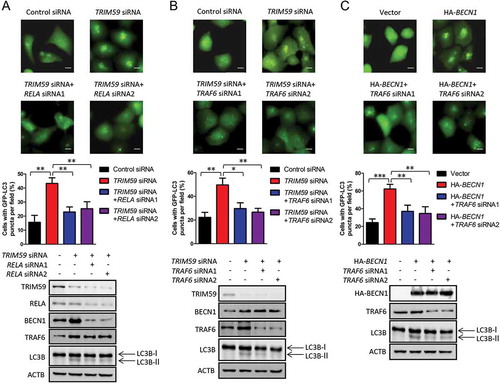
Noting that the expression level of BECN1 remained unchanged between the cells transfected with TRIM59 siRNA and the cells co-transfected with TRIM59 and TRAF6 siRNAs, we proposed that both the expression and the ubiquitination of BECN1 were important for autophagy induction. To further demonstrate this, we overexpressed BECN1 in H1299 cells stably expressing GFP-LC3B with or without TRAF6 knockdown. We found that BECN1 overexpression strongly induced autophagy as determined by increased cell numbers with GFP-LC3B puncta and enhanced expression of LC3-II, whereas TRAF6 knockdown decreased the effects ()). A previous study demonstrated that the Lys117 of BECN1 was the major ubiquitination site for K63-linkage and the Glu299 of BECN1 was important for the recruitment of TRAF6 to BECN1 [Citation34]. We then mutated these 2 residues of BECN1 and assessed the autophagy induction effects. As shown in Figure S7A-S7C, the autophagy induction effects for BECN1K117R and BECN1E299A were significantly attenuated compared with BECN1 wild-type. These results demonstrated that both the transcription and the ubiquitination of BECN1 were important for autophagy induced by TRIM59 knockdown.
Discussion
BECN1 plays a central role in autophagy that triggers a cascade of proteins involved in autophagosome formation [Citation1]. The regulation of BECN1 can occur at the level of transcription, translation and post-translational modifications [Citation8]. At the transcription level, the transcription factor E2F1 and NFKB can bind to the promoter region of the BECN1 gene to enhance transcription [Citation32,Citation42]. However, another transcription factor, SMAD2, negatively regulates the transcription of BECN1 and thus represses autophagy in endothelial cells [Citation43]. These studies demonstrate that modulating the expression of BECN1 is an important event in autophagy induction. In the current study, we found that the expression of TRIM59 and BECN1 were inversely correlated in NSCLC. Further studies demonstrated that TRIM59 repressed the transcription of BECN1. Although TRIM59 is not a transcription factor, it acted as a negative regulator of the NFKB pathway, and thus affected BECN1 expression. This was one of the mechanisms for TRIM59 to regulate autophagy determined in our study. TRIM59 has now been accepted as an important oncoprotein, and the molecular mechanisms for its tumor-promoting functions are very preliminary. Previous studies proved that TRIM59 could also affect the TP53 pathway, SMAD2/3 pathway and AKT pathway [Citation11,Citation44–Citation46]. These findings together with ours indicated that TRIM59 has many profound functions including the effect on autophagy, and these functions are important for cancer progression.
Post-translational modifications (PTMs) of BECN1 including phosphorylation, acetylation and ubiquitination are essential for autophagy induction [Citation47]. Ubiquitination of BECN1 affects either its protein stability or functions. NEDD4 was reported to ubiquitinate BECN1 with a K11-linkage for proteasomal degradation when PIK3C3/VPS34 is depleted [Citation48]. In another study, USP19 was reported to rescue the proteasomal degradation of BECN1 through removing the K11-linked ubiquitination [Citation49]. K63-linked ubiquitination of BECN1 is critical for TLR4-induced autophagy, and TRAF6 functions as an E3 ligase in this process [Citation34]. In our study, we found that except for the effect on the transcription of BECN1, TRIM59 knockdown induced autophagy accompanied by the upregulation of BECN1 ubiquitination. As TRIM59 functions as an E3 ligase, this result was in contrast with our initial idea that TRIM59 inhibited autophagy by ubiquitination and degradation of BECN1. What interesting results we found were that TRIM59 regulated K63-linked ubiquitination of BECN1 through direct ubiquitination and degradation of TRAF6, and the ring domain of TRIM59 was essential in this process. We finally demonstrated that both the transcriptional effects on BECN1 by RELA and the binding of TRAF6 to BECN1 were essential for autophagy induced by TRIM59 knockdown. In conclusion, we discovered the dual roles of TRIM59 in autophagy regulation by regulating both the transcription and K63-linked ubiquitination of BECN1.
Until now, many studies have proved that TRIM proteins are associated with autophagy. The expression of TRIM55 parallels that of autophagy proteins NBR1, SQSTM1 and LC3 during cardiac myofibril assembly and turnover [Citation15]. TRIM30 localizes in cytoplasmic bodies with ubiquitin chains and the autophagy marker LC3, and is degraded through both proteasome and autophagy pathways [Citation16]. TRIM11 can act as a receptor to recruit AIM2 to SQSTM1 for selective autophagy degradation upon DNA virus infection [Citation17]. Another study demonstrated that TRIM23 is essential for autophagy in response to viral infection and a TRIM23-TBK1-SQSTM1 axis is a key component in selective autophagy [Citation18]. TRIM13 regulates the initiation of autophagy during ER stress and TRIM13-induced autophagy is essential for ER stress-induced caspase activation and cell death [Citation19,Citation20]. The interaction between TRIM16 and LGALS3 orchestrates the recruitment of core autophagic factors and activates selective autophagy in response to damaged endomembranes [Citation21]. Besides, TRIM16 can also act as a receptor for IL1B and interact with SEC22B to recruit cargo to LC3-II-positive sequestration membrane in response to lysosomal damage, a process termed secretory autophagy [Citation22]. In ATG5- or ATG7-deficient cells, TRIM31 directly interacts with phosphatidylethanolamine (PE) in a palmitoylation-dependent manner, leading to the induction of autophagy. In this process, the B-box domain of TRIM31 is required for the formation of TRIM31-positive puncta and colocalization with PE [Citation23]. The E3 ubiquitin ligase complex of TRIM28 with its regulators MAGEA3 or MAGEA6 (MAGEA3/6-TRIM28) can target AMPK for ubiquitination and proteasome-mediated degradation, resulting in significantly reduced autophagy. However, in cells not expressing MAGEA3 or MAGEA6, TRIM28 functions as a pro-autophagy factor through sumoylating PIK3C3/VPS34 and promoting the formation of the PIK3C3-BECN1 complex [Citation24]. In another study, TRIM17 can inhibit certain types of selective autophagy by stabilizing the MCL1-BECN1 complex. However, TRIM17 can also promote the removal of midbodies through autophagy because of the absence of MCL1 from TRIM17-BECN1 complexes at midbodies [Citation25]. These 2 TRIM proteins (TRIM17 and TRIM28) further expand the roles of TRIM proteins in regulating autophagy by showing that a single TRIM can either positively or negatively regulate autophagy depending upon different conditions.
A recent comprehensive analysis demonstrated that some TRIM proteins act as platforms assembling activated ULK1 and BECN1 to induce autophagy. Besides, TRIM5/TRIM5α can also act as an autophagy receptor by directly recognizing its cognate target without a need for ubiquitin tagging [Citation26]. Another comprehensive study demonstrated that a subset of TRIM proteins (such as MEFV/TRIM20 and TRIM21) function as regulators and receptors for autophagy-targeting components of the inflammasome and type Ⅰ IFN response systems [Citation27]. These studies demonstrate that the functions of TRIM proteins in autophagy induction are diverse and each TRIM protein may have a specific function in autophagosome formation, suggesting that a particular TRIM protein does not compensate for the function of other TRIM proteins in autophagy induction. Although the function of TRIM59 in cancer progression has been intensively studied, the role of TRIM59 in autophagy induction has not been clarified yet. Thus, our studies point out another new way for the TRIM proteins to regulate autophagy though regulating the transcription and modulating the post-translational modifications of autophagy-related proteins.
Materials and methods
Cell culture and NSCLC patient samples
The non-small cell lung cancer (NSCLC) cell lines H1299, A549, H292, H358, H1975, H23, HCC827 from ATCC were cultured in RPMI 1640 (Invitrogen, C11875500BT), containing 10% fetal bovine serum (Excel, FCS100). The SPC-A1 cells from National Infrastructure of Cell Line Resource were cultured in RPMI 1640 (Invitrogen, C11875500BT), containing 10% fetal bovine serum (Excel, FCS100). The 293T cells were cultured in Dulbecco modified Eagle medium (DMEM, high glucose; Hyclone, SH30022.01B), containing 10% fetal bovine serum (Excel, FCS100). All the cells were incubated at 37℃ with 5% CO2. The NSCLC patient samples were kindly provided by Dr. Bentong Yu (Department of Cardiovascular Surgery, The First Affiliated Hospital of Nanchang University).
Reagents and plasmids
MG132 was purchased from Biovision (1791–5). Cycloheximide (CHX; C7698) and chloroquine (C7698) were bought from Sigma. Rapamycin was acquired from Gene Operation (IPA1021-0010MG). IPTG (isopropyl-β-D-thiogalactopyranoside) was purchased from Amresco (0487). TRIM59 siRNAs, RELA siRNAs and TRAF6 siRNAs were synthesized by Thermo Fisher Scientific. The plasmids pCMV-HA-BECN1, pEGFP-C2-LC3B, pEGFP-C2-TRAF6, pCMV-HA-TRAF6, pCMV-HA-TRIM59, pCMV-HA-ΔR, pEGFP-C2-ΔR, pcDNA3.0-RELA, pGEX-GST-TRIM59 were constructed for this study as follows: the genes were amplified by PCR from cDNA prepared from H1299 cells (the primers used were: pCMV-HA-BECN1 [EcoRI, KpnI], 5ʹ-GCGAATTCGGATGGAAGGGTCTAAGACGT-3ʹ [forward], 5ʹ-GCGGTACCTCATTTGTTATAAAATTGTG-3ʹ [reverse]; pEGFP-C2-LC3B [XhoI, KpnI], 5ʹ-GCCTCGAGCATGCCGTCGGAGAAGACCTT-3ʹ [forward], 5ʹ-GCGGTACCTTACACTGACAATTTCATCC-3ʹ [reverse]; pCMV-HA-TRAF6 [EcoRI, KpnI], 5ʹ-GCGAATTCGGATGAGTCTGCTAAACTGTGA-3ʹ [forward], 5ʹ-GCGGTACCCTATACCCCTGCATCAGTAC-3ʹ [reverse]; pEGFP-C2-TRAF6 [EcoRI, KpnI], 5ʹ-GCGAATTCATGAGTCTGCTAAACTGTGA-3ʹ [forward], 5ʹ-GCGGTACCCTATACCCCTGCATCAGTAC-3ʹ [reverse]; pCMV-HA-TRIM59 [SalI, KpnI], 5ʹ-GCGTCGACCATGCACAATTTTGAGGAAGAG-3ʹ [forward], 5ʹ-GCGGTACCTCAATGGGAAACTATTTTCC-3ʹ [reverse]; pGEX-GST-TRIM59 [BamHI, SalI], 5ʹ-GCGGATCCATGCACAATTTTGAGGAAGAG-3ʹ [forward], 5ʹ-GCGTCGACTCAATGGGAAACTATTTTCC-3ʹ [reverse]; pCMV-HA-ΔR [SalI, KpnI], 5ʹ-GCGTCGACCAGAAGTATTACTGAAATTGC-3ʹ [forward], 5ʹ-GCGGTACCTCAATGGGAAACTATTTTCC-3ʹ [reverse]; pEGFP-C2-ΔR [SalI, BamHI], 5ʹ-GCGTCGACCAGAAGTATTACTGAAATTGC-3ʹ [forward], 5ʹ-GCGGATCCTCAATGGGAAACTATTTTCC-3ʹ [reverse]; pcDNA3.0-RELA [KpnI, XhoI], 5ʹ-GCGGTACCATGGACGAACTGTTCCCCCT-3ʹ [forward], 5ʹ-GCCTCGAGTTAGGAGCTGATCTGACTCA-3ʹ [reverse]); the PCR products was purified using a Gel Extraction Kit (Omega Bio-tek, D2500-02), digested with the indicated restriction endonucleases, re-purified and ligated into the digested and purified vectors (pCMV-HA [Clontech, 631,604], pEGFP-C2 [Clontech, 6083–1], pcDNA3.0 [Invitrogen, A-150,228], pEGX-6P-1 [Amersham Pharmacia Biotech, 27–4597-01]) using T4 DNA ligase (New England Biolabs, M0202L). The pGL3-enhancer vectors containing different fragments of the BECN1 promoter were constructed for this study as follows: the BECN1 promoter fragments were amplified by PCR from human genomic DNA extracted from H1299 cells and cloned into the KpnI-XhoI restriction sites of pGL3-enhancer vectors (Promega, E1771). The plasmid pcDNA3.1-FLAG-PIK3C3 was kindly provided by Dr. Qimin Zhan (Chinese Academy of Medical Sciences and Peking Union Medical College). The plasmid pCMV6-AC-GFP-TRIM59 was purchased from OriGene (PS100010). The mutant plasmids (BECN1-K117R, BECN1-E299A) were made by Generay Biotech.
Antibody against BECN1 ordered from OriGene (TA502127) was used to detect the expression of BECN1 in human tissues and cell lines. The BECN1 antibody ordered from Proteintech (11,306–1-AP) was used to perform immunoprecipitation. The normal rabbit IgG and normal mouse IgG were purchased from Santa Cruz Biotechnology (sc-2027, sc-2025). Mouse anti-ACTB and anti-GFP were purchased from Proteintech (66,009–1-lg, 66,002–1-lg). Mouse anti-tGFP and anti-SQSTM1 monoclonal antibody were from OriGene (TA150041, TA502127). Mouse anti-HA monoclonal antibody was ordered from Thermo Fisher Scientific (26,183). Commercial rabbit polyclonal antibodies anti-LC3B, anti-TRAF6, anti-GST Tag and anti-UB were purchased from Proteintech (18,725–1-AP, 12,809–1-AP, 10,000–0-AP, 10,201–2-AP). The TRAF6 antibody purchased from Cell Signaling Technology (8028) was used for immunoprecipitation. Antibody against TRIM59 was purchased from Sigma (HPA017759). RELA antibody was ordered from Cell Signaling Technology (8242). The NFKB pathway sample kit was used to detect the activation of the NFKB pathway (Cell Signaling Technology, 9936). The K48-linkage-specific polyubiquitin antibody and K63-linkage-specific polyubiquitin antibody were purchased from Cell Signaling Technology (8081, 5621). The ULK1 and p-ULK1 (Ser757) antibodies were ordered from Cell Signaling Technology (6439, 14,202).
The recombinant human ubiquitin and His-tag ubiquitin activating enzyme (His-UBA1/UBE1) were purchased from UB-biotech (UB-100H-5M, UBE-024). The recombinant His-tag UBE2D1 was ordered from Sino Biological (11,432-H07E-50). The recombinant protein of TRAF6 was purchased from OriGene (TP319528). The Dual-Luciferase Reporter Assay System was purchased from Promega (E1910).
Western blot
After transfection with plasmids or siRNAs for 48 h, cells were lysed in RIPA buffer (Beyotime, P0013), containing protease inhibitor cocktail (Sigma, P2714) and phenylmethylsulfonyl fluoride (DINGGUO, WB0180), and the protein concentrations were measured using the BCA Protein Assay Kit (Pierce Biotechnology, 23,225). Total proteins were subjected to 10% or 12% SDS-PAGE and transferred to PVDF membranes (Millipore, IPVH00010). The membranes were blocked with 5% skim milk (BD, 232,100) for 1 h at room temperature and incubated with the indicated antibodies overnight at 4°C. The membranes were washed 3 times at room temperature with 1× TBST (20 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20) for 10 min every time, followed by incubation for 1 h at room temperature with horseradish peroxidase-conjugated anti-mouse (Thermo Fisher Scientific, 31,430) or anti-rabbit (Thermo Fisher Scientific, 31,460) secondary antibodies. Protein bands were visualized after incubation using the Pro-Light chemiluminescence detection kit (TIANGEN, PA112-01) and a digital gel image analysis system (TANON 5500), and the band intensities were quantified with Tanon GIS software.
Immunoprecipitation
After cells were lysed with RIPA buffer (Beyotime, P0013) containing PMSF (DINGGUO, WB0181) and cocktail, the cell lysates were precleaned with protein G agarose (Roche, 11,243,233,001) at 4℃ for 1 h, then the supernatant were incubated with the indicated antibodies and protein G agarose beads at 4℃ overnight. On the second day, immunocomplexes combined with beads were washed with lysis buffer, followed by western blot.
GST affinity isolation
The GST-TRIM59 protein was induced in E. coli using 0.5 mM IPTG (isopropyl-β-D-thiogalactopyranoside [Amresco, 0487]) at 37℃ for 5 h. The cells were harvested through centrifugation at 10000g for 10 min. After that, the cells were resuspended in phosphate-buffered saline (PBS [Solarbio, P1020] containing 1% Triton X-100 [Solarbio, T8200], 1× protease inhibitor), and lysozyme (Solarbio, L1080) was added to reach a concentration of 1mg/ml. The cells were then incubated on ice for 1 h and frozen completely at −80℃ overnight. Then, samples were thawed on ice and the pellets were broken by sonication. After centrifugation at 12,000g for 20 min, the supernatants were transferred to fresh tubes and glutathione Sepharose beads (GE, 17–0756-01) were added and incubated at 4℃ overnight. The GST fusion proteins conjugated to glutathione Sepharose beads were washed with PBS 3 times and then mixed with cell lysate at 4℃ for 8 h. After that, the beads were washed with PBS 3 times and boiled with loading buffer. The binding proteins were detected using western blot.
Luciferase activity assay
The human genomic DNA was extracted from H1299 cells, the BECN1 promoter fragments were amplified by PCR and cloned into the KpnI/XhoI restriction sites of pGL3-enhancer vectors. The primers used were:
Forward primer: 5ʹ-GCGGTACCTTGCCCAGGCTGGAGTGCAG-3ʹ (+ 1000 to −934),
Reverse primer: 5ʹ-GCCTCGAGCTCTGTGCTCTTGCTGTTCC-3ʹ (+ 1000 to −934);
Forward primer: 5ʹ-GCGGTACCGTGTCCAGTTTCAGGGGCTG-3ʹ (+ 500 to −934),
Reverse primer: 5ʹ-GCCTCGAGCTCTGTGCTCTTGCTGTTCC-3ʹ (+ 500 to −934);
Forward primer: 5ʹ-GCGGTACCCGCCGAGACCGGACGTGACG-3ʹ (+ 1 to −934),
Reverse primer: 5ʹ-GCCTCGAGCTCTGTGCTCTTGCTGTTCC-3ʹ (+ 1 to −934);
Forward primer: 5ʹ-GCGGTACCCAACAACAAAAGGCCGGGCA-3ʹ (−500 to −934),
Reverse primer: 5ʹ-GCCTCGAGCTCTGTGCTCTTGCTGTTCC-3ʹ (−500 to −934);
Forward primer: 5ʹ-GCGGTACCGTGTCCAGTTTCAGGGGCTG-3ʹ (+ 500 to −130),
Reverse primer: 5ʹ-GCCTCGAGGTGGTAACCTTGTTCATCCG-3ʹ (+ 500 to −130).
For transient transfection, pGL3-enhancer vector containing BECN1 promoter fragments were transfected into H1299 cells using SuperFectin II in vitro DNA transfection reagent (Pufei, Shanghai, 2102–100). Forty-eight h after transfection, cells were lysed and luciferase activity was detected using the Dual-Luciferase reporter assay kit (Promega, E1910). The relative levels of luciferase activity were normalized to the levels of luciferase activity of the Renilla control plasmid.
RNA purification and Q-PCR analysis
Total RNA was extracted by TRIzol reagent (Invitrogen, 15,596–026). The cDNA was synthesized using PrimeScript RT reagent kit (Takara, RR047A). Q-PCR experiments were performed with a SYBR Green Premix Ex Taq II kit (Takara, RR820A), then we used the RT-PCR System-Applied Bio-system to detect mRNA expression of target genes using GAPDH as a control. The comparative Ct method was used to calculate the relative amount of mRNA expression of target genes. All Q-PCR data were obtained using an ABI ViiATM 7 Real-Time PCR System.
In vitro ubiquitination assay
The in vitro ubiquitination assay was performed as previously described with minor modifications [Citation50]. The assay was performed in a 30-μl reaction volume containing some or all of the following components: 50 ng of UBA1/UBE1, 200 ng of UBE2D1, 500 ng of purified TRAF6, 5 μg of ubiquitin and 1.5 μl 20× reaction buffer (1 M Tris, pH 7.5, 40 mM ATP [Sigma, A1852], 100 mM MgCl2, 40m M DTT). The reaction was initiated by incubation with GST-tagged TRIM59 purified from bacteria with glutathione Sepharose beads. The reaction was performed at 30℃ for 1.5 h with rotation and stopped by adding 2× loading buffer. The assay was analyzed by western blot.
Gene overexpression and knockdown
For gene knockdown experiments, the cells were seeded 18–24 h prior to transfection. A nonspecific oligonucleotide from Thermo Fisher Scientific was used as a negative control. The siRNAs were transiently transfected using SuperFectin siRNA Transfection Reagent (Pufei, 2103–100). After 48 h, the knockdown efficiency was determined by western blot using appropriate antibodies. For gene overexpression, the cells were transfected with the indicated plasmids using SuperFectin DNA Transfection Reagent kit (Pufei, 2102–100). After 48 h, the transfection efficiency was determined by western blot using relevant antibodies.
Statistical analysis
Data are presented as means ± SD. The unpaired t-test was used to make the statistical comparisons, P-value≤ 0.05 was considered to be statistically significant.
Supplemental Material
Download MS Word (2.6 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41.
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662.
- Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676.
- Aita VM, Liang XH, Murty VV, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65.
- Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep. 2015;7:18.
- Jung YY, Lee YK, Koo JS. The potential of beclin 1 as a therapeutic target for the treatment of breast cancer. Expert Opin Ther Targets. 2016;20:167–178.
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nature Reviews Molecular Cell Biology. 2007;8:931–937.
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 2005;27:1147–1157.
- Zhou Z, Ji Z, Wang Y, et al. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology. 2014;147:1043–1054.
- Pathiraja TN, Thakkar KN, Jiang S, et al. TRIM24 links glucose metabolism with transformation of human mammary epithelial cells. Oncogene. 2015;34:2836–2845.
- Cambiaghi V, Giuliani V, Lombardi S, et al. TRIM proteins in cancer. Adv Exp Med Biol. 2012;770:77–91.
- Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, et al. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity. 2013;38:384–398.
- Perera S, Holt MR, Mankoo BS, et al. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev Biol. 2011;351:46–61.
- Choi UY, Choi WY, Hur JY, et al. Polyubiquitin chain-dependent protein degradation in TRIM30 cytoplasmic bodies. Exp Mol Med. 2015;47:e159.
- Liu T, Tang Q, Liu K, et al. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 2016;16:1988–2002.
- Sparrer KMJ, Gableske S, Zurenski MA, et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat Microbiology. 2017;2:1543–1557.
- Tomar D, Singh R, Singh AK, et al. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim Biophys Acta. 2012;1823:316–326.
- Tomar D, Prajapati P, Sripada L, et al. TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim Biophys Acta. 2013;1833:3134–3144.
- Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 Co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27.
- Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60.
- Ra EA, Lee TA, Won Kim S, et al. TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat Commun. 2016;7:11726.
- Pineda CT, Potts PR. Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy. 2015;11:844–846.
- Mandell MA, Jain A, Kumar S, et al. TRIM17 contributes to autophagy of midbodies while actively sparing other targets from degradation. J Cell Sci. 2016;129:3562–3573.
- Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30:394–409.
- Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210:973–989.
- Zhan W, Han T, Zhang C, et al. TRIM59 promotes the proliferation and migration of non-small cell lung cancer cells by upregulating cell cycle related proteins. PloS one. 2015;10:e0142596.
- Valiyeva F, Jiang F, Elmaadawi A, et al. Characterization of the oncogenic activity of the novel TRIM59 gene in mouse cancer models. Mol Cancer Ther. 2011;10:1229–1240.
- Khatamianfar V, Valiyeva F, Rennie PS, et al. TRIM59, a novel multiple cancer biomarker for immunohistochemical detection of tumorigenesis. BMJ open. 2012;2:e001410.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. (3rd edition). Autophagy. 2016;12:1–222.
- Copetti T, Bertoli C, Dalla E, et al. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29:2594–2608.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42.
- Miggin SM, O’Neill LA. New insights into the regulation of TLR signaling. J Leukoc Biol. 2006;80:220–226.
- Zhou L, Ma Q, Shi H, et al. NUMBL interacts with TRAF6 and promotes the degradation of TRAF6. Biochem Biophys Res Commun. 2010;392:409–414.
- Zhao W, Wang L, Zhang M, et al. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J Immunology. 2012;188:2567–2574.
- Lin XW, Xu WC, Luo JG, et al. WW domain containing E3 ubiquitin protein ligase 1 (WWP1) negatively regulates TLR4-mediated TNF-alpha and IL-6 production by proteasomal degradation of TNF receptor associated factor 6 (TRAF6). PloS one. 2013;8:e67633.
- Muroi M, Tanamoto K. IRAK-1-mediated negative regulation of toll-like receptor signaling through proteasome-dependent downregulation of TRAF6. Biochim Biophys Acta. 2012;1823:255–263.
- Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–38108.
- Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234.
- Wang B, Ling S, Wc L. 14-3-3Tau regulates beclin 1 and is required for autophagy. PloS one. 2010;5:e10409.
- Pan CC, Kumar S, Shah N, et al. Endoglin regulation of smad2 function mediates beclin1 expression and endothelial autophagy. J Biol Chem. 2015;290:14884–14892.
- Zhang Y, Yang WB. Down-regulation of tripartite motif protein 59 inhibits proliferation, migration and invasion in breast cancer cells. Biomed Pharmacotherapy. 2017;89:462–467.
- Chen W, Zhao K, Miao C, et al. Silencing Trim59 inhibits invasion/migration and epithelial-to-mesenchymal transition via TGF-beta/Smad2/3 signaling pathway in bladder cancer cells. Onco Targets Ther. 2017;10:1503–1512.
- Sun Y, Ji B, Feng Y, et al. TRIM59 facilitates the proliferation of colorectal cancer and promotes metastasis via the PI3K/AKT pathway. Oncol Rep. 2017;38:43–52.
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25:354–363.
- Platta HW, Abrahamsen H, Thoresen SB, et al. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem J. 2012;441:399–406.
- Cui J, Jin S, Wang RF. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy. 2016;12:1210–1211.
- Wertz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699.